

Abstract
Autism Spectrum Disorder (ASD) demonstrates high heritability and familial clustering, yet the genetic causes remain only partially understood as a result of extensive clinical and genomic heterogeneity. Whole-genome sequencing (WGS) shows promise as a tool for identifying ASD risk genes as well as unreported mutations in known loci, but an assessment of its full utility in an ASD group has not been performed. We used WGS to examine 32 families with ASD to detect de novo or rare inherited genetic variants predicted to be deleterious (loss-of-function and damaging missense mutations). Among ASD probands, we identified deleterious de novo mutations in six of 32 (19%) families and X-linked or autosomal inherited alterations in ten of 32 (31%) families (some had combinations of mutations). The proportion of families identified with such putative mutations was larger than has been previously reported; this yield was in part due to the comprehensive and uniform coverage afforded by WGS. Deleterious variants were found in four unrecognized, nine known, and eight candidate ASD risk genes. Examples include CAPRIN1 and AFF2 (both linked to FMR1, which is involved in fragile X syndrome), VIP (involved in social-cognitive deficits), and other genes such as SCN2A and KCNQ2 (linked to epilepsy), NRXN1, and CHD7, which causes ASD-associated CHARGE syndrome. Taken together, these results suggest that WGS and thorough bioinformatic analyses for de novo and rare inherited mutations will improve the detection of genetic variants likely to be associated with ASD or its accompanying clinical symptoms.
Introduction
Autism spectrum disorder (ASD; MIM 209850) is a lifelong developmental condition affecting one in 88 children and is considered one of today’s most urgent public health challenges. ASD is characterized by atypical development of social communication and by the presence of repetitive behaviors and restricted interests; onset occurs in early childhood. ASD is also often accompanied by intellectual disability, anxiety, sleep problems, seizures, and gastrointestinal or other medical problems.1 Given that diagnosis relies on clinical observation, families often experience a “diagnostic odyssey” as they try to find an explanation for their child’s condition.1
Individuals with ASDs are increasingly presenting for clinical genetics evaluation, and 10% have an identifiable genetic condition. Fragile X (MIM 300524) (∼1%–2% of ASD cases), Rett (MIM 312750) (∼0.5%), microdeletion and microduplication, Cowden (MIM 158350), Timothy (MIM 601005), and other syndromes are occasionally diagnosed.2 A recent study identified more than 103 genes and 44 genomic loci with mutations among individuals with some form of ASD.3 These genes have all been causally implicated in intellectual disability, indicating that these two neurodevelopmental disorders often share a common genetic basis. A genetic overlap between ASD and epilepsy,4 as well as between ASD and neuropsychiatric conditions5, is also apparent in many cases.
Family studies indicate a significant genetic basis for ASD. Clinically identifiable copy-number variations (CNVs) in ∼10% of ASD-affected individuals5 has led to the recommendation that microarrays be considered a "standard of care" test for any ASD diagnostic evaluation.6 Curation of large-scale CNV and other genomic data sets has produced an evidence-based collection of more than 100 genes implicated in ASD risk and the risk for related disorders (we refer to these as known ASD-linked genes).3,7,8 The first beneficiaries of genetic tests are young children, in whom formal diagnosis based on early behavioral signs can be challenging but who benefit most from earlier behavioral intervention. Understanding the genetic causes of ASD in an individual also potentially informs prognosis, medical management, and familial-recurrence risk assessment, and it can potentially facilitate drug-intervention trials through stratification based on genetic-pathway profiles.1,9
As one approach to identify additional ASD risk genes, researchers have used genome-wide sequencing of the coding regions (collectively, the exome) of human DNA to search for de novo mutations.10–13 Reports from such projects have indicated (1) a 2- to 4-fold increase in de novo nonsense variants among ASD individuals in comparison to their unaffected siblings, (2) some unrecognized ASD risk genes, and (3) the possibility that hundreds of genes are involved in the ASD risk.14 However, other genomic investigations have identified mutations in individuals with ASD in nongenic segments of DNA15 and in noncoding RNAs,16,17 and in these examples and others, inherited risk variants for ASD are known.18,19 Individuals with multiple genetic risk factors7,20–22 for ASD and/or associated medical conditions3 are also documented.
There has been significant progress in identifying unrecognized ASD risk variants as each technology of increasing resolution (cytogenetics, microarrays, exome sequencing) has become available. Even so, when one combines all of the data from these complementary approaches, the picture remains incomplete: the etiological basis of about 20% of cases of ASD, at most, can be explained to date.5 High heritability estimates (37%–90%)23,24 and family studies reconfirm strong genetic contributions18 and suggest that more genetic variants remain to be discovered.
We therefore implemented WGS in an effort to capture all classes of genetic variation in a single experiment and potentially more fully describe the genetic architecture of ASD. We developed an analysis plan to search for de novo or rare inherited mutations in previously recognized, as well as unrecognized, genes linked to ASD and for variants in genes that might not be specific to ASD (Figure 1). For the latter group, the genes could be related to core autism symptoms, such as social communication, or conditions strongly associated with ASD, such as intellectual disability, seizures, or other medical complications. Given that this is a pilot study for WGS in ASD and is based on a relatively small number of families, we stress the need for replication of our findings. However, promising results thus far indicate that WGS could eventually help direct personalized approaches to clinical management in individuals and families affected by ASD.
Figure 1.
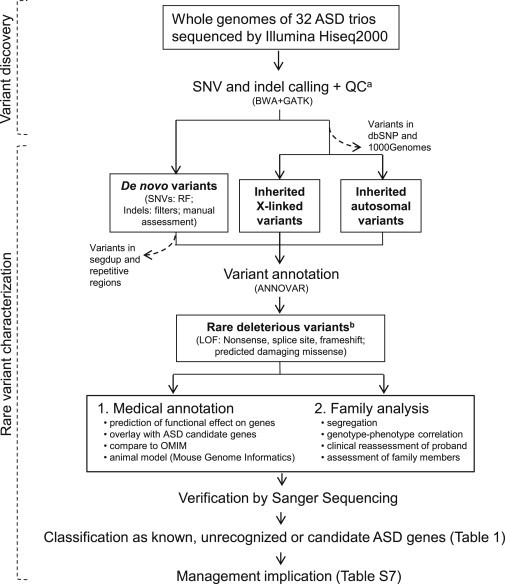
Detection and Classification of Medically Relevant Genetic Variants
Rare SNV and indel variants were assessed as putative etiologic factors in ASD. Samples were also run on high-resolution microarray for CNV calling as indicated in the Material and Methods. Dashed arrows indicate variants not included in downstream analyses. In addition to applying random forests (RF) algorithms and filtering methods, we used Sanger sequencing to perform a manual assessment of potential de novo variants annotated by GATK.
aLow-quality variants were eliminated with GATK’s filters.
bRare variants were defined as those not found in dbSNP or the 1000 Genomes Project. Each gene bearing rare deleterious variants was classified as (1) a "known" (also called "linked") ASD gene if it was previously identified to be involved in ASD, according to lists developed by the Autism Genome Project Consortium,3,7 (2) an "unrecognized" ASD gene if it was not previously recognized to carry a loss-of-function mutation (nonsense, splice site, or frameshift) and if it showed an ASD-related phenotype, as reported in OMIM and/or the MGI mouse knockout database, or (3) a "candidate" ASD gene if it was affected by a de novo deleterious variant. In the case of rare inherited X-linked deleterious variants found in males, the same gene also needed to be implicated in other sequencing studies. Abbreviations are as follows: GATK, Genome Analysis Toolkit; BWA, Burrows-Wheeler Aligner; and segdup, segmental duplication
Material and Methods
Samples for ASD-Affected Families
Thirty-two unrelated Canadian individuals with ASD (25 males and seven females) were diagnosed with the Autism Diagnostic Interview-Revised and the Autism Diagnostic Observation Schedule-Generic protocols, and their family members were studied. They were selected from a cohort of ASD families with at least one unaffected sibling at the time of the proband’s diagnosis, 25 and selection was based on available blood genomic DNA and completeness of phenotype information. The samples were also part of the Autism Genetic Resource Exchange (AGRE). We obtained informed consent, as approved by the research ethics boards at The Hospital for Sick Children and McMaster University. We assessed ASD probands with standardized measures of intelligence, language, and adaptive function and collected information on developmental, medical, and physical measures and family history. Out of the 30 families for which we assessed IQ, there were six probands with intellectual disability (IQ < 70). We genotyped all probands on high-resolution microarray platforms to rule out obvious pathogenic CNVs26 (Figure S1 in the Supplemental Data available online).
Sequencing and Variant Discovery
Genomic DNA of probands and parents (trios) was extracted from blood and sequenced with Hiseq2000 technology (Illumina). DNA was extracted and sheared into fragments, which were then purified by gel electrophoresis. DNA fragments were ligated with adaptor oligonucleotides to form paired-end DNA libraries with an insert size of 500 bp. To enrich libraries for sequences with 5′ and 3′ adaptors, we used ligation-mediated PCR amplification with primers complimentary to the adaptor sequences. The DNA libraries were sequenced to generate 90 bp pair-end reads with at least 30× average genome coverage per sample (Table S1). Filtered reads were aligned to the reference genome (build GRCh37) with the Burrows-Wheeler Aligner (BWA),27 allowing detection of single-nucleotide variants (SNVs) and small insertions/deletions (indels; <65 nucleotides).
Local realignment and quality recalibration were done with the Genome Analysis Toolkit (GATK-1.4-30).28 Sequence Alignment/Map tools (SAMtools)29 was used for removal of duplicated reads. The effects (e.g., missense, nonsense, or frameshift mutations) and classifications (e.g., in exonic, intronic, or intergenic regions) of variants across the genome were annotated by ANNOVAR.30 The number of SNVs and indels detected per sample is summarized in Table S1.
Variant Confirmation
Primers were designed to span at least 100 bp upstream and downstream of the putative variant. For validation, 91 candidate mutations in the whole genomes of one trio (2-1266) were selected randomly, and all the 38 exonic de novo and ASD-relevant variants were validated experimentally by Sanger sequencing (see below). Candidate regions from whole-blood-derived DNA were amplified by PCR for all trios, as well as unaffected siblings or other available family members.
De Novo SNV Detection
A potential de novo mutation was identified if there was a heterozygous genotype in the offspring and homozygous reference genotypes in both parents. First, we removed heterozygous calls in the proband when more than 70% of reads were the reference call. Next, we eliminated cases where a parent had more than 5% nonreference reads that matched the proband’s heterozygous call. To eliminate spurious calls that formed when the proband had a homozygously deleted segment, we removed sites where the proband’s read depth was <10% of the total parental read depth. We refined further by applying filters based on the genotype likelihood, denoted as a "phred-scale" (PL). We used PL of ≥ 30 for the genotypes of the proband and both parents (i.e., the assigned genotype is 1,000 times more likely to be true than the alternative genotypes). Finally, we removed all the sites found in the dbSNP (dbSNP135), given that common SNPs are unlikely to be de novo events.
We evaluated the approach by using the validation results of (1) all genomic de novo SNVs in one family (2-1266) and (2) Sanger sequencing of de novo exonic SNVs in all families (Table S2). For genomic SNVs, 91 de novo SNVs were selected randomly for validation by Sanger sequencing (86 PCR assays were designed to detect 91 de novo SNVs); 64 of the 86 (74.4%) PCR assays worked. Of the 64 putative de novo SNVs, 60 were confirmed as being true positives (i.e., 94% validated). We detected 40 exonic de novo SNVs, and 32 were confirmed as true positives (i.e., 80% validated) (Table S2).
Refinement of de novo SNVs by the Machine-Learning Method
To refine the de novo SNV detection approach, we used a machine-learning-based classifier31 called forestDNM (RF-2). The method was designed to distinguish de novo variants from false-positive de novo mutations (DNMs) that arose from errors in sequencing, alignment, or variant calling. We used a combination of thresholds on site-quality metrics to determine which DNMs were likely to be true positives. In order to facilitate the optimization process and minimize bias, we adopted a data-driven approach for the machine-learning process. First, we created a training data set by using validation results from Sanger sequencing (including calls validated as true and false positives) for all putative de novo SNVs detected by the filter method in one trio (family 2-1266) and for all exonic DNMs in 32 trios. Then, we trained the RF-2 to differentiate the validated de novo SNVs from the invalidated putative de novo SNVs on the basis of a combination of associated quality metrics. Finally, the trained RF-2 detected DNMs in the remaining 31 trios.
We detected 63 genomic de novo SNVs in family 2-1266 and 38 exonic de novo mutations in all families by using this approach. In family 2-1266, 60 of the 63 genomic de novo SNVs were those found by the filter method (Table S2). For de novo exonic SNVs, 36 were confirmed as true positives (i.e., 95% validated). One validated de novo exonic SNV from the filter method was missed by RF-2.
De Novo Indel Detection
We applied similar filtering criteria for de novo indel detection: the proband had to be heterozygous for the indel call, whereas the parents both had to be homozygous reference at the same position. The read depth at the variant position of each family member was more than 10×. Furthermore, the variant calls in the trio data were selected with the following filters: a phred-scaled quality score (QUAL) of more than 30; a QualByDepth (QD: variant confidence from the QUAL field/unfiltered depth) of more than 10; a HomopolymerRun (Hrun: Largest contiguous homopolymer run of the variant allele in either direction on the reference) less than 5; and a MappingQualityZero (MQ0: total count across all samples that had reads with a mapping quality of zero) less than 4. In addition to the essential variant-quality filters, the indel call from the proband needed to be supported by at least 30% of the reads, and there needed to be at least a 17× read depth. Also, parents could have no reads with an indel at the same position where an indel was detected in the proband. The filter matrix was optimized by evaluating the performance on simulated trio data containing de novo mutations generated by pIRS.32 On the basis of the simulated trio data, the method showed 84% sensitivity and a 5% false-detection rate.
Because de novo indels found in dbSNP and simple repeats were prone to be false positives, we filtered out indels reported in dbSNP135 and located in simple repeat regions (Table S3). We designed PCR assays for 3 of the 4 indels identified in family 2-1266. Sanger sequencing confirmed that they were all true positives. We also validated the two de novo exonic indels and confirmed that both were true positives (Table S3).
Manual Assessment of Potential De Novo Exonic Variants
Because the automated de novo detection methods only take into account de novo variants that are homozygous in parents and heterozygous in the proband, which is not applicable for variants on the X chromosomes in males, we manually curated X-linked hemizygous variants that occurred in male probands but were not present at the same position in the X chromosome of their respective mothers. This identified two additional de novo mutations (in KAL1 and WWC3). In addition, we prioritized a list of potential de novo exonic indels that were assigned by GATK (23 in total) but were missed by the automated de novo detection methods described above, and we validated them by using Sanger sequencing. We thus confirmed one additional de novo indel (found in MICALCL [MIM 612355]).
CNV Detection by Genomic Array and WGS
Although each of the 32 trios had been previously examined by microarrays, we also tested them by using the CytoScan HD Array (Affymetrix, Santa Clara, CA). These CNV calls provided an additional resource for comparison against the WGS data. We performed CNV analysis by using four algorithms: Chromosome Analysis Suite software (Affymetrix, Santa Clara, CA), iPattern,7 Nexus (BioDiscovery, CA), and Partek (Partek, MO). CNVs were called when they were detected by at least two of the four algorithms and spanned at least five or more consecutive array probes.33
CNVnator was used for detection of CNVs from WGS as previously described.34 CNV calls with a p value < 0.05 and a size >1 kb were used, and calls with >50% of q0 (zero mapping quality) reads within the CNV regions were removed (q0 filter). Rare CNVs were defined as not present in the Database of Genomic Variants (DGV)35. Putative de novo CNVs for each trio were detected with the following criteria: (1) the normalized average RD signal in the child was <1.4 (for deletions) or >2.6 (for duplications); and (2) the normalized average RD signal in each parent was between 1.6 and 2.4. CNVs located in segmental duplications and repetitive regions were removed.
Variant Characterization
When assessing inherited variants, we removed those that are common in the population (i.e., found in dbSNP135 or the 1000 Genomes Project). We determined X-linked or autosomal location and the parent of origin by comparing to the genotypes of the parents. All variants were annotated for their effects on the basis of their sequence alterations. Rare (< 0.1% frequency) deleterious variants including nonsense, splice site, frameshift, and damaging missense mutations were selected for medical annotation and family analysis. Variants of interest were verified by Sanger sequencing for the trios and additional family members.
On average, there were 8,982 nonsynonymous SNVs per proband (Table S4); we defined rare variants as those not found in dbSNP135 or the 1000 Genomes Project,36 and there were 231 of such SNVs per proband (Table S4). We defined damaging missense SNVs as those that both SIFT37 and PolyPhen-238 predicted to be damaging via the Variant Effect Predictor.39 There were 40 rare damaging missense mutations, 75 rare splice-site mutations, and six rare nonsense mutations per proband (Table S4). We found 444 rare indels per proband; of these, 126 were denoted as rare frameshifts (Table S4). We examined the frequency of all the ASD "clinically relevant" variants as identified in the exome sequencing project (ESP6500) from NHLBI and the Human Gene Mutation Database (Table S5).
To evaluate the performance of functional prediction by the combination of SIFT and PolyPhen-2, we selected all the de novo and inherited ASD-relevant missense mutations and performed function prediction by using two other variant-prediction tools: PANTHER40 and MutationTaster.41 Although all of these tools have taken into account the conservation score, there are substantial differences in their prediction strategies, in which their outcomes were not necessarily concordant. However, the prediction results we found in these variants were significantly correlated (PANTHER p = 0.003; MutationTaster p = 0.0008).
Medical Classification
We assessed the medical relevance of the detected genetic variants to ASD by (1) predicting whether they were likely to have a deleterious effect on splicing of the gene or functional properties of the protein product; (2) assessing the frequency of predicted deleterious variants in a population database (NHLBI GO Exome Sequencing Project); (3) comparing these variants to known ASD candidate genes;3,7 (4) looking at segregation in families and comparing predicted mutations to disease genes in the Online Mendelian Inheritance in Man (OMIM) database and (5) comparing predicted mutations to orthologous genes in the Mouse Genome Informatics (MGI) phenotype database. We tested X-inactivation as described by Allen et al.42
Medical Annotation and Family Analysis
We examined the function of genes that carried rare deleterious mutations in the documented neurodevelopmental/behavioral phenotype of humans or mice. For humans, we used the human phenotype ontology (HPO) resources and selected all genes annotated for “behavioural/psychiatric abnormality” (HP:0000708) and/or “cognitive impairment” (HP:0100543). We considered genes with HPO phenotype annotation derived from OMIM because only for these could we reliably infer the mode of inheritance. For mice, we used the mammalian phenotype ontology (MPO) annotations provided by Mouse Genome Informatics (MGI). After removing quantitative trait loci (QTLs), complex genotypes (i.e., requiring a combination of genotypes of different genes), and phenotypes associated with transgenes, we selected all genes annotated for “behavior/neurological phenotype” (HP:0000708) and/or “nervous system phenotype” (HP:0100543).
We also determined the mode of inheritance for these genes. For human phenotypes, we used the phenotype OMIM-number notation to determine the mode of inheritance: for OMIM IDs starting with 1, we assumed an autosomal-dominant mode of inheritance; for those starting with 2, we assumed an autosomal-recessive mode of inheritance, and for those starting with 3, we assumed an X-linked mode of inheritance; OMIM identifiers corresponding to official gene symbols were derived from the Bioconductor package org.Hs.egOMIM2EG, version 2.7.1, packaged on March 13, 2012. For mouse phenotypes, we inferred the mode of inheritance on the basis of the genotype associated with the phenotype of interest: (1) genes associated with the phenotypes of interest through at least one autosomal-heterozygous genotype were inferred to be dominant, (2) genes associated with the phenotypes of interest only through an autosomal-homozygous genotype(s) were inferred to be recessive, (3) genes with human orthologs outside of the pseudo-autosomal regions on the X chromosome were inferred to be X-linked. In the case of discordance between mouse and human modes of inheritance (2.94% of the genes), we retained only the human information. Medical annotation results for each ASD-relevant variant are summarized in Table S5. A summary of family analysis and assessment results is provided in Table S6.
Comparison between WGS and Whole-Exome Sequencing
We selected ten whole-genome samples (from five males and five females) from six families; we sampled probands as well as mothers and fathers, and average autosome depth per sample ranged from 33× to 39× (the average across samples was 35.6×, which is very close to the overall average of 35.7×). We also selected ten whole-exome samples (from five males and five females), sequenced with HiSeq 2000 and the SureSelect capture kit (SSV4) (Agilent Technologies, Santa Clara, CA), that were not from the ASD cohort, and we used an analysis pipeline (preprocessing, alignment) similar to that used for the whole genome. The average autosome depth per sample of whole-exome samples ranged from 78.3× to 144.3× (average across samples: 115.9×).
Read depth was calculated for all exons according to the hg19 RefSeq June 14, 2012 version; the longest transcript was considered for each gene. For each exon, we calculated the average sequencing depth for the entire exon as well as for five genomic intervals overlapping the exon, as follows: (1) 10 intronic bp upstream of the start of the exon (labeled “5′ splice”); (2) 10 exonic bp downstream from the start of the exon (labeled “start”); (3) from 11 bp downstream of the exon-start to 11 bp upstream of the exon end (labeled “middle”); (4) 10 exonic bp upstream of the end of the exon (labeled “end”), and (5) 10 intronic bp downstream from the end of the exon (labeled “3′ splice”).
We also analyzed the SSV4 capture regions and the percent overlap with the coding sequence. We reported the coverage statistics for known ASD risk genes on the basis of the number of exons that had insufficient (d < 5×) or low (5× ≤ d < 10×) coverage or that were not on the exome capture target (Figure S2). Coding exons were defined as 100% overlapping the CDS; all other exons were considered to be noncoding. To assess depth distribution, we first calculated the average depth across exome and whole-genome samples and then plotted the number of coding and noncoding exons whose depth was equal to or greater than a given threshold (5×, 10×, 20×); coding exons were defined as those that were 100% overlapping with the CDS, and all other exons were considered to be noncoding. This analysis was done for all chromosomes together, as well as for the X chromosome alone. We also generated a segmented distribution curve, resembling a density curve, based on the number of exons within 10 depth unit bins (e.g., −5:5, 5:15, …). Keeping male and female samples separate, we generated a more detailed bar plot to display the number of coding and noncoding exon intervals for the same depth thresholds (Figure S2).
Results
Genetic Variants in Whole-Genome Sequence
Thirty-two trio families comprising a child with ASD (index case or proband) and both biological parents were selected for WGS. Our primary criteria for selection were that the index case fulfilled a formal diagnosis of ASD according to gold-standard tests and our previous studies, that no member of the trio carried a known pathogenic CNV,7,20,26,43,44 and that all trio members had blood-derived genomic DNA available. Otherwise, the families were randomly selected from a larger research cohort of more than 180 families of children with ASD, for which similar criteria were met (The Autism Simplex Collection). Beyond the trio, in many cases additional siblings and relatives (with varying levels of phenotype data) were also available for examination (Figure 2 and Table S6).
Figure 2.
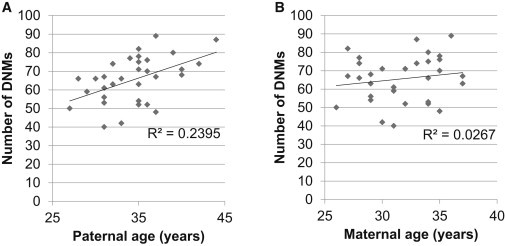
Correlation between Number of De Novo Mutations and Parental Ages
The total number of de novo mutations (DNMs) in the proband was plotted against the age of (A) the father and (B) the mother. The number of mutations increases with the age of the father (p = 0.0045) at a rate of 1.5 de novo mutations per year (slope).
The average coverage relative to the human genome reference sequence was 99.8%, and average sequence depth was 38.4× (Table S1). We detected an average of 3,210,237 SNVs and 804,635 indels per genome. Of these, an average of 19,241 SNVs and 510 indels were in exonic regions. The concordance of SNVs between WGS and microarray calls was high and ranged from 99.1% to 99.9% per sample.45 The overall validation rate for indels was not determined but was expected to be low, as previously reported.46 In ASD probands, there were 8,982 nonsynonymous SNVs (missense and nonsense mutations) and splice-site mutations and 444 indels per individual (Table S4); 231 SNVs and 328 indels, on average, had not been identified previously. As in other studies, herein we use the term “mutation” to describe rare variants (defined as those not found in the dbSNP or 1000 Genomes Project) that were predicted to be deleterious (loss-of-function and predicted damaging missense) by DNA sequence analysis (Figure 1). 10–13,31,47–49
We used the program CNVnator and called an average of 5,705 CNVs per individual from the WGS data; this number is 100 times more than the number of CNVs detected by the CytoScan HD microarray (which detected 45 CNVs per individual) (Figure S3A). This is in part due to the limited probe density for smaller CNVs (e.g., CNV size < 10 kb) in the microarray platform (Figure S3B). However, when restricted to the regions where microarray has sufficient coverage (more than 5 probes in > 10 kb), we found that CNVnator has a specificity of only 12% and a sensitivity of 75% (Figures S3C and S3D). Despite the suboptimal CNV detection performance in particular for smaller unbalanced changes,50 we analyzed all available data and confirmed that there was no obvious rare exonic CNV in the known pathogenic regions.
De Novo Mutations
Detection of de novo mutations (mutations presumably arising in a parental germline) represents an effective approach to identifying genes linked to ASD.10–13 Using a systemic approach to identify de novo variants (Figure 1), we detected 2,097 de novo SNVs (65.5 per individual; Table S2) and 164 de novo indels (5.1 per individual; Table S3). There were 42 de novo variants detected in exons (1.3 per individual) (Table S5); of these variants, 34 were nonsynonymous, and five were synonymous (ratio 6.8, which exceeds the ratio of 1 seen across a single genome51). There were three nonsense and 31 missense mutations. Functional analysis with both PolyPhen-2 and SIFT predicted that 13 of the 31 missense mutations would be deleterious. The three de novo indels detected were all predicted to cause frameshift mutations in the affected genes. Consistent with previous studies,31,49 we found that the number of de novo mutations was significantly correlated with paternal age (p < 0.005), but not with maternal age (p = 0.37) (Figure 2).
Of these 32 probands, 15 (47%) were found to carry at least one de novo deleterious mutation (Tables S5 and S6). By evaluating the severity and medical relevance of these mutations (Figure 1), we determined that, in six of 32 (19%) probands, these de novo events possibly contributed to clinical symptoms (Figures 3A–3F; Table 1). In family 2-1186, the proband (II-1 in Figure 3A) had a de novo nonsense mutation (c.219T>A [p.Tyr73∗]) that disrupts the VIP (MIM 192320; RefSeq accession number NM_003381.3) on chromosome 6. The proband has average intelligence by standardized testing and a clinical diagnosis of Asperger syndrome. The younger male sibling of the proband (I-2 in Figure 3A) also carried the mutation. Upon revisiting this individual’s records, the referring clinician (without knowledge of the molecular result) indicated that he had subclinical features of ASD and needed to be called back for a full assessment (clinical details are given in Table S6). He presented with expressive language delay, above-average receptive vocabulary, and a history of “clumsiness.” The mutation was not found in either parent, which might reflect gonadal mosaicism in one of them. VIP is involved in circadian rhythm, and mutant mice exhibit social behavioral and cognitive deficits.52,53 In family 2-1305, the proband (II-1 in Figure 3B) had a nonsense de novo mutation (c.1195C>T [p.Gln399∗]) in CAPRIN1 (MIM 601178; RefSeq NM_005898.4), which is functionally related to the fragile X protein (FMRP).54 According to medical history, this proband had fetal distress and a respiratory problem. These features have similarly been seen in Caprin1-null mice55 (Table S7). In family 2-1337, the proband with intellectual disability (II-2 in Figure 3C) had a de novo 32 bp deletion (c.1317–1348del [p.440–450del]) that causes a frameshift in an ASD-linked gene, SCN2A (RefSeq NM_001040143.1), which encodes the type II alpha subunit of a voltage-gated sodium channel.3,10,13 A female sibling with ASD but normal IQ did not have this mutation, suggesting that the phenotype associated with this mutation might be intellectual disability rather than ASD per se. On the other hand, Sanders et al. reported that two ASD probands from the Simons Simplex Collection had de novo loss-of-function mutations in SCN2A, but information about IQ was not provided.10 Rauch et al. reported three probands with severe intellectual disability and de novo mutations in SCN2A; two of these individuals had autistic features.56 A possible scenario in our family is that the SCN2A mutation exerts a greater effect on overall intellectual function and that the ASD phenotype is incompletely penetrant. Alternatively, multiple incompletely penetrant genetic variants might contribute to autism in these siblings, which would be consistent with the often multigenic nature of familial ASD and has been observed in other families.22 The proband in family 2-1370 (II-2 in Figure 3D) had a de novo 1 bp insertion (c.1194dup [p.Ser399Glufs∗10]) in USP54 (RefSeq NM_152586.3), which codes for ubiquitin peptidase in the brain.57 There is no other supporting genetic evidence of a role for this gene in ASD, so its relevance remains to be determined. The male proband in family 2-1303 (II-1 in Figure 3E), diagnosed with ASD and borderline intellect at age 14 years, was found to have a nonsense de novo mutation (c.1267C>T [p.Arg423∗]) in the X-linked KAL1 (MIM 300836; RefSeq NM_000216.2), associated with Kallmann syndrome (MIM 308700). The boy was clinically diagnosed with Kallmann syndrome prior to WGS (but after ASD diagnosis) because of hypogonadism and anosmia. Therefore, in this case we are confident that the p.Arg423∗ substitution in KAL1 causes his Kallmann syndrome, and possibly contributes to his ASD on the basis of the protein’s role in neural development.58,59 However, we are not aware of previous reports of autistic features in KAL1-mutation-positive individuals with Kallmann syndrome, so this is speculative pending further study. In family 2-1276, a de novo frameshift insertion (c.1193-1194insA [p.Ser399Leufs∗10]) in MICALCL (MIM 612355; RefSeq NM_032867.2) was identified in the proband (II-1 in Figure 3F). This gene is largely uncharacterized, but exome sequencing had revealed a de novo missense mutation in an unrelated individual with ASD.12 Proband 2-1276-03 (II-1 in Figure 3F) also has a de novo deleterious missense mutation (c.1903C>T [p.Arg635Trp]) in the DNA methyltransferase, encoded by DNMT3A (MIM 602769; RefSeq NM_175629.2).
Figure 3.
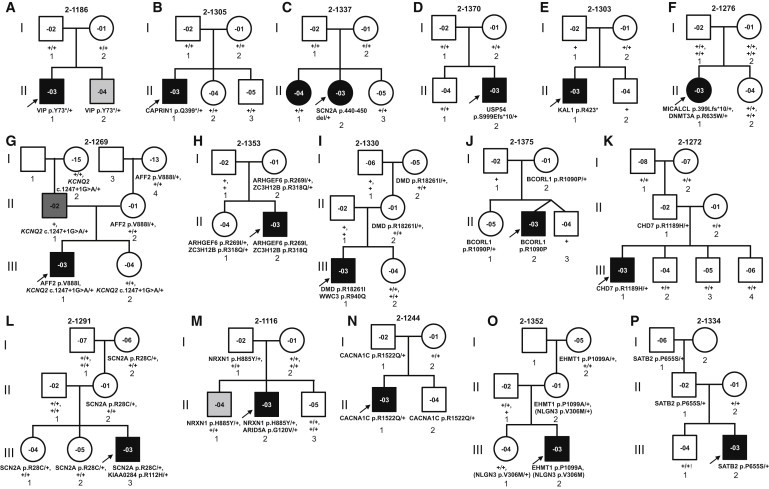
Pedigrees of Families with ASD-Relevant Genetic Variants
(A–F) Families with de novo deleterious variants as potential causal events.
(G–P) Families with inherited deleterious variants as potential causes of ASD (the sibling in Family A was also found to be a mutation carrier, suggesting gonadal mosaicism).
The de novo or inherited variant alleles are shown below each family member. “+” indicates the allele containing the reference (presumably wild-type) sequence (“+/+” for genes on the autosomal chromosome or the X chromosome in female; “+” for genes on the X chromosome in male). Males are denoted by squares and females by circles. Symbols with no inside number indicate that no DNA sample was available for testing. Black symbols indicate individuals diagnosed with ASD. Individual 2-1269-03 (dark gray symbol) was diagnosed with Asperger disorder in adulthood. Symbols filled in light gray (2-1186-04 [A; II-2] and 2-1116-04 [M; II-1]) denote individuals with subclinical features of ASD. As discussed in the Results, individual 2-1269-04 (G; III-2) was also reported by the parents to exhibit autistic-like behavior, and a full ASD evaluation is ongoing. Open symbols denote unaffected individuals, according to currently available information, but status for some may change upon retrospective evaluation. Arrows indicate ASD probands in each family.
Table 1.
Summary of ASDa-Associated Genes Found with Mutations in this Study
|
Known Genes |
Unrecognized Genes |
Candidate Genes |
||||
|---|---|---|---|---|---|---|
| X-Linked | Autosomal | X-Linked | Autosomal | X-Linked | Autosomal | |
| Missense (damaging) | AFF2, ARHGEF6, DMD | CACNA1C, CHD7, EHMT1, SATB2, SCN2A, NRXN1 | - | - | BCORL1, WWC3,bZC3H12B | ARID5A,bDNMT3A,bKIAA0284b |
| Nonsense | - | - | KAL1b,c | CAPRIN1,b,dVIPb,d | - | - |
| Frameshift | - | SCN2Ab | - | - | - | USP54,bMICALCLb |
| Splice Site | - | - | - | KCNQ2c | - | - |
Mutations of interest were found in 16 families, of which five families carried more than one mutation. Deleterious mutations were found in four unrecognized genes in four families, in nine known genes in ten families, and in eight candidate genes in seven families.
Genes with de novo mutations in the proband.
Supported by human phenotype from OMIM.
Supported by mouse phenotype from MGI.
Inherited Mutations in X-Linked Genes
Given the strong male-to-female gender bias in ASD60 and recent progress in finding X-linked forms of ASD and intellectual disability,61–63 we sought to detect additional X-linked mutations that males had inherited from their unaffected mothers. We identified 12 rare X-linked deleterious variants among eight male ASD probands and their mothers. Of these, five mutations (in four families) affected previously identified ASD candidate genes (Figures 3G–3J). In family 2-1269 (Figure 3G), a rare missense mutation (c.2662G>A [p.Val888Ile]) was found in AFF2 (MIM 300806; RefSeq NM 001170628.1), mutations in which cause fragile X E syndrome (MIM 309548).64 The male proband in family 2-1353 (II-2 in Figure 3H) has missense mutations (c.806G>T [p.Arg269Ile]; c.953G>A [p.Arg318Gln]) in ARHGEF6 (MIM 300267; RefSeq NM_004840.2) and ZC3H12B (MIM 300889; RefSeq NM 001010888.3); the former gene is implicated in X-linked intellectual disability (MIM 300436),3 and the latter has been found to be mutated in another unrelated individual with ASD.11 The male proband of family 2-1330 (III-1 in Figure 3I) inherited an X-linked missense mutation (c.5477G>T [p.Arg1826Ile]) in DMD (MIM 300377; RefSeq NM_004009.3), mutations in which cause dystrophinopathies Duchenne and Becker muscular dystrophy (DMD and BMD; MIM 310200 and 300376, respectively). Previous reports have suggested an increased incidence of ASDs in boys with DMD.3,21 The individual in question is now 14 years old and does not show obvious signs of muscular dystrophy, but he will need to be assessed further. In addition, a de novo missense mutation (c.2819G>A [p.Arg940Gln]) was identified in the uncharacterized X-linked gene WWC3 (NM_015691.3). Finally, we found a rare missense BCORL1 (MIM 300688; RefSeq NM_021946.4) mutation (c.3269G>C [p.Arg1090Pro]) that was predicted to be damaging in the male ASD-affected proband of family 2-1375 (II-2 in Figure 3J). A de novo missense mutation (c.2390G>A [p.Cys797Tyr]) of the same gene was also reported in another unrelated individual with ASD.10
Inherited Dominant Mutations in Autosomal Genes
Heritability estimates strongly support a significant role for autosomal inherited factors,18,23,24 but genome-wide studies of inherited autosomal mutations in ASD have so far been limited to the model of recessive inheritance;63,65,66 some mutations have been identified.19,63,65,66 Given the expanding number of rare variants in the human population,67–69 we also explored the possibility of accumulation of deleterious variants in the known ASD-linked genes with haploinsufficiency, in addition to the recessive ones in these 32 families.
We found that seven out of 32 (22%) families carried rare mutations that are predicted to have deleterious effects, that have been associated with ASD, and that segregate in an autosomal-dominant manner (Figures 3G and 3K–3P). Family 2-1269 (Figure 3G), described above with regard to the X-linked variant in AFF2, also carried a rare paternally inherited splice-site mutation (c.1247+1G>A) in KCNQ2 (MIM 602235; RefSeq NM_172109.1). Defects in KCNQ2 are implicated in benign familial neonatal seizures type 1 (MIM 121200).70 Microdeletions encompassing KCNQ2 have been reported in ASD cases.71 Mutations in this gene and its other gene family member, KCNQ3 (MIM 602232), have also recently been associated with intellectual disability in a few families.56,72 Medical history indicates that seizures were present in both the proband (III-1) and father (II-1) during early infancy.4 In fact, as a result of a follow-up interview motivated by our genetic findings, the female sibling, who carried the KCNQ2 mutation, was also reported by parents to exhibit autistic-like behavior, and she is now undergoing formal examination. In family 2-1272, a CHD7 (MIM 608892; RefSeq NM 017780.3) missense mutation (c.3566G>A [p.Arg1189His]) that is predicted to be damaging was found in the proband (III-1 in Figure 3K), and although this alteration was inherited from his father (II-1), it was not observed in biological grandparents (I-1 and I-2). Mutations of CHD7 are associated with CHARGE syndrome (MIM 214800), a developmental condition in which 27.5%–42% of the affected individuals are reported to have autistic-like behavior.73,74 The proband carrying the mutation has some symptoms, such as sleep apnea and frequent ear infections,75,76 that can be found in CHARGE syndrome, but determining whether he fits full clinical criteria for the diagnosis (Table S7) will require assessment by a clinical geneticist. Fewer clinical data are available for the father, who should also be assessed for CHARGE syndrome and/or ASD features.
The proband (III-3 in Figure 3G) and two unaffected sisters (III-1 and III-2 in Figure 3G) of family 2-1291 inherited, from the non-ASD mother, a rare p.Arg28Cys missense substitution (c.82C>T) in SCN2A, which was also mutated in family 2-1337 and has been found in other recent studies to be involved in ASD, as well as intellectual disability.10,13,48,56 The ASD proband of family 2-1116 (II-2 in Figure 3M) and one brother (II-1 in Figure 3M) inherited a rare missense mutation (c.2653C>T [p.His885Tyr]) in NRXN1 (MIM 600565; RefSeq NM_001135659.1) from their father (I-1 in Figure 3M). Although the brother has not been diagnosed with ASD, he has learning disability. Mutations in CACNA1C (MIM 114205; RefSeq NM_001129842.1) can cause ASD-associated Timothy syndrome (MIM 601005), and a rare missense mutation (c.4565G>A [p.Arg1522Gln) of this gene in family 2-1244 is carried by the proband (II-1 in Figure 3N) and his unaffected father (I-1 in Figure 3N) and brother (II-2 in Figure 3N). The familial mutation is not the same as that typically associated with Timothy syndrome, so it is unclear whether the family is at risk for arrhythmias (MIM 600919) that are typically associated with this condition. The proband in family 2-1352 (III-2 in Figure 3O) inherited from his mother (II-2 in Figure 3O) rare missense mutations (c.3295C>G [p.Pro1099Ala]; c.916G>A [p.Val306Met]) in both EHMT1 (MIM 607001; RefSeq NM_024757.4) and X-linked NLGN3 (MIM 300336; RefSeq NM_001166660.1) (although the latter alteration is predicted to be benign by bioinformatic analysis). Finally, the proband in family 2-1334 (III-2 in Figure 3P) inherited a rare missense mutation (c.1963C>T [p.Pro655Ser]) in SATB2 (MIM 608148; RefSeq NM_015265.3) from his unaffected father (II-1 in Figure 3P) and grandfather (I-1 in Figure 3P). No biallelic mutations were identified in ASD candidate genes previously shown to demonstrate autosomal-recessive inheritance patterns3,63,77.
Coverage of Coding Exons by WGS
To evaluate the performance of variant detection by WGS, we compared the sequence coverage in the exonic regions between WGS and whole-exome sequencing (WES). In the autosomal regions, we found that WGS covered at least 10.8% more annotated exons than WES did (Figure 4). Furthermore, 9.2% (11/119) of ASD-linked genes have at least one exon untargeted by the most widely used capture kit currently available for exome sequencing (Figure S4; see also the Discussion). A more detailed comparison between the two technologies' coverage in the coding regions showed that WGS captured 2.7% more annotated coding exons with coverage sufficient for variant calling (>5x) than did WES (Figure 4). The coverage difference was even more prominent for the X chromosome and gene splice sites (Figure S2), the former of which carries a disproportionate number of ASD susceptibility genes.
Figure 4.
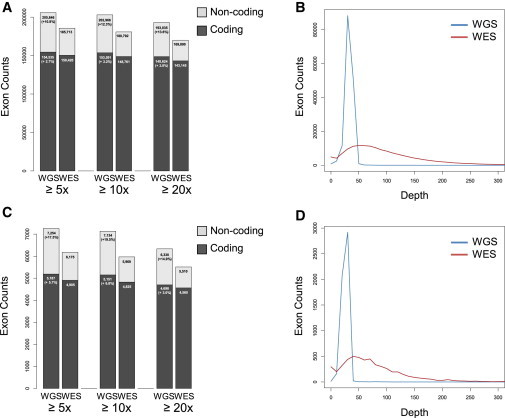
Sequencing Coverage in Exonic Regions
(A) Comparison of sequencing depth in autosomal exonic regions.
(B) Comparison of sequencing read distribution in autosomal exonic regions.
(C) Comparison of sequencing depth in X chromosome exonic regions.
(D) Comparison of sequencing read distribution in X chromosome exonic regions.
WGS captured (A) 10.8% more annotated exons (2.7% more coding) in autosomes and (C) 17.5% more annotated exons (5.7% more coding) in the X chromosome with coverage >5x than WES did. WGS also showed a more uniform capturing of coding regions than WES (B and D).
Discussion
Multiple lines of evidence demonstrate the importance of complex genetic factors in ASD (chromosomal abnormalities, CNVs, indels, SNVs).5,6 At the same time, careful clinical characterization has revealed that ASD is extremely heterogeneous, in that it ranges from milder Asperger disorder to more complex forms associated with profound intellectual disability, epilepsy and other medical problems.1,3 As a pilot study, we used WGS in a group of families with at least one child affected by ASD to provide more comprehensive detection of the wide-ranging genetic variants that might contribute to ASD risk. We, as others have, sought de novo variants,10–13,31,49 but given the well-established role of inherited factors in ASD,18,23,24 we also considered rare autosomal and X-linked inherited variants in families. Following this design, we ascertained potentially significant variants in 16 of 32 families (50%); this potential significance was either because the genes involved have an established association with clinical syndromes found among individuals with ASD or because they are linked to ASD and/or related disabilities, such as intellectual disability, or medical co-morbidities, such as seizures. Further investigation of the clinical relevance of these variants is ongoing. Some of the detected mutations could reasonably be considered pathogenic and/or have possible implications for clinical management or genetic counseling for the proband or family members. For other mutations, their role in the condition needs to be closely followed in the medical literature (Table S7; Figure 3). Such putative mutations were identified in nine genes previously linked to ASD, as well as in four unrecognized and eight candidate ASD risk genes (defined in Figure 1 legend) in these 16 families (some carried more than one mutation) (Table 1).
Thirteen out of 32 (41%) probands (and some family members) would be prioritized for comprehensive clinical assessment on the basis of the presence of the mutation and the predicted functional impact of the mutation(s) and gene(s) (Table S7). These include all probands who carry mutations in known and unrecognized ASD-linked genes (Table 1). The c.5477G>T mutation in DMD in a boy with ASD (family 2-1330; III-1 in Figure 3I) indicated that he should be examined for dystrophinopathy, which is associated with progressive muscle weakness and cardiomyopathy. In family 2-1303, the ASD proband (II-1 in Figure 3E) later received a diagnosis of Kallmann syndrome (mirror movements, microphallus, and unilateral renal agenesis [MIM 308700]).78 WGS detected a de novo mutation in KAL1, an X-linked gene that accounts for ∼10% of cases of Kallmann syndrome (see GeneReviews entry in Web Resources), and after genetic confirmation of the disorder, management can include hormone treatment to initiate puberty. Detection of a c.3566G>A mutation in CHD7 in the proband (III-1 in Figure 3K) and father (II-1 in Figure 3K) in family 2-1272 indicates that they should be assessed for CHARGE syndrome.75,76 CHD7 mutations are also found in rare cases of Kallmann syndrome, which, like CHARGE syndrome, can feature sensory impairment and hypogonadism.79 In families 2-1337, 2-1291, 2-1244, and 2-1352, carriers of mutations of SCN2A, CACNA1C, and EHMT1 should be assessed for features of epilepsy, Timothy syndrome, and Kleefstra syndrome (MIM 610253), respectively. These individuals, along with the sibling who was a carrier of the VIP mutation and reportedly exhibited symptoms of ASD, should also undergo full assessment for ASD and/or related neurodevelopmental disorders.
In the future, results of WGS might allow earlier diagnosis of ASD, especially in siblings, for whom recurrence rates are approximately 18%.80 For families of probands with de novo mutations in genes such as SCN2A and CAPRIN1, recurrence risk might be similar to that of the general population (but note that, as in family 2-1186, germline mosaicism can confound estimates).81 The presence of rare inherited variants in ASD candidate genes such as CACNA1C, CHD7, and KCNQ2, as well as X-linked mutations in genes such as AFF2 and ARGEF6 in males, might confer increased risk that would lead to a recommendation of increased developmental monitoring to allow earlier identification. The average age of ASD diagnosis in this group of 16 families was 4.4 years old; for some of these families mentioned above, the genetic findings could have informed diagnosis, thus shortening the diagnostic path and allowing children to benefit more fully from early interventions.82 The X-linked gene mutations also help to explain the male gender bias observed in ASD.
The reported genetic variants also highlight potential molecular targets for pharmacologic manipulation and open the way for personalized therapeutics in ASD. For example, personal mutations in CAPRIN1, AFF2, and SCN2A (families 2-1305, 2-1337, 2-1269, and 2-1291) identify carriers as potential candidates for drug trials, involving allosteric modulators of GABA receptors, which have ameliorated autism-like symptoms in mice.83–85 Mutation of KCNQ2 in family 2-1269 identifies a candidate pathway for treatment by Kv7 channel openers, such as retigabine.86 The VIP variant highlights a potential drug target pathway, in that the family of receptors (VPAC1 and VPAC2) that mediate its effects, along with related peptides such as pituitary adenylate cyclase-activating polypeptide (PACAP), are considered to be drug targets in neurodegenerative and inflammatory diseases.87
With respect to sequencing, we detected a higher rate of de novo mutations than was previously found in ASD exome projects,10–13 which could be due to the more uniform capturing that WGS affords of the entire coding regions (Figure 4). Although paternal age difference between studies might contribute to the increase in yield, we did not find a significant difference between the paternal age in our study and that in others10–13 (Figure 2; average age 34, range: 27–44). We also note that six out of 56 (11%) of the de novo and clinically relevant mutations, including the mutations in VIP and KCNQ2, found in this study would not have been captured by exome sequencing (Table S5), although this might depend on the capturing method used and the coverage achieved. WGS also demonstrated a more uniform coverage; the majority of the exon samples had a coverage of ∼30× (ranging from 0 to ∼50x), whereas WES had a distribution of coverage ranging from 0 to ∼200× (Figure 4B). A similar pattern of coverage difference was observed on the X chromosome (Figures 4C and 4D), but there was an even higher extent of difference: at least 17.5% more annotated exons (5.7% more coding) were covered by WGS than by WES. Such a coverage difference was even more pronounced when regions such as gene splice sites were considered (Figure S2). Supporting our findings, a recent study reported that WES could not detect 11% of the variants detected by WGS.88 Although there are also WES-specific variants that could be missed by WGS, the number was less significant.89 Of course, WGS also covers the entire genome. However, calling CNVs from WGS remains a challenge.90 Using a read-depth-based approach for CNV detection, we found that as many as 88% of the calls could be false positives (Figure S3). As we test other existing and improved annotation algorithms, the as-yet-undiscovered indels and CNVs in both genic and nongenic intervals will also become available for genotype and phenotype studies in these and other ASD families.
We have presented a comprehensive analysis of the genomes of individuals with ASD by using WGS. Although limited by the small sample size (32 unrelated trios), we have attempted to fully utilize the public databases on allelic frequency and functional information to delineate the underlying genetic variants contributing to ASD risk. The genetic variants reported in this study are extremely rare because they are not present in more than thousands of non-ASD individuals from public databases. Because these mutations were specific to the families, genotype-phenotype correlation could only be performed in the context of the corresponding families, particularly for the inherited mutations. All autosomally inherited deleterious mutations we present here reside in ASD candidate genes whose established mechanism of action is likely to be haploinsufficiency. Thus, they were likely to be expressed in a dominant manner, in some cases possibly with incomplete penetrance and/or variable expressivity. Although each individual carries hundreds of deleterious variants, many have been previously documented to be recessive and therefore are not known to be pathogenic. A recent study of rare deleterious variants in the whole genomes of 179 individuals revealed that very few known pathogenic variants were found in genes associated with dominant disorders after very stringent filters were applied, and the associated phenotypes were either late onset or no overt disease,47 suggesting that rare deleterious mutations found in genes associated with known dominant disorders are rare. Indeed, some families in our study showed a correlation of phenotype with the associated known outcome of the variants (e.g., family 2-1269, showing the KCNQ2 mutation, among other families in Table S7), but the reason that some carriers were unaffected remains to be determined. Further population-genetic and functional studies of the variants, and reassessments of the carriers, are needed to address this issue.
In conclusion, our study provides evidence that WGS data can aid in the detection and clinical evaluation of individuals with ASD and their families. The diagnostic yield and clinical utility should increase as more undetected structural genetic variants are discovered and characterized and as additional individuals with ASD are studied. Although the cost per sample for WGS is currently ∼2.5 times more than that for WES, these results suggest that more comprehensive coverage of the exome and noncoding regions offers higher yield in detected mutations which, in some cases, have significant diagnostic and clinical utility for individuals with ASD and their families. As more samples are analyzed by WGS, a thorough health-technology assessment study will need to be performed. Importantly, our genomic data reinforce the growing number of observations that significant clinical and genetic heterogeneity and pleiotropy exist in ASD. WGS offers one tool that can help to advance the understanding of the genetic architecture of ASD.
Acknowledgments
We thank Dr. Bernie Devlin for guidance in study design, the families for their participation, The Centre for Applied Genomics, and BGI-Shenzhen. This work was supported by Autism Speaks, Autism Speaks Canada, the National Institutes of Health (NIMH R01MH098114-01), the National Science and Technology Ministry Project 973 program (2011CB809200), the National Science and Technology Ministry Project 863 program (2012AA02A201), the National Natural Science Foundation of China Major Program (30890032), the Shenzhen Municipal Government of China (CXB201108250094A, CXB201108250096A, ZYC200903240080A, ZYC201105170397A, and ZYC201105170394A), the Enterprise Key Laboratory supported by Guangdong Province (the Guangdong Enterprise Key Laboratory of Human Disease Genomics), Shenzhen Key Laboratory of Gene Bank for National Life Science (National Gene Bank Project of China), NeuroDevNet, the Canadian Institutes for Advanced Research, the University of Toronto McLaughlin Centre, Genome Canada/Ontario Genomics Institute, the government of Ontario, the Canadian Institutes of Health Research, and The Hospital for Sick Children Foundation. R.K.C.Y. holds an Autism Speaks Postdoctoral Fellowship in Translational Research. S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children. S.W.S. is on the Scientific Advisory Board of Population Diagnositics (a US Company) and is a founding scientist of YouNique Genomics, both of which could use data from this study. E.A. has received consultation fees from Novartis and Seaside Therapeutics. She has also received an unrestricted grant from Sanofi Canada and has received research funding from SynaptDx. G.D. is on the Professional Advisory Board for Integragen Inc.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Yingrui Li, Email: liyr@genomics.cn.
Stephen W. Scherer, Email: stephen.scherer@sickkids.ca.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Exome Variant Server, http://evs.gs.washington.edu/EVS/
GeneReviews, Pallais, J.C., Au, M., Pitteloud, N., Seminara, S., and Crowley, W.F. (2011). Kallmann Syndrome, http://www.ncbi.nlm.nih.gov/books/NBK1334/
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Accession Numbers
The raw sequencing data reported in this paper have been deposited at the Autism Genetic Resource Exchange and the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/), which is hosted at the European Bioinformatics Institute, under accession number EGAS00001000556.
References
- 1.Scherer S.W., Dawson G. Risk factors for autism: translating genomic discoveries into diagnostics. Hum. Genet. 2011;130:123–148. doi: 10.1007/s00439-011-1037-2. [DOI] [PubMed] [Google Scholar]
- 2.Carter M.T., Scherer S.W. Autism spectrum disorder in the genetics clinic: a review. Clin. Genet. 2013;83:399–407. doi: 10.1111/cge.12101. [DOI] [PubMed] [Google Scholar]
- 3.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 4.Tuchman R., Cuccaro M. Epilepsy and autism: neurodevelopmental perspective. Curr. Neurol. Neurosci. Rep. 2011;11:428–434. doi: 10.1007/s11910-011-0195-x. [DOI] [PubMed] [Google Scholar]
- 5.Devlin B., Scherer S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Basu S.N., Kollu R., Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37(Database issue):D832–D836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen Y., Dies K.A., Holm I.A., Bridgemohan C., Sobeih M.M., Caronna E.B., Miller K.J., Frazier J.A., Silverstein I., Picker J., Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–e735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxbaum J.D., Daly M.J., Devlin B., Lehner T., Roeder K., State M.W., Autism Sequencing Consortium The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron. 2012;76:1052–1056. doi: 10.1016/j.neuron.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiśniowiecka-Kowalnik B., Nesteruk M., Peters S.U., Xia Z., Cooper M.L., Savage S., Amato R.S., Bader P., Browning M.F., Haun C.L. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010;153B:983–993. doi: 10.1002/ajmg.b.31064. [DOI] [PubMed] [Google Scholar]
- 16.Noor A., Whibley A., Marshall C.R., Gianakopoulos P.J., Piton A., Carson A.R., Orlic-Milacic M., Lionel A.C., Sato D., Pinto D., Autism Genome Project Consortium Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3001267. 49ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerin T., Ramanathan A., Rivas K., Grepo N., Coetzee G.A., Campbell D.B. A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003479. 28ra40. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X., Leotta A., Kustanovich V., Lajonchere C., Geschwind D.H., Law K., Law P., Qiu S., Lord C., Sebat J. A unified genetic theory for sporadic and inherited autism. Proc. Natl. Acad. Sci. USA. 2007;104:12831–12836. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato D., Lionel A.C., Leblond C.S., Prasad A., Pinto D., Walker S., O’Connor I., Russell C., Drmic I.E., Hamdan F.F. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012;90:879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pagnamenta A.T., Holt R., Yusuf M., Pinto D., Wing K., Betancur C., Scherer S.W., Volpi E.V., Monaco A.P. A family with autism and rare copy number variants disrupting the Duchenne/Becker muscular dystrophy gene DMD and TRPM3. J Neurodev Disord. 2011;3:124–131. doi: 10.1007/s11689-011-9076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leblond C.S., Heinrich J., Delorme R., Proepper C., Betancur C., Huguet G., Konyukh M., Chaste P., Ey E., Rastam M. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey A., Le Couteur A., Gottesman I., Bolton P., Simonoff E., Yuzda E., Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 24.Hallmayer J., Cleveland S., Torres A., Phillips J., Cohen B., Torigoe T., Miller J., Fedele A., Collins J., Smith K. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Constantino J.N., Todorov A., Hilton C., Law P., Zhang Y., Molloy E., Fitzgerald R., Geschwind D. Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol. Psychiatry. 2013;18:137–138. doi: 10.1038/mp.2012.9. [DOI] [PubMed] [Google Scholar]
- 26.Prasad A., Merico D., Thiruvahindrapuram B., Wei J., Lionel A.C., Sato D., Rickaby J., Lu C., Szatmari P., Roberts W. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michaelson J.J., Shi Y., Gujral M., Zheng H., Malhotra D., Jin X., Jian M., Liu G., Greer D., Bhandari A. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X., Yuan J., Shi Y., Lu J., Liu B., Li Z., Chen Y., Mu D., Zhang H., Li N. pIRS: Profile-based Illumina pair-end reads simulator. Bioinformatics. 2012;28:1533–1535. doi: 10.1093/bioinformatics/bts187. [DOI] [PubMed] [Google Scholar]
- 33.Pinto D., Darvishi K., Shi X., Rajan D., Rigler D., Fitzgerald T., Lionel A.C., Thiruvahindrapuram B., Macdonald J.R., Mills R. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011;29:512–520. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abyzov A., Urban A.E., Snyder M., Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Feuk L., Duggan G.E., Khaja R., Scherer S.W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet. Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 36.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 38.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas P.D., Kejariwal A., Campbell M.J., Mi H., Diemer K., Guo N., Ladunga I., Ulitsky-Lazareva B., Muruganujan A., Rabkin S. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 42.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 43.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L., Autism Genome Project Consortium Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lionel A.C., Crosbie J., Barbosa N., Goodale T., Thiruvahindrapuram B., Rickaby J., Gazzellone M., Carson A.R., Howe J.L., Wang Z. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002464. 95ra75. [DOI] [PubMed] [Google Scholar]
- 45.Kirkness E.F., Grindberg R.V., Yee-Greenbaum J., Marshall C.R., Scherer S.W., Lasken R.S., Venter J.C. Sequencing of isolated sperm cells for direct haplotyping of a human genome. Genome Res. 2013;23:826–832. doi: 10.1101/gr.144600.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam H.Y., Clark M.J., Chen R., Chen R., Natsoulis G., O’Huallachain M., Dewey F.E., Habegger L., Ashley E.A., Gerstein M.B. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 2012;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., Tyler-Smith C., 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 49.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pang A.W., MacDonald J.R., Pinto D., Wei J., Rafiq M.A., Conrad D.F., Park H., Hurles M.E., Lee C., Venter J.C. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng P.C., Levy S., Huang J., Stockwell T.B., Walenz B.P., Li K., Axelrod N., Busam D.A., Strausberg R.L., Venter J.C. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaudhury D., Loh D.H., Dragich J.M., Hagopian A., Colwell C.S. Select cognitive deficits in vasoactive intestinal peptide deficient mice. BMC Neurosci. 2008;9:63. doi: 10.1186/1471-2202-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill J.M., Cuasay K., Abebe D.T. Vasoactive intestinal peptide antagonist treatment during mouse embryogenesis impairs social behavior and cognitive function of adult male offspring. Exp. Neurol. 2007;206:101–113. doi: 10.1016/j.expneurol.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 54.El Fatimy R., Tremblay S., Dury A.Y., Solomon S., De Koninck P., Schrader J.W., Khandjian E.W. Fragile X mental retardation protein interacts with the RNA-binding protein Caprin1 in neuronal RiboNucleoProtein complexes. PLoS ONE. 2012;7:e39338. doi: 10.1371/journal.pone.0039338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiina N., Yamaguchi K., Tokunaga M. RNG105 deficiency impairs the dendritic localization of mRNAs for Na+/K+ ATPase subunit isoforms and leads to the degeneration of neuronal networks. J. Neurosci. 2010;30:12816–12830. doi: 10.1523/JNEUROSCI.6386-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 57.Su A.I., Cooke M.P., Ching K.A., Hakak Y., Walker J.R., Wiltshire T., Orth A.P., Vega R.G., Sapinoso L.M., Moqrich A. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cariboni A., Pimpinelli F., Colamarino S., Zaninetti R., Piccolella M., Rumio C., Piva F., Rugarli E.I., Maggi R. The product of X-linked Kallmann’s syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum. Mol. Genet. 2004;13:2781–2791. doi: 10.1093/hmg/ddh309. [DOI] [PubMed] [Google Scholar]
- 59.Bülow H.E., Berry K.L., Topper L.H., Peles E., Hobert O. Heparan sulfate proteoglycan-dependent induction of axon branching and axon misrouting by the Kallmann syndrome gene kal-1. Proc. Natl. Acad. Sci. USA. 2002;99:6346–6351. doi: 10.1073/pnas.092128099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillberg C., Cederlund M., Lamberg K., Zeijlon L. Brief report: “the autism epidemic”. The registered prevalence of autism in a Swedish urban area. J. Autism Dev. Disord. 2006;36:429–435. doi: 10.1007/s10803-006-0081-6. [DOI] [PubMed] [Google Scholar]
- 61.Piton A., Gauthier J., Hamdan F.F., Lafrenière R.G., Yang Y., Henrion E., Laurent S., Noreau A., Thibodeau P., Karemera L. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry. 2011;16:867–880. doi: 10.1038/mp.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nava C., Lamari F., Héron D., Mignot C., Rastetter A., Keren B., Cohen D., Faudet A., Bouteiller D., Gilleron M. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transcult. Psychiatry. 2012;2:e179. doi: 10.1038/tp.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim E.T., Raychaudhuri S., Sanders S.J., Stevens C., Sabo A., MacArthur D.G., Neale B.M., Kirby A., Ruderfer D.M., Fromer M., NHLBI Exome Sequencing Project Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gecz J., Gedeon A.K., Sutherland G.R., Mulley J.C. Identification of the gene FMR2, associated with FRAXE mental retardation. Nat. Genet. 1996;13:105–108. doi: 10.1038/ng0596-105. [DOI] [PubMed] [Google Scholar]
- 65.Chahrour M.H., Yu T.W., Lim E.T., Ataman B., Coulter M.E., Hill R.S., Stevens C.R., Schubert C.R., Greenberg M.E., Gabriel S.B., Walsh C.A., ARRA Autism Sequencing Collaboration Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu T.W., Chahrour M.H., Coulter M.E., Jiralerspong S., Okamura-Ikeda K., Ataman B., Schmitz-Abe K., Harmin D.A., Adli M., Malik A.N. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keinan A., Clark A.G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh N.A., Charlier C., Stauffer D., DuPont B.R., Leach R.J., Melis R., Ronen G.M., Bjerre I., Quattlebaum T., Murphy J.V. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 71.Mosca-Boidron A.L., Valduga M., Thauvin-Robinet C., Lagarde N., Marle N., Henry C., Pinoit J.M., Huet F., Béri-Deixheimer M., Ragon C. Additional evidence to support the role of the 20q13.33 region in susceptibility to autism. Am. J. Med. Genet. A. 2013;161:1505–1507. doi: 10.1002/ajmg.a.35878. [DOI] [PubMed] [Google Scholar]
- 72.Weckhuysen S., Mandelstam S., Suls A., Audenaert D., Deconinck T., Claes L.R., Deprez L., Smets K., Hristova D., Yordanova I. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012;71:15–25. doi: 10.1002/ana.22644. [DOI] [PubMed] [Google Scholar]
- 73.Johansson M., Gillberg C., Råstam M. Autism spectrum conditions in individuals with Möbius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: diagnostic aspects. Res. Dev. Disabil. 2010;31:9–24. doi: 10.1016/j.ridd.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 74.Vervloed M.P., Hoevenaars-van den Boom M.A., Knoors H., van Ravenswaaij C.M., Admiraal R.J. CHARGE syndrome: relations between behavioral characteristics and medical conditions. Am. J. Med. Genet. A. 2006;140:851–862. doi: 10.1002/ajmg.a.31193. [DOI] [PubMed] [Google Scholar]
- 75.Trider C.L., Corsten G., Morrison D., Hefner M., Davenport S., Blake K. Understanding obstructive sleep apnea in children with CHARGE syndrome. Int. J. Pediatr. Otorhinolaryngol. 2012;76:947–953. doi: 10.1016/j.ijporl.2012.02.061. [DOI] [PubMed] [Google Scholar]
- 76.Edwards B.M., Van Riper L.A., Kileny P.R. Clinical manifestations of CHARGE Association. Int. J. Pediatr. Otorhinolaryngol. 1995;33:23–42. doi: 10.1016/0165-5876(95)01188-h. [DOI] [PubMed] [Google Scholar]
- 77.Morrow E.M., Yoo S.Y., Flavell S.W., Kim T.K., Lin Y., Hill R.S., Mukaddes N.M., Balkhy S., Gascon G., Hashmi A. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hardelin J.P., Levilliers J., Blanchard S., Carel J.C., Leutenegger M., Pinard-Bertelletto J.P., Bouloux P., Petit C. Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum. Mol. Genet. 1993;2:373–377. doi: 10.1093/hmg/2.4.373. [DOI] [PubMed] [Google Scholar]
- 79.Bergman J.E., Janssen N., Hoefsloot L.H., Jongmans M.C., Hofstra R.M., van Ravenswaaij-Arts C.M. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J. Med. Genet. 2011;48:334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 80.Ozonoff S., Young G.S., Carter A., Messinger D., Yirmiya N., Zwaigenbaum L., Bryson S., Carver L.J., Constantino J.N., Dobkins K. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128:e488–e495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.State M.W., Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rogers S.J., Estes A., Lord C., Vismara L., Winter J., Fitzpatrick A., Guo M., Dawson G. Effects of a brief Early Start Denver model (ESDM)-based parent intervention on toddlers at risk for autism spectrum disorders: a randomized controlled trial. J. Am. Acad. Child Adolesc. Psychiatry. 2012;51:1052–1065. doi: 10.1016/j.jaac.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Silverman J.L., Smith D.G., Rizzo S.J., Karras M.N., Turner S.M., Tolu S.S., Bryce D.K., Smith D.L., Fonseca K., Ring R.H., Crawley J.N. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003501. 31ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Han S., Tai C., Westenbroek R.E., Yu F.H., Cheah C.S., Potter G.B., Rubenstein J.L., Scheuer T., de la Iglesia H.O., Catterall W.A. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henderson C., Wijetunge L., Kinoshita M.N., Shumway M., Hammond R.S., Postma F.R., Brynczka C., Rush R., Thomas A., Paylor R. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3004218. 152ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rundfeldt C., Netzer R. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells tranfected with human KCNQ2/3 subunits. Neurosci. Lett. 2000;282:73–76. doi: 10.1016/s0304-3940(00)00866-1. [DOI] [PubMed] [Google Scholar]
- 87.Chu A., Caldwell J.S., Chen Y.A. Identification and characterization of a small molecule antagonist of human VPAC(2) receptor. Mol. Pharmacol. 2010;77:95–101. doi: 10.1124/mol.109.060137. [DOI] [PubMed] [Google Scholar]
- 88.O’Rawe J., Jiang T., Sun G., Wu Y., Wang W., Hu J., Bodily P., Tian L., Hakonarson H., Johnson W.E. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5:28. doi: 10.1186/gm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clark M.J., Chen R., Lam H.Y., Karczewski K.J., Chen R., Euskirchen G., Butte A.J., Snyder M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teo S.M., Pawitan Y., Ku C.S., Chia K.S., Salim A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics. 2012;28:2711–2718. doi: 10.1093/bioinformatics/bts535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.