

Abstract
Retinitis pigmentosa (RP) is a genetically heterogeneous retinal degeneration characterized by photoreceptor death, which results in visual failure. Here, we used a combination of homozygosity mapping and exome sequencing to identify mutations in ARL2BP, which encodes an effector protein of the small GTPases ARL2 and ARL3, as causative for autosomal-recessive RP (RP66). In a family affected by RP and situs inversus, a homozygous, splice-acceptor mutation, c.101−1G>C, which alters pre-mRNA splicing of ARLBP2 in blood RNA, was identified. In another family, a homozygous c.134T>G (p.Met45Arg) mutation was identified. In the mouse retina, ARL2BP localized to the basal body and cilium-associated centriole of photoreceptors and the periciliary extension of the inner segment. Depletion of ARL2BP caused cilia shortening. Moreover, depletion of ARL2, but not ARL3, caused displacement of ARL2BP from the basal body, suggesting that ARL2 is vital for recruiting or anchoring ARL2BP at the base of the cilium. This hypothesis is supported by the finding that the p.Met45Arg amino acid substitution reduced binding to ARL2 and caused the loss of ARL2BP localization at the basal body in ciliated nasal epithelial cells. These data demonstrate a role for ARL2BP and ARL2 in primary cilia function and that this role is essential for normal photoreceptor maintenance and function.
Main Text
Retinitis pigmentosa (RP [MIM 268000]), the most frequent inherited retinal degeneration, comprises a group of conditions characterized by the initial loss of rod photoreceptors and the resulting impaired night vision followed by progressive visual-field constriction as both rod and cone photoreceptors die.1,2 The disorder exhibits striking genetic heterogeneity and can be inherited as an autosomal-dominant, autosomal-recessive, or X-linked trait. More than 45 distinct RP-associated genes and/or loci have been reported to date and include 23 genes associated with nonsyndromic autosomal-recessive RP (RetNet, see Web Resources). Notably, mutations in cilia-associated genes account for at least 36% of genetically diagnosed cases of RP.3 Within this ciliopathy category, mutations can cause a broad spectrum of phenotypes ranging from isolated RP to more complex syndromic ciliopathies (of which RP is a component), such as Usher syndrome, Bardet-Biedl syndrome, Meckel-Gruber syndrome, and Senior-Loken syndrome.4,5
We investigated RP-affected families with evidence of parental consanguinity. The study was approved by the local research ethics committees at Moorfields Eye Hospital and the Hadassah Hebrew Medical Center, and all investigations were conducted in accordance with the principles of the Declaration of Helsinki. Informed consent was obtained from all participating individuals. One family of Arab-Muslim origin (family MOL0807; Figure 1A) consisted of three siblings affected by autosomal-recessive RP. The clinical diagnosis of RP was made in their twenties, after complaints of night vision and visual-field impairment. Individual IV-2 (Figure 1A), at the age of 33 years, had a visual acuity of 0.2 in the right eye (RE) and 0.4 in the left eye (LE), and his Goldmann visual fields were constricted to 10°–12° with the V4e target. When she was 36 years old, his older sister (IV-1; Figure 1A) had severely impaired vision: she could detect hand motion in the RE and had bare light perception in the LE. There were no significant refractive errors. Mild posterior subcapsular cataracts were present, and fundoscopy revealed optic-disc pallor, mild-to-moderate bone-spicule-like pigmentation in the midperiphery, and attenuated retinal blood vessels, all typical signs of RP. Interestingly, all three siblings displayed a marked component of macular atrophy, including smaller and larger atrophic patches, as well as epiretinal membranes with wrinkling of the retina (Figures 2A–2D). Neither photopic nor scotopic electroretinogram (ERG) responses were detectable. Audiometric testing was within normal limits in all three siblings, and computed-tomography imaging revealed full thoracic and abdominal situs inversus in individuals IV-1 and IV-2 but normal situs composition (situs solitus) in individual IV-3.
Figure 1.
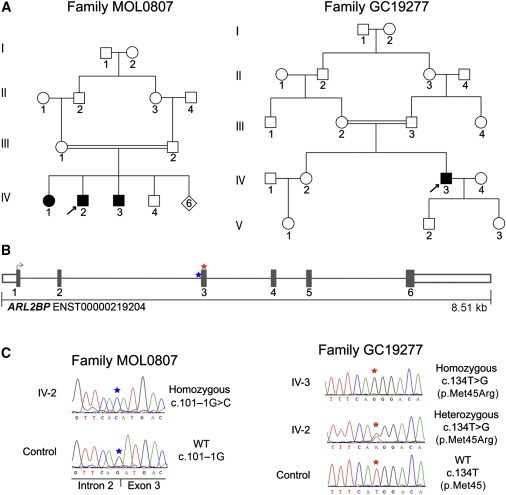
Identification of Homozygous ARL2BP Variants in Two Unrelated Consanguineous Families Affected by Autosomal-Recessive RP
(A) Pedigrees analyzed in this study. On the left is family MOL0807; homozygosity mapping was performed on individuals IV-1 and IV-2, and whole-exome sequencing (WES) was performed with DNA from individual IV-2. On the right is family GC19277; homozygosity mapping and WES were performed with DNA from individual IV-3.
(B) A schematic of the genomic structure of ARL2BP depicts the position of the mutations identified in this study. A homozygous splice-site mutation, c.101–1G>C (IVS2–1G>C), was identified in individuals IV-1, IV-2, and IV-3 from family MOL0807 and is highlighted with a blue star. A homozygous missense mutation, c.134T>G (p.Met45Arg), was identified in individual IV-3 from family GC19277 and is highlighted with a red star.
(C) Electropherograms of the mutations identified in this study. On the left, traces are shown for individual IV-2 (from family MOL0807), homozygous for the c.101–1G>C splice mutation, and a control individual. On the right, traces are shown for individual IV-3 (from family GC19277), homozygous for the c.134T>G (p.Met45Arg) missense variant. Traces are also shown for his unaffected sister (IV-2), who is heterozygous for the same variant, and an unrelated control sample. Both parents (III-2 and III-3) were also found to be heterozygous for the variant. All traces are shown in the forward orientation.
Figure 2.

Retinal Imaging of Individuals with RP
(A–D) Fundus imaging of the RE and LE of individual IV-3 from family MOL0807 at the age of 31 years. Heidelberg MultiColor imaging (A), infrared photos (B and D, left panels), and fundus autofluorescence images (C) show significant involvement of the macular area with patches of atrophy surrounding the fovea, as well as smaller spots of retinal pigment epithelium (RPE) dropout throughout the posterior pole. Optical coherence tomography (OCT) scans (B and D, right panels) show a relatively preserved photoreceptor layer in the fovea, and marked thinning of this layer is evident in the parafoveal regions.
(E) Fundus autofluorescence imaging of the right and left retinas of individual IV-3 (family GC19277) at the age of 48 years. Irregular peripheral autofluorescence consistent with RPE dysfunction or loss is evident. A parafoveal ring of increased density is noted; a similar appearance has been previously described in individuals with RP and defects in photoreceptor-cilium-related genes.6
(F) Linear OCT scans of the left and right retinas of IV-3 in family GC19277. The hyperreflective line corresponding to the inner segment ellipsoid is relatively preserved at the fovea but is absent more peripherally, suggesting loss of photoreceptor outer segments. Outside the fovea, observed thinning of the photoreceptor cell layer is consistent with extensive photoreceptor degeneration.
All scale bars represent 200 μm.
DNA samples from individuals IV-1 and IV-2 (family MOL0807) were analyzed with high-density genome-wide SNP microarrays (Affymetirx SNP 6.0) according to the manufacturer’s recommendations. Seven shared regions of homozygosity greater than 5 Mb were identified and were found to encompass multiple genes associated with retinal disease (Table S1, available online). Subsequent whole-exome sequencing (WES) using a Roche NimbleGen V2 preparation kit and the HiSeq2000 sequencer (Illumina) was performed with DNA from individual IV-2. The DNAnexus software package was used for aligning reads to the human reference sequence (UCSC Genome Browser hg19) and for calling and annotating sequence variants. No variants in any known retinal-disease-associated genes, including those present within large regions of homozygosity, were identified (Table S1).
On the basis of the hypothesis that RP-associated mutations are rare, calls with a minor allele frequency over 0.5% in the 1000 Genomes database and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS) were filtered. Furthermore, on the basis of the consanguineous ancestry observed, variants identified within regions of homozygosity were examined initially. A homozygous splice-site variant, c.101−1G>C (IVS2−1G>C; Figure 1C), in ARL2BP (RefSeq accession number NM_012106.3) was identified in a large homozygous interval (Table S1). This variant alters the invariant AG dinucleotide at the splice acceptor site of intron 2 and segregates with disease in the family (Figure 1C). This sequence change is absent from both dbSNP and 1000 Genomes and is not present in DNA from 100 ethnically matched Arab-Muslim control individuals. Furthermore, the variant is absent from 12,996 control haplotypes from the NHLBI EVS.
RT-PCR analysis of RNA extracted from blood samples of the three affected siblings and their unaffected sibling, who did not carry the mutation, revealed that the normal spliced ARL2BP transcript was absent in all individuals homozygous for the c.101−1G>C variant. Instead, multiple PCR products were found to be present (Figure S1) and were investigated by Sanger sequencing. The five amplified products were all identified to represent abnormal splicing events resulting in transcripts with a frameshift after 33 or 34 codons of the ARL2BP-coding sequence and the introduction of premature termination codons (Figure S1). We therefore predict that the identified splicing mutation results in the lack of full-length ARL2BP in vivo.
In parallel, a 48-year-old white male (IV-3 in family GC19277) of European descent, with evidence of parental consanguinity (Figure 1A), and diagnosed with RP and primary ciliary dyskinesia (PCD [MIM 244400]; Table S2 and Movie S1) was included in the study. He had respiratory failure from an early age and required physiotherapy regularly through the school years. He had recurrent otitis media, progressively affecting his hearing. In his twenties, he noticed night-vision problems, which led to a diagnosis of RP, and an ERG confirmed severe rod and cone dysfunction. There was progressive loss of peripheral vision and, more recently, of central vision. There was no family history of any of these conditions. When he was 48 years old, his visual acuities were 0.25 in the RE and 0.5 in the LE and visual fields were constricted to 10°. Fundus examination revealed widespread retinal degeneration with only sparse bone-spicule pigmentation (Figures 2E and 2F).
On the basis of the proband’s consanguineous ancestry, DNA from individual IV-3 (family GC19277) was analyzed by homozygosity mapping, as described previously. Thirteen chromosomal segments over 5 Mb were identified and were found to encompass multiple retinal-disease-associated genes (RetNet), in addition to two recently identified PCD-associated genes, DNAAF3 (MIM 614566)7 and HYDIN (MIM 610812)8 on chromosomes 19 and 16, respectively (Table S3). WES was performed with the Illumina TruSeq Exome Enrichment Kit and the HiSeq 2000 sequencer (Illumina). Reads were aligned to the human reference sequence (UCSC Genome Browser hg19) with Novoalign (Novocraft) version 2.05. The ANNOVAR tool (OpenBioinformatics) was used for annotating SNPs and small indels. ExomeDepth9 was used for calling copy-number variants.
A rare (1/12,331 control alleles [NHLBI EVS]) homozygous splice-site variant was identified in HYDIN, a gene recently associated with PCD8 (Figure S2). The variant alters the invariant splice acceptor site of intron 24, c.3786–1G>T (RefSeq NM_001270974.1). RT-PCR analysis of RNA extracted from blood samples of individual IV-3 (family GC19277) and an unrelated control individual revealed that the variant leads to aberrant pre-mRNA splicing (Figure S3). To date, two loss-of-function HYDIN variants have been reported in nine individuals affected by PCD and hearing loss but with a normal situs composition (situs solitus) and absence of any retinal dystrophy, congenital heart disease, or hydrocephalus.8 This is thought to be because HYDIN-deficient cilia lack the C2b projection of the central-pair apparatus, which is important for 9+2 motile cilia function, but not for 9+0 primary cilia.8 Electron microscopy of nasal epithelial cells of individual IV-3 (family GC19277) confirmed the absence of the C2b projection of the motile cilia (Figure S4). The PCD phenotype and hearing loss observed in this subject are therefore consistent with loss of HYDIN function and resemble the phenotype of individuals already reported. However, this did not explain his retinal degeneration.
We examined the WES data for possible causes of RP. Overall, 22,778 exonic sequence alterations were identified in individual IV-3’s (family GC19277 in Figure 1) exome compared to the human reference sequence (UCSC Genome Browser hg19). The average sequencing depth on target was 61×, and 87.9% of the targeted region was covered with a minimum read depth of 10. No disease-associated variants, including those present within large regions of homozygosity, were identified in genes previously associated with RP or any other degenerative retinal phenotypes (Table S3). Sanger sequencing excluded the presence of causal variants in ORF15, the terminal exon of the retina-enriched RPGR (MIM 312610) transcript. Mutations in this X-linked gene are associated with RP and, in some instances, additional ciliopathy-related features including sinorespiratory infections and deafness.10
WES data were filtered as described for family MOL0807. In total, six rare nonsynonymous variants were identified in the homozygous regions. A homozygous missense variant (c.134T>G [p.Met45Arg]) present in ARL2BP within the largest region of homozygosity on chromosome 16 was considered most likely to be pathogenic (Figure 1C and Table S3). The variant was found to segregate with disease in the family (Figure 1C). This sequence change is absent from both dbSNP and 1000 Genomes and is not present in DNA from 192 ethnically matched control individuals (Human Random Control DNA Panels). Furthermore, the variant is absent from 12,996 control haplotypes (NHLBI EVS). The Met45 residue is located in an evolutionarily conserved region of the protein (Figure S5), and bioinformatics programs SIFT, PolyPhen-2, and Blosum62 support the likely pathogenicity of the identified ARL2BP missense variant (Table S4).
ARL2BP represents an attractive candidate for cilia-related disorders, such as RP, because ARF-like GTPases, their effectors, and GTPase-activating protein (GAP) play critical roles in cilia formation and signaling.11,12 ARL3-knockout mice have a phenotype consistent with primary cilia dysfunction,13 and alterations in the ARL3 GAP, encoded by RP2, cause X-linked RP (MIM 300757).14,15 ARL3 shares a high degree of structural, and possibly functional, conservation with ARL2, and both proteins share a highly overlapping set of effector proteins, such as ARL2BP.16 However, neither ARL2 nor ARL2BP has been previously implicated in human disease.
The identification of rare homozygous ARL2BP variants in four RP-affected individuals from two unrelated families led us to assess whether mutations in ARL2BP are a common cause of adult-onset autosomal-recessive retinal degeneration. Bidirectional Sanger sequencing of ARL2BP was performed for 298 unrelated individuals diagnosed with autosomal-recessive retinal degeneration, but no disease-associated variants were identified. Additionally, no potential disease-associated variants were identified in WES data generated from 60 additional individuals diagnosed with retinal degeneration.
ARL2BP has been previously identified as a centrosome-associated protein in a variety of mammalian cell lines; however, no previous studies have described retinal localization or association with a human phenotype.17 The retinal localization of ARL2BP was determined in cryofixed C57B16 mouse eye sections18,19 stained with ARL2BP antibodies. ARL2BP was detected in the region of the photoreceptor connecting cilium (Figures 3A and 3B). Higher magnifications of photoreceptor connecting cilia triple stained for ARL2BP, the cilia, basal body, and centriole marker centrin-3 and the periciliary marker PCM-1 verified the localization of ARL2BP to the basal body, adjacent centriole, and ciliary rootlet (Figure 3C). This was confirmed with a second antibody to ARL2BP (Figure S6). Moreover, localization at the distal connecting cilia was corroborated by immunofluorescence (Figure 3C) and immunoelectron microscopy (Figure 3D). For immunoelectron microscopy, we applied a previously described pre-embedding labeling protocol.20,21 Transmission electron microscopy further revealed ARL2BP localization in the periciliary extension of the inner segment and confirmed its localization at the basal body, at the adjacent centriole, and in the ciliary rootlet (Figures 3C and 3D).
Figure 3.
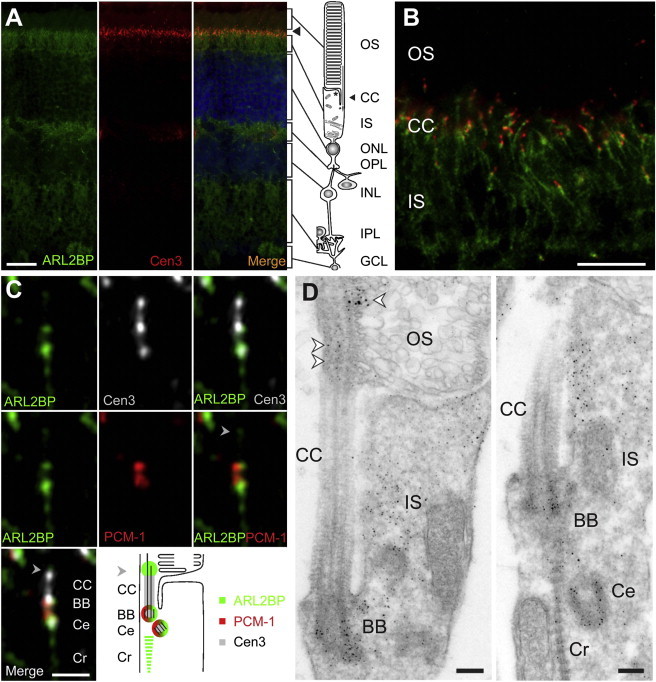
ARL2BP Is a Basal Body Protein
(A) Mouse retina cryosections were stained for ARL2BP (green, 1:1,000 in-house antibody RK16), counterstained for the ciliary marker centrin-3 (Cen3, red, 1:100 antibody20), and stained with DAPI (blue) for the outer nuclear layer (ONL) and inner nuclear layer (INL). ARL2BP colocalized with Cen3 in the ciliary region of photoreceptor cells (arrowhead).
(B) A close-up view of the junction of the photoreceptor inner segment (IS), connecting cilia (CC), and outer segment (OS) shows ARL2BP (green) overlap with Cen3 (red).
(C) High-resolution immunofluorescence of the ciliary region in photoreceptors of the mouse retina. Triple labeling of ARL2BP (green), Cen3 (white), and pericentrin (PCM-1, red, 1:400 Abcam) revealed localization of ARL2BP in the distal CC (arrowhead), basal body (BB), ciliary-associated centriole (Ce), and ciliary rootlet (Cr) of photoreceptor cells. On the bottom right is a color-coded schematic of ARL2BP localization relative to that of Cen3 and PCM-1.
(D) Immunoelectron microscopy of mouse photoreceptors shows ARL2BP ultrastructural localization at the junction of the OS and IS; labeling of the BB, Ce, and distal CC is marked with arrowheads. In addition, some labeling was observed in the Cr projecting into the IS.
Other abbreviations are as follows: OPL, outer plexiform layer; IPL, inner plexiform layer; and GCL, ganglion cell layer. Scale bars represent 25 μm (A), 10 μm (B), 1 μm (C), and 0.2 μm (D).
The basal body complex and the periciliary extension of the inner segment are important for targeting and regulating protein entry into the primary cilium, thus acting as docking sites for pericentriolar transport vesicles and intraflagellar transport (IFT) particles.20–23 Therefore, it is possible that ARL2BP functions in the targeting, docking, or loading of proteins and/or vesicles in the periciliary region for cilia-associated traffic. The presence of ARL2BP in distal connecting cilia, where new outer-segment disks are formed, affirms the close relation between ARL2BP and IFT molecules, which show analogous spatial distribution in ciliary subcompartments,20 and indicates a potential role in disk neogenesis.
Localization of ARL2BP in the basal body was confirmed in mouse fibroblast cells (NIH 3T3) and human retina pigment epithelial cells (ARPE19) (Figures 4A and 4B) by immunocytochemistry (ICC) as described previously.19,24 After treatment with ARL2BP siRNA, cytoplasmic and basal body staining of ARL2BP was reduced to undetectable levels in over 70% of cells, and immunoblotting confirmed that the protein level was reduced by approximately 90% (Figure 4B and Figure S7). Interestingly, staining of ARL2BP at the basal body was also lost in over 70% of cells transfected with ARL2 siRNA, even though the overall levels of ARL2BP were not affected (Figure 4B and Figure S7). In contrast, siRNAs targeting ARL3 or the ARL3 GAP RP2 had no effect on ARL2BP localization or protein levels (Figures S7C and S7D). The specificity of one ARL2BP antibody has been previously reported16 and was confirmed for both antibodies by ARL2BP siRNA with ICC and immunoblotting (Figure 4 and Figures S6 and S7). The efficiency of siRNAs against ARL2BP, ARL2, and ARL3 was confirmed by immunoblotting or RT-PCR (Figure S7), and the specificity of the siRNA against RP2 has been described previously.19
Figure 4.
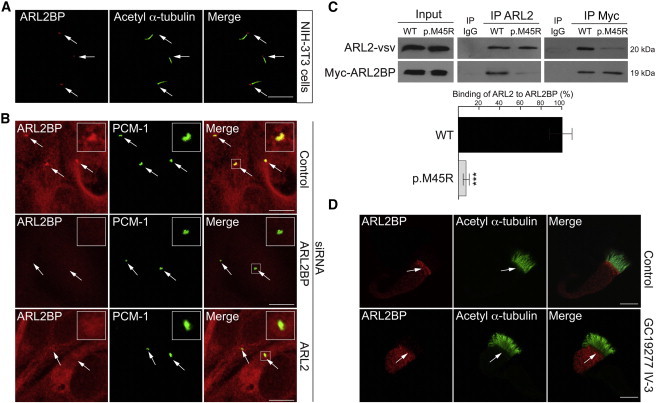
ALR2BP Cilia Targeting Is Dependent on ARL2 and Is Disrupted by the p.Met45Arg Amino Acid Substitution
(A) ARL2BP localized to the base of the cilium in mouse fibroblast cells (NIH 3T3). Cilia (green) were stained with acetylated α-tubulin antibody (acetyl α-tubulin, 1:1,000, Sigma Aldrich) and for ARL2BP (red) (1:500 antibody, Protein Tech Group).
(B) In human RPE cells (ARPE19), ARL2BP (red, 1:1,000 in-house antibody16) colocalized with the basal body marker PCM-1 (green, 1:1,000). ARL2BP localization at the basal body was abolished by ARL2BP or ARL2 siRNA, but not by control siRNA. Basal body localization is highlighted by an arrow. Inserts show higher magnification of ARL2BP basal body localization. siRNAs for ARL2, ARL3, RP2, or ARL2BP and a nontargeting control siRNA were obtained from Dharmacon.
(C) Reciprocal coimmunoprecipitation (co-IP) of vsv-tagged ARL2 with wild-type (WT) or p.Met45Arg (p.M45R) Myc-tagged ARL2BP immunoblotted for ARL2 and ARL2BP, as indicated. Compared to that of WT ARL2BP, the binding of p.Met45Arg ARL2BP to ARL2 was reduced by 90%. Immunoblots from three independent experiments were analyzed with ImageJ software (National Institutes of Health). Human Myc-tagged ARL2BP in pcDNA4/TO/Myc-HIS A was from Abgent, and bovine ARL2 in pcDNA4/TO/vsv was a kind gift from Jane Evans (UCL Institute of Ophthalmology). For co-IPs, equal amounts of ARL2BP and ARL2 were transfected into SK-N-SH cells. The ARL2BP p.Met45Arg variant was introduced by site-directed mutagenesis (QuickChange, QIAGEN). The goat Arl2 antibody was purchased from Abcam (1:200) and used for immunoprecipitation (IP), and the vsv antibody (1:50), a kind gift from Karl Matter, was used for immunoblotting. The Myc antibody (1:1,000, Sigma Aldrich) was used for IPs and immunoblotting of Myc-tagged ARL2BP. n = 3 co-IPs for quantification of ARL2 binding to ARL2BP. Values are means ± SEM; ∗∗∗p ≤ 0.001.
(D) ARL2BP localizes to the basal body region in human nasal epithelial cells, but not in cells derived from individual IV-3 from family GC19277 (lower panel). Cilia (green) were stained for acetylated α-tubulin (1:1,000 antibody, Sigma Aldrich) and for ARL2BP (red, 1:100 antibody16).
Scale bars represent 10 μm.
These findings suggest that the interaction between ARL2BP and ARL2 is important for recruitment or anchoring of ARL2BP at the basal body. ARL2BP and ARL2 siRNAs appeared to have no major adverse effect on ARPE19 cell morphology, survival, or division (data not shown). To determine the effect that loss of ARL2BP at the basal body has on cilia morphology, we examined cilia incidence and length in ARPE19 cells by staining for the cilia membrane protein ARL13B after treatment with ARL2BP, ARL2, and control siRNA. The incidence of cells displaying cilia and normal cilia morphology was unaffected by treatment with control siRNA (Figure S8). However, in cells treated with ARL2BP or ARL2 siRNA, there was a significant reduction in cilia length (Figure S8).
To investigate the effect of the p.Met45Arg amino acid substitution (IV-3 in family GC19277; Figure 1) on ARL2BP, we overexpressed wild-type and p.Met45Arg ARL2BP in human SK-N-SH neuroblastoma cells. The substitution did not affect expression levels of ARL2BP (Figure 4C), suggesting no major effect on protein stability. Therefore, we tested the hypothesis that the amino acid substitution affects the interaction with ARL2 by using coimmunoprecipitation, as previously described.24 The interaction between wild-type Myc-tagged ARL2BP and vsv-tagged ARL2 (Figure 4C) was confirmed, but the level of binding between vsv-tagged ARL2 and p.Met45Arg Myc-tagged ARL2BP was 90% lower than the level of binding between vsv-tagged ARL2 and wild-type Myc-tagged ARL2BP (Figure 4C).
To determine the significance of this finding in vivo, we investigated ARL2BP expression in nasal epithelial cells from individual IV-3 (family GC19277) and healthy controls. Nasal epithelial cells from nasal brush biopsies were sampled from individual IV-3 and three control individuals without ciliary disease with the use of a modified cytology brush, and immunofluorescence staining of the cells was performed as previously described.25,26 In control epithelial cells, ARL2BP was enriched at the basal body region of the cell beneath the motile cilia, stained for acetylated α-tubulin (Figure 4D) or γ-tubulin (Figure S9). However, in individual IV-3’s (family GC19277) epithelial cells, which contained only the p.Met45Arg amino acid substitution, ARL2BP was distributed diffusely in the cytoplasm and was not enriched at the basal bodies (Figure 4D and Figure S9). This supports the hypothesis that the binding of ARL2BP to ARL2 is critical for localization of ARL2BP to the basal body. We hypothesize that because the level of binding between ARL2 and p.Met45Arg ARL2BP is lower than the level of binding between ARL2 and wild-type ARL2BP, ARL2 is not able to sufficiently recruit or anchor the altered ARL2BP to the base of the cilium.
ARL2 shares a high degree of structural, and possibly functional, conservation with ARL3, given that ARL2 and ARL3 have common binding partners, including UNC119,27,28 a subunit of phosphodiesterase (PDE6D),29,30 and ARL2BP.16 PDE6D and UNC119 share extensive sequence homology and cooperate with ARL2 and/or ARL3 to regulate the membrane association of posttranslationally modified acylated and prenylated proteins, such as the α-subunit of transducin, NPHP4, and Rheb.31–33 RP2 localizes to the ciliary apparatus at the base of the photoreceptor cilium and, along with ARL3, facilitates the traffic and membrane association of NPHP4, transducin, and potentially other cilia-associated proteins.24,32,34–36 Therefore, the cooperation between ARL3 and its regulatory proteins (such as RP2) and effector proteins (such as UNC119) is important for the correct function of ciliated cells and photoreceptors in particular. However, little is known about the role of ARL2 in cilia function. For example, ARL2 binds to UNC119 but does not release cargo from UNC119,31 suggesting that despite their similarity, ARL2 and ARL3 have different functions in the retina. In addition, ARL2BP binds the same site in ARL2 as do the effectors UNC119 and PDE6D37 and would compete for binding to the activated GTPase. Importantly, we have shown that the targeting of ARL2BP to cilia is dependent on ARL2, not ARL3. Therefore, the role of ARL2BP in cilia is likely to be distinct from that of the ARL3-RP2-UNC119 complex and ARL2-PDE6D and ARL3-PDE6D. In this study, we describe ARL2BP mutations associated with autosomal-recessive RP, demonstrating that ARL2BP plays an essential role in the retina, and show that this role is mediated in part by the ARL2BP-ARL2 interaction but does not involve ARL3.
Acknowledgments
We acknowledge the cooperation and help provided by the family members in this study. We are grateful to colleagues who referred affected individuals to us at Moorfields Eye Hospital and those who contributed to the assembly of large panels of probands, particularly Bev Scott, Genevieve Wright, Sophie Devery, Michel Michaelides, Tony Moore, Eva Lenassi, Graham Holder, and Tony Robson. We acknowledge the nationally commissioned Royal Brompton Hospital Primary Ciliary Dyskinesia diagnostic service led by Claire Hogg and Steve Rothery at the Imperial College Imaging Facility (London, UK). We acknowledge Alexy Obolensky for retinal-imaging assistance. We are grateful to Nick Cowan, Karl Matter, and Jane Evans for providing antibodies or plasmids. We acknowledge the following sources of funding: RP Fighting Blindness, Fight for Sight, Moorfields Eye Hospital Special Trustees, the Moorfields Eye Charity, the National Institute for Health Research (NIHR) (Moorfields Eye Hospital and Institute of Ophthalmology [London, UK]), the Foundation Fighting Blindness (BR-GE-0510-0490-HUJ to D.S.), a Yedidut 1 research grant (to E.B.), European Community’s Seventh Framework Programme FP7/2009/241955 (SYSCILIA), and the FAUN Foundation (Nuremburg, Germany) (to U.W.). V.P. is partly funded by the UK Medical Research Council (G1001158) and by the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and Institute of Ophthalmology at University College London. R.A.K. is supported in part by a grant from the National Institutes of Health (GM090158).
Contributor Information
Michael E. Cheetham, Email: michael.cheetham@ucl.ac.uk.
Dror Sharon, Email: dror.sharon1@gmail.com.
Supplemental Data
High-speed video microscopy shows that individual IV-3’s nasal epithelial cilia lack coordinated beating activity. A disorganized, stiff, and ineffective ciliary beat pattern was observed. The ciliary beat frequency was 9.86 Hz (the normal range is 7.8–15.5 Hz).
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/
BioMart, http://www.biomart.org
Blossum62, http://www.ncbi.nlm.nih.gov/Class/FieldGuide/BLOSUM62.txt
DNAnexus, https://www.dnanexus.com/
ExomeDepth, http://cran.r-project.org/web/packages/ExomeDepth/index.html
GeneDistiller 2, http://www.genedistiller.org/
HomozygosityMapper, http://www.homozygositymapper.org
Interactive Genomics Viewer, http://www.broadinstitute.org/software/igv/
NHLBI Exome Variant Server (EVS) Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Novocraft, http://www.novocraft.com
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Retinal Information Network (RetNet), https://sph.uth.edu/retnet/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SAMtools, http://samtools.sourceforge.net/
Savant Genome Browser, http://genomesavant.com/
SIFT, http://sift.jcvi.org/
The R Project for Statistical Computing, http://www.r-project.org/
Unigene, http://www.ncbi.nlm.nih.gov/UniGene/
UniProt, http://www.uniprot.org/
References
- 1.Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 2.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Estrada-Cuzcano A., Roepman R., Cremers F.P., den Hollander A.I., Mans D.A. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet. 2012;21(R1):R111–R124. doi: 10.1093/hmg/dds298. [DOI] [PubMed] [Google Scholar]
- 4.Mockel A., Perdomo Y., Stutzmann F., Letsch J., Marion V., Dollfus H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011;30:258–274. doi: 10.1016/j.preteyeres.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 6.Robson A.G., Lenassi E., Saihan Z., Luong V.A., Fitzke F.W., Holder G.E., Webster A.R. Comparison of fundus autofluorescence with photopic and scotopic fine matrix mapping in patients with retinitis pigmentosa: 4- to 8-year follow-up. Invest. Ophthalmol. Vis. Sci. 2012;53:6187–6195. doi: 10.1167/iovs.12-10195. [DOI] [PubMed] [Google Scholar]
- 7.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zito I., Downes S.M., Patel R.J., Cheetham M.E., Ebenezer N.D., Jenkins S.A., Bhattacharya S.S., Webster A.R., Holder G.E., Bird A.C. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J. Med. Genet. 2003;40:609–615. doi: 10.1136/jmg.40.8.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Y., Esmail M.A., Ansley S.J., Blacque O.E., Boroevich K., Ross A.J., Moore S.J., Badano J.L., May-Simera H., Compton D.S. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet. 2004;36:989–993. doi: 10.1038/ng1414. [DOI] [PubMed] [Google Scholar]
- 12.Cantagrel V., Silhavy J.L., Bielas S.L., Swistun D., Marsh S.E., Bertrand J.Y., Audollent S., Attié-Bitach T., Holden K.R., Dobyns W.B., International Joubert Syndrome Related Disorders Study Group Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrick J.J., Vogel P., Abuin A., Hampton B., Rice D.S. ADP-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am. J. Pathol. 2006;168:1288–1298. doi: 10.2353/ajpath.2006.050941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwahn U., Lenzner S., Dong J., Feil S., Hinzmann B., van Duijnhoven G., Kirschner R., Hemberger M., Bergen A.A., Rosenberg T. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 15.Hardcastle A.J., Thiselton D.L., Van Maldergem L., Saha B.K., Jay M., Plant C., Taylor R., Bird A.C., Bhattacharya S. Mutations in the RP2 gene cause disease in 10% of families with familial X-linked retinitis pigmentosa assessed in this study. Am. J. Hum. Genet. 1999;64:1210–1215. doi: 10.1086/302325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharer J.D., Kahn R.A. The ARF-like 2 (ARL2)-binding protein, BART. Purification, cloning, and initial characterization. J. Biol. Chem. 1999;274:27553–27561. doi: 10.1074/jbc.274.39.27553. [DOI] [PubMed] [Google Scholar]
- 17.Zhou C., Cunningham L., Marcus A.I., Li Y., Kahn R.A. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol. Biol. Cell. 2006;17:2476–2487. doi: 10.1091/mbc.E05-10-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfrum U., Schmitt A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil. Cytoskeleton. 2000;46:95–107. doi: 10.1002/1097-0169(200006)46:2<95::AID-CM2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Evans R.J., Schwarz N., Nagel-Wolfrum K., Wolfrum U., Hardcastle A.J., Cheetham M.E. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum. Mol. Genet. 2010;19:1358–1367. doi: 10.1093/hmg/ddq012. [DOI] [PubMed] [Google Scholar]
- 20.Sedmak T., Wolfrum U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell Biol. 2010;189:171–186. doi: 10.1083/jcb.200911095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maerker T., van Wijk E., Overlack N., Kersten F.F., McGee J., Goldmann T., Sehn E., Roepman R., Walsh E.J., Kremer H., Wolfrum U. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 22.Reiter J.F., Blacque O.E., Leroux M.R. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall W.F. Basal bodies platforms for building cilia. Curr. Top. Dev. Biol. 2008;85:1–22. doi: 10.1016/S0070-2153(08)00801-6. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz N., Novoselova T.V., Wait R., Hardcastle A.J., Cheetham M.E. The X-linked retinitis pigmentosa protein RP2 facilitates G protein traffic. Hum. Mol. Genet. 2012;21:863–873. doi: 10.1093/hmg/ddr520. [DOI] [PubMed] [Google Scholar]
- 25.Rutland J., Dewar A., Cox T., Cole P. Nasal brushing for the study of ciliary ultrastructure. J. Clin. Pathol. 1982;35:357–359. doi: 10.1136/jcp.35.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Omran H., Loges N.T. Immunofluorescence staining of ciliated respiratory epithelial cells. Methods Cell Biol. 2009;91:123–133. doi: 10.1016/S0091-679X(08)91007-4. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi A., Kubota S., Mori N., McLaren M.J., Inana G. Photoreceptor synaptic protein HRG4 (UNC119) interacts with ARL2 via a putative conserved domain. FEBS Lett. 2003;534:26–32. doi: 10.1016/s0014-5793(02)03766-3. [DOI] [PubMed] [Google Scholar]
- 28.Ismail S.A., Chen Y.X., Miertzschke M., Vetter I.R., Koerner C., Wittinghofer A. Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J. 2012;31:4085–4094. doi: 10.1038/emboj.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renault L., Hanzal-Bayer M., Hillig R.C. Coexpression, copurification, crystallization and preliminary X-ray analysis of a complex of ARL2-GTP and PDE delta. Acta Crystallogr. D Biol. Crystallogr. 2001;57:1167–1170. doi: 10.1107/s0907444901009556. [DOI] [PubMed] [Google Scholar]
- 30.Linari M., Hanzal-Bayer M., Becker J. The delta subunit of rod specific cyclic GMP phosphodiesterase, PDE delta, interacts with the Arf-like protein Arl3 in a GTP specific manner. FEBS Lett. 1999;458:55–59. doi: 10.1016/s0014-5793(99)01117-5. [DOI] [PubMed] [Google Scholar]
- 31.Ismail S.A., Chen Y.X., Rusinova A., Chandra A., Bierbaum M., Gremer L., Triola G., Waldmann H., Bastiaens P.I., Wittinghofer A. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 2011;7:942–949. doi: 10.1038/nchembio.686. [DOI] [PubMed] [Google Scholar]
- 32.Wright K.J., Baye L.M., Olivier-Mason A., Mukhopadhyay S., Sang L., Kwong M., Wang W., Pretorius P.R., Sheffield V.C., Sengupta P. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011;25:2347–2360. doi: 10.1101/gad.173443.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H., Constantine R., Vorobiev S., Chen Y., Seetharaman J., Huang Y.J., Xiao R., Montelione G.T., Gerstner C.D., Davis M.W. UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci. 2011;14:874–880. doi: 10.1038/nn.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz N., Hardcastle A.J., Cheetham M.E. Arl3 and RP2 mediated assembly and traffic of membrane associated cilia proteins. Vision Res. 2012;75:2–4. doi: 10.1016/j.visres.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz N., Hardcastle A.J., Cheetham M.E. The role of the X-linked retinitis pigmentosa protein RP2 in vesicle traffic and cilia function. Adv. Exp. Med. Biol. 2012;723:527–532. doi: 10.1007/978-1-4614-0631-0_66. [DOI] [PubMed] [Google Scholar]
- 36.Veltel S., Gasper R., Eisenacher E., Wittinghofer A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct. Mol. Biol. 2008;15:373–380. doi: 10.1038/nsmb.1396. [DOI] [PubMed] [Google Scholar]
- 37.Zhang T., Li S., Zhang Y., Zhong C., Lai Z., Ding J. Crystal structure of the ARL2-GTP-BART complex reveals a novel recognition and binding mode of small GTPase with effector. Structure. 2009;17:602–610. doi: 10.1016/j.str.2009.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
High-speed video microscopy shows that individual IV-3’s nasal epithelial cilia lack coordinated beating activity. A disorganized, stiff, and ineffective ciliary beat pattern was observed. The ciliary beat frequency was 9.86 Hz (the normal range is 7.8–15.5 Hz).