

Abstract
Defects of motile cilia cause primary ciliary dyskinesia (PCD), characterized by recurrent respiratory infections and male infertility. Using whole-exome resequencing and high-throughput mutation analysis, we identified recessive biallelic mutations in ZMYND10 in 14 families and mutations in the recently identified LRRC6 in 13 families. We show that ZMYND10 and LRRC6 interact and that certain ZMYND10 and LRRC6 mutations abrogate the interaction between the LRRC6 CS domain and the ZMYND10 C-terminal domain. Additionally, ZMYND10 and LRRC6 colocalize with the centriole markers SAS6 and PCM1. Mutations in ZMYND10 result in the absence of the axonemal protein components DNAH5 and DNALI1 from respiratory cilia. Animal models support the association between ZMYND10 and human PCD, given that zmynd10 knockdown in zebrafish caused ciliary paralysis leading to cystic kidneys and otolith defects and that knockdown in Xenopus interfered with ciliogenesis. Our findings suggest that a cytoplasmic protein complex containing ZMYND10 and LRRC6 is necessary for motile ciliary function.
Main Text
Primary ciliary dyskinesia (PCD) is a rare, Mendelian, autosomal-recessive disorder caused by defective structure and function of cilia or flagella and has an incidence of 1 in 16,000 individuals (MIM 244400). Ciliary dysfunction causes respiratory distress in term neonates, impaired mucociliary clearance, chronic cough, sinusitis, bronchiectasis, and male infertility1,2 (also see GeneReviews in Web Resources). Defective motility of embryonic nodal cilia (required for normal organ asymmetry) leads to situs abnormalities in ∼50% of cases.1–4 The diagnosis of PCD usually relies on documentation of axonemal ultrastructural defects, but this complex technique is not widely available and an increasing number of individuals with PCD have only functional defects with normal ultrastructure.5,6 Currently, mutations in 19 genes are known to cause PCD, but these account for only ∼65% of cases2,3,7–10 (also see GeneReviews in Web Resources). Therefore, additional genetic causes of PCD have yet to be identified.
The finding that each of the recently identified genes associated with PCD causes only a small number of cases necessitates an ability to identify additional PCD-causing mutations in unknown genes in unrelated families. We therefore developed a strategy that combines whole-exome resequencing (WER) and homozygosity mapping in single families and thereby reduces the high number of false-positive variants resulting from WER.11–14 We searched for PCD-causing mutations in a worldwide cohort of sibling pairs and single cases with PCD from 31 different families (Figure S1, available online). We obtained blood samples and pedigrees after acquiring informed consent from individuals with PCD and their family members. Approval for human subjects research was obtained from the institutional review boards at the Universities of North Carolina, Michigan, Freiburg, and Münster and the other institutions involved. We detected both PCD-causing alleles of known genes in 16 families, thereby solving 51.6% (16/31) of PCD cases by using this combined approach of homozygosity mapping and WER (Figure S1 and Tables S1 and S2). We identified mutations in previously PCD-associated genes, including DNAH5 (MIM 603335), the most frequently mutated in monogenic PCD15 (five families), CCDC39 (MIM 613798; three families), CCDC40 (MIM 613799; three families), DNAH11 (MIM 603339; two families), DNAI2 (MIM 605483; one family), CCDC103 (MIM 614677; one family), and LRRC6 (MIM 614930; one family).
Homozygosity mapping in an Israeli individual (A4231) with situs inversus yielded 13 candidate regions of homozygosity by descent (Figure 1A). By WER we detected in this individual a homozygous missense variant (c.1136A>G [p.Tyr379Cys], conserved to D. melanogaster) in ZMYND10 (zinc finger, MYND-domain-containing 10, RefSeq accession number NM_015896.2) (Figure 1B and Table 1). In order to investigate whether ZMYND10 mutations occur in additional individuals with a full PCD phenotype, we examined a worldwide cohort of over 300 individuals with PCD by exon resequencing of all 12 ZMYND10 exons by using a high-throughput, barcoded, next-generation-sequencing technique that we recently developed (Figure 1C).16 In total, we identified both disease-causing alleles of ZMYND10 in 14 families affected by PCD and/or situs inversus and detected 11 different homozygous or compound-heterozygous ZMYND10 mutations without any predilection for specific exons (Table 1, Figures 1C–1E, and Figure S2). We thereby identified recessive mutations in ZMYND10 as a cause of PCD. ZMYND10 (also known as BLU) contains a C-terminal MYND (myeloid, nervy and DEAF-1) domain (Figure 1D). ZMYND10 is highly enriched in ciliated cells compared to nonciliated cells,17 but little is known about its function. ZMYND10 is known to act as a tumor suppressor, inhibits clonogenic growth of nasopharyngeal carcinoma cells, arrests the cell cycle at the G1 phase, downregulates JNK (c-Jun N-terminal kinase) and cyclin D1 promoter activities, and inhibits phosphorylation of c-Jun.18
Figure 1.
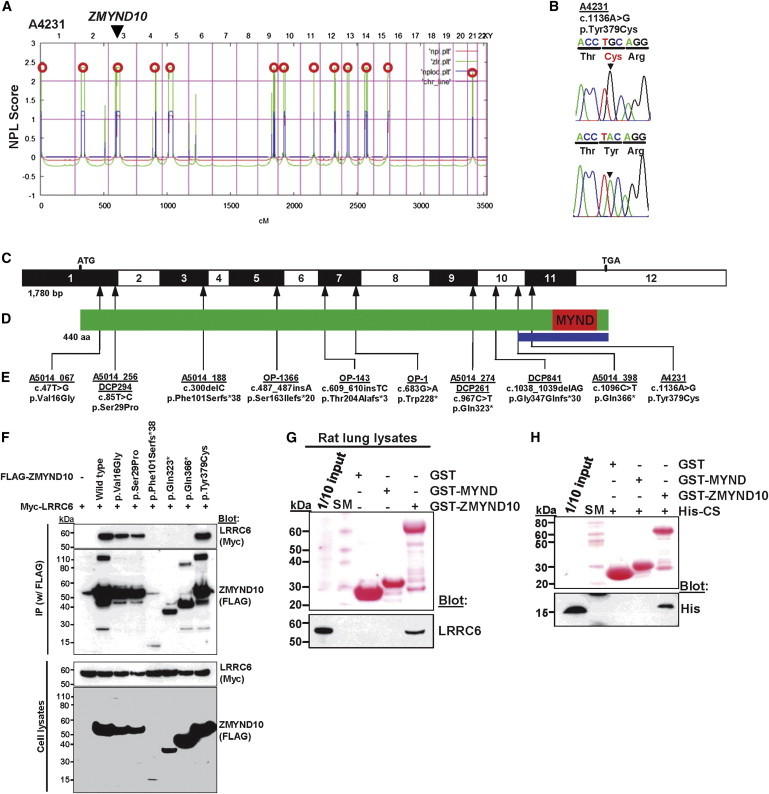
Homozygosity Mapping and WER in Family A4231 and Identification of 11 Different Homozygous or Compound-Heterozygous ZMYND10 Mutations in 13 Additional Unrelated PCD-Affected Families
(A) For individual A4231, who has situs inversus, nonparametric LOD (NPL) scores from whole-genome mapping are plotted across the human genome. The x axis shows Affymetrix 250K StyI array SNP positions on human chromosomes concatenated from pter (left) to qter (right). Genetic distance is given in cM. Thirteen maximum NPL peaks (red circles) indicate candidate regions of homozygosity by descent. Note that the ZMYND10 locus (arrow head) is positioned within one of the maximum NPL peaks on chromosome 3p.
(B) Homozygous ZMYND10 mutation detected in family A4231. Family number (underlined), mutation (arrowhead), and predicted translational changes are indicated (see also Table 1). Sequence trace is shown for the mutation above the normal control.
(C) Exon structure of human ZMYND10 cDNA. The positions of the start codon (ATG) and the stop codon (TGA) are indicated.
(D) Domain structure of ZMYND10. The C terminus contains a MYND (myeloid, Nervy and DEAF-1) domain. The blue bar delineates the region necessary for interaction with LRRC6 as identified in Figure 1F.
(E) Ten homozygous or compound-heterozygous ZMYND10 mutations detected in 12 PCD-affected families. Family number (underlined), mutation, and predicted translational changes are indicated (see Table 1 and Figure S2).
(F) Interaction between the wild-type (WT) and six ZMYND10 variants detected in human PCD (see Table 1) and LRRC6. FLAG-tagged ZMYND10 and Myc-tagged LRRC6 constructs were transfected into human embryonic kidney 293T (HEK293T) cells and coimmunoprecipitated with a FLAG antibody. Note that the three truncating protein alterations (p.Phe101Serfs∗38, p.Gln323∗, and p.Gln366∗) abrogated interaction with LRRC6, whereas the three alterations resulting from missense mutations did not.
(G) GST pull-down of purified ZMYND10 (MYND domain and full-length). Note that LRRC6 in rat lung lysates was pulled down by the full-length ZMYND10, but not by the MYND domain alone.
(H) The purified CS domain of LRRC6 binds to the full-length ZMYND10, but not to the MYND domain of ZMYND10. Therefore, the MYND domain alone is not sufficient for the interaction with LRRC6.
SM denotes size marker.
Table 1.
Mutations of ZMYND10 in 14 Families Affected by PCD or Situs Inversus
| Familya | Individual(s)a | Ethnic Origin | Nucleotide Mutationb,c(Segregation) | Deduced Protein Change | Exon (Zygosity) | AA Sequence Conservationd | PolyPhen-2/MutationTaster | Parental Consanguinity | IF (Axonemal IDA or ODA Protein Localization) | TEM | Video Microscopy | Other (Clinical Features) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A5014_067 (#111) | 21 (#658) | white | c.47T>G (−11: ND, −12: het) | p.Val16Gly | 1 (hom) | Mus musculuse | 0.99/DC | no | ND | ODA and IDA defects | immotile cilia | SI |
| A5014_252 (#355) | 21 (#1347) | white | c.47T>G | p.Val16Gly | 1 (het) | Mus musculuse | 0.99/DC | no | ND | ODA defects | ND | SS |
| c.300delC | p.Phe101Serfs∗38 | 3 (het) | NA | NA/DC | ||||||||
| A5014_256 (#360) | 21 (#1365) | Hispanic | c.85T>C | p.Ser29Pro | 1 (hom) | Drosophilaf | 0.99/pol | yes | ND | ODA and IDA defects | ND | SI |
| DCP294 | - | French | c.85T>C | p.Ser29Pro | 1 (hom) | Drosophilaf | 0.99/pol | unknown | ND | ODA and IDA defects | ND | SI, infertility |
| A5014_188 (#278) | 21 (#1209) | white | c.300delC | p.Phe101Serfs∗38 | 3 (hom) | NA | NA/DC | no | ND | ODA and IDA defects | immotile cilia | SS |
| OP-1366 | II1 | Turkish | c.486dupA | p.Ser163Ilefs∗20 | 5 (hom) | NA | NA/DC | yes | DNAH5: defect DNALI1: defect |
- | immotile cilia | SS |
| OI-143 | II1 and II2 | Israeli | c.dup608_609dupTC | p.Thr205Alafs∗3 | 7 (hom) | NA | NA/DC | yes | DNAH5: defect DNALI1: defect |
ODA and IDA defects | ND | SI |
| OP-1 | - | Turkish | c.683G>A (−11: het, −12: het) | p.Trp228∗ | 7 (hom) | NA | NA/DC | ND | DNAH5: defect DNALI1: defect |
ODA and IDA defects | ND | - |
| OP-55 | II1 | Turkish | homozygous deletion, exons 7-12 | NA | 7–12 (hom) | NA | NA/DC | yes | DNAH5: defect DNALI1: defect |
ODA and IDA defects | immotile cilia | SS |
| A5014_274 (#383) | 21 (#1420) | Hispanic | c.967C>T | p.Gln323∗ | 9 (hom) | NA | NA/DC | no | ND | ODA and IDA defects | ND | SI |
| DCP261 | - | French | c.967C>T | p.Gln323∗ | 9 (hom) | NA | NA/DC | no | ND | ODA and IDA defects | immotile cilia | SI |
| DCP841 | - | Portuguese | c.1038_1039delAG | p.Gly347Glnfs∗30 | 10 (hom) | NA | NA/DC | no | ND | ODA and IDA defects | immotile cilia | SI |
| A5014_398 (#523) | 21 (#1665) | white | c.1096C>T | p.Gln366∗ | 10 (hom) | NA | NA/DC | no | ND | ODA and IDA defects | immotile cilia | SS |
| A4231 (#568) | 21 (#1761)g | Israeli | c.1136A>G (−11: het, −12: het) | p.Tyr379Cys | 11 (hom) | Drosophila | 1.0/DC | yes | ND | normalg | ND | SI and congenital athyroid (no PCD) |
Abbreviations are as follows: AA, amino acid; DC, disease causing; het, heterozygous; hom, homozygous; IDA, inner dynein arm; IF, immunofluorescence; NA, not applicable; ND, no data; ODA, outer dynein arm; pol, polymorphism; SI, situs inversus; SS, situs solitus; and TEM, transmission electron microscopy.
Numbers in parentheses are sample identifiers of the University of North Carolina.
The mutation in the index family is in bold. Recurrent mutations are underlined.
cDNA mutations are numbered according to human cDNA reference sequence NM_015896.2 (ZMYND10); +1 corresponds to the A of ATG start translation codon.
Amino acid residue is continually conserved throughout evolution as indicated.
Variant in dbSNP database (rs138815960), allele frequency EVS Server: CC = 0/CA = 7/AA = 6491.
Except Xenopus has Asp.
This individual did not have a full PCD phenotype but had situs inversus only.
Transmission electron microscopy (TEM) of samples from individuals (A5014_188, OI-143 II2, and OP-55 II1) with ZMYND10 mutations showed a lack of outer dynein arms (ODAs) and inner dynein arms (IDAs) (Figures 2A–2D). These findings prompted us to perform immunofluorescence studies for the presence of IDA light-chain protein DNALI1 and ODA heavy-chain protein DNAH5. We found that ODA component DNAH5 (Figures 2E–2G and Figure S3) and IDA component DNALI1 (Figure S4) were absent in ZMYND10 mutant ciliary axonemes from respiratory epithelia obtained by nasal brushings.
Figure 2.

Loss of ZMYND10 Function Causes Structural and Functional Defects of Motile Cilia in Human Lung and in a Zebrafish Model of zmynd10 Knockdown
(A–D) Compared to those of a normal control (A), ciliary axonemes from individuals A5014_188 (B), OI-143II2 (C), and OP-55II1 (D), who have PCD and ZYMYND10 mutations, lack IDAs and ODAs (arrows).
(E–G) Images of respiratory epithelial cells from a healthy control and from PCD-affected individuals who carry ZMYND10 loss-of-function mutations. Cells were costained with antibodies against acetylated α-tubulin (green) and DNAH5 (red). Nuclei were stained with Hoechst 33342 (blue). In cells from a healthy control (E), DNAH5 localized to the axonemes of respiratory cilia. The yellow costaining within the ciliary axoneme indicates that both proteins colocalized within respiratory cilia. In respiratory cells of individuals OI-143II2 (F) and OP-55II1 (G), DNAH5 was not detectable in the ciliary axonemes, suggesting that ZMYND10 loss-of-function mutations led to defects in the ODA heavy-chain DNAH5. Rabbit polyclonal DNAH5 and DNALI1 antibodies were described previously.7 See also Figures S3 and S4.
(H–M) zmynd10 knockdown in zebrafish replicated a ciliopathy phenotype. Compared to control-injected embryos (H), embryos injected with zmynd10-translation-blocking MOs (I) showed three otoliths (arrowheads), cystic pronephric glomeruli (arrows), and distended pronephric tubules (white bar).
(J) A still image of a high-speed microvideo of pronephric cilia (J) shows the tubule outline (dashed line) and position of pixels (black line) sampled for the kymograph (J′; one second total duration). The double arrowhead denotes the approximate tubule lumen diameter.
(K) A still image of a high-speed microvideo of zmynd10-morphant pronephric cilia shows a distended tubule outline (dashed line), pixels sampled for the kymograph (black line; K′, 1 s total duration), and a distended tubule lumen dimension (double-arrowhead line).
(L) A still image of a high-speed microvideo of control olfactory placode cilia and position of pixels sampled for the kymograph (black line; L′, 1 s total duration). See also Movies S1, S2, S3, and S4.
(M) A still image of high-speed microvideo of zmynd10-morphant olfactory placode cilia and position of pixels sampled for the kymograph (black line; M′, 1 s total duration). See also Movies S1, S2, S3, and S4.
Scale bars represent 5 μm in (J)–(M).
We further confirmed that loss of ZMYND10 impairs motile ciliary function by knockdown of its zebrafish ortholog, zmynd10 (Figures 2H–2M′). Specifically, targeting the zmynd10 ortholog in zebrafish by either translation blocking (ATG) or exon 3 splice donor (e3i3) antisense morpholinos (MOs) replicated characteristic ciliopathy phenotypes, including the appearance of three otoliths, kidney cysts, and dilated kidney tubules at 2.5 days postfertilization (Figures 2H and 2I, Figure S5, and Table S3). High-speed-microvideo and kymograph analyses of motile cilia in the kidney (Figures 2H and 2I′) showed disorganized cilia bundles with either severely reduced beat amplitude or paralysis in zmynd10 ATG morphants (Movies S1 and S2). Similarly, olfactory motile cilia (Figures 2J and 2K′) were nearly completely paralyzed in zmynd10 ATG morphants (Movies S3 and S4). Similar but milder defects were observed in zmynd10 e3i3 morphants (Figure S3). In addition, we performed MO-knockdown studies in epidermal multiciliated cells of Xenopus laevis embryos and injected Xenopus embryos with either a start-site (ATG) or a splice-site (SPL) morpholino targeting zmynd10. Control embryos contained multiciliated cells with thick tufts of cilia (Figure S6A). Interestingly, although a large percentage of cells failed to generate cilia in both morphants (ATG and SPL) because of a substantial defect in ciliogenesis, the numbers of centrioles appeared normal (Figures S6B and S6C). Although it is known that cilia on the surface of Xenopus embryos work to generate a vigorous flow oriented toward the posterior side of the embryo,19 we found that morphant embryos generated a significantly weaker flow as measured by the displacement of fluorescent beads across the surface of the embryo (Figure S6D). Expression studies of fluorescently tagged human LRRC6 and ZMYND10 in the Xenopus epidermal ciliated epithelia revealed localization to both the basal body and the striated rootlet, an appendage that projects away from the basal body into the cytoplasm (Figures S6E and S6F). This localization also supports a possible role for zmynd10 in ciliogenesis or cilia function.
Mutations in DNAAF1, DNAAF2, DNAAF3, and LRRC6 cause PCD with ODA and IDA defects, and their encoded proteins have previously been found to localize to the cytoplasm of respiratory epithelial cells.20–23 Among them, DNAAF1, DNAAF2, and DNAAF3 have been proposed to be involved in the cytoplasmic preassembly of dynein-arm complexes, which are later loaded to the ciliary compartment by intraflagellar-transport (IFT) complexes.21,22 Because we found that ZMYND10 mutations caused ODA and IDA defects and because ZMYND10 localizes predominantly to the cytoplasm in the Human Protein Atlas, we investigated whether ZYMND10 interacts with these proteins. The interaction between ZMYND10 and DNAAF1, DNAAF2, or DNAAF3 was below the threshold of detection (data not shown). Interestingly, however, ZMYND10 bound to LRRC6 when it was overexpressed in human embryonic kidney 293T (HEK293T) cells (Figure 1F). We also confirmed the interaction between ZMYND10 and LRRC6 in human tracheal epithelial cells (HTEpCs) (Figure S7). We further examined whether mutations detected in individuals with PCD would disrupt the interaction between ZMYND10 and LRRC6. The three truncating protein alterations (p.Phe101Serfs∗38, p.Gln323∗, and p.Gln366∗)—which delete varying C-terminal portions, including the MYND domain—abrogated the interaction between ZMYND10 and LRRC6, whereas the three alterations resulting from missense mutations did not (Figure 1F). By GST pull-down of rat lung lysates, we confirmed the interaction between LRRC6 and full-length ZMYND10 and showed that the MYND domain alone was not sufficient for pull-down of LRRC6 (Figure 1G). LRRC6 contains the four recognizable domains: LRR (leucine-rich repeat), LRRCT (C-terminal to LRR), CC (coiled-coil), and CS (CHORD-containing proteins and SGT1) (Figure S8). We found that the purified CS domain of LRRC6 alone was sufficient for interaction with the full-length ZMYND10 but that the MYND domain of ZMYND10 was not sufficient for interaction with the CS domain of LRRC6 (Figure 1H). Thus, a C-terminal fragment that extends beyond the MYND domain is necessary for interaction between ZMYND10 and LRRC6 (see blue bar in Figure 1D).
Besides the recent reports about LRRC6,23,24 we independently identified nine different homozygous or compound-heterozygous LRRC6 mutations in 13 PCD-affected families: c.169_173delinsTCCCAAT (pGly57Serfs∗3), c.[259T>C];[436G>C] (p.[Cys87Arg];[Asp146His]), c.562C>T (pGln188∗), c.630delG (pTrp210Cysfs∗12), c.598_599delAA (p.Lys200Glufs∗3), c.653+1G>A, c.710_711delCA (p.Thr237Lysfs∗7), and c.891delA (pAla298Profs∗2) (Table S2 and Figures S9 and S10). By homozygosity mapping and WER in an inbred Pakistani family (A4213) consisting of two siblings with the clinical features of PCD and one sibling with situs inversus (Table S2 and Figures S8A and S8B) (all individuals exhibited defective ciliary ODA and IDA upon TEM), we detected a homozygous frameshift mutation (c.630delG) in LRRC6. We identified variant c.630delG homozygously in five additional unrelated families of Pakistani descent by the restriction-fragment-length-polymorphism (RFLP) method with BsrG1, indicating that it represents a Pakistani founder mutation (Tables S2 and S4). Similar to ZMYND10 mutations, LRRC6 loss-of-function mutations also caused the absence of the ODA and IDA components DNAH5 (Figure S10) and DNALI1 (Figure S11) from ciliary axonemes. These results are congruent with the previous reports that showed that ODA components DNAI2 and DNAI1 and IDA components DNALI1 and DNAH7 are defective in airway epithelial cells from individuals with LRRC6 mutations.23,24
We further examined whether variant proteins resulting from LRRC6 mutations reciprocally abrogate the interaction with ZMYND10 because we found that they interact (Figure 1). When studying the interaction between wild-type (WT) and mutant LRRC6 constructs and ZMYND10, we found that the five truncating protein alterations (p.Gln188∗, p.Lys200Glufs∗3, p.Trp210Cysfs∗12, p.Thr237Lysfs∗7, and p.Ala298Profs∗2) abrogated interaction with ZMYND10 but that the two alterations resulting from missense mutations did not (Figure 3A). By overexpression of progressive truncating constructs of LRRC6, we showed that LRRC6’s N-terminal amino acid residues 1–204, which include the LRR, LRRCT, and CC domains, were not necessary for the interaction with ZMYND10 (Figure 3B) but that the CS domain of LRRC6 alone was sufficient for pull-down of ZMYND10 in rat lung lysates (Figure 3C). We thus demonstrated that the C termini of LRRC6 and ZMYND10 engaged in a protein-protein interaction, which was abrogated by truncating mutations detected in individuals with PCD.
Figure 3.
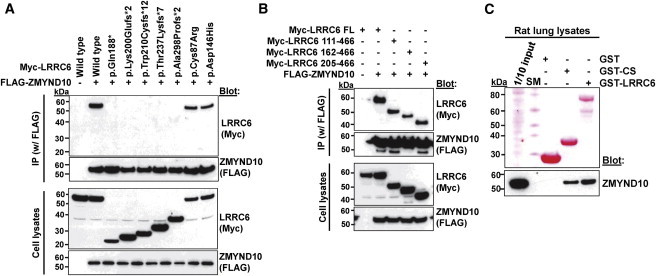
Truncating Variants of LRRC6 Abrogate Interaction with ZMYND10
(A) For testing the effect of seven LRRC6 mutations detected in individuals with PCD, FLAG-tagged ZMYND10 and Myc-tagged LRRC6 constructs were transfected into HEK293T cells and coimmunoprecipitated with a FLAG antibody. Note that the five truncating protein alterations (p.Gln188∗p.Lys200Glufs∗3, p.Tro210Cysfs∗12, p.Thr237Lysfs∗7, and p.Ala298Profs∗2) abrogated interaction with ZMYND10, whereas the two alterations resulting from missense mutations did not.
(B) LRRC6 residues 1–204, which include the LRR, LRRCT, and CC domains, were not necessary for the interaction with ZMYND10. Coimmunoprecipitation was done as in Figure 3A.
(C) GST pull-down of purified LRRC6 (CS domain and full-length). Note that the CS domain of LRRC6 was sufficient for pull-down of ZMYND10 in rat lung lysates.
“SM” denotes size marker.
Because both genetic defects shared the features of ODA and IDA defects demonstrated by TEM and IF, we hypothesized that ZMYND10 and LRRC6 might be part of a shared protein complex that is necessary for motile ciliary function. We therefore performed immunofluorescence studies in rat trachea. We found that ZMYND10 (Figures 4A–4C) localized to sites proximal to the axoneme (Figure 4A). It also fully colocalized to cytoplasmic puncta of varying sizes with SAS6 (spindle assembly abnormal protein 6) (Figure 4B), which is required for centriole assembly during ciliogenesis,25 and PCM1 (pericentriolar material 1) (Figure 4C), which is a component of centriolar satellites. Therefore, these results suggest that ZMYND10 localizes to centriolar satellites. However, considering its varying size, similar structures, described as “fibrous granules,” have been shown to play a role in the biogenesis of motile cilia.26,27 PCM1 is shown to localize to fibrous granules of ciliogenic cells, but not to deuterosomes.28 Recently, ZMYND10 has been shown to localize to puncta also in the cytoplasm of human retinal pigment epithelial cells.29
Figure 4.

ZMYND10 and LRRC6 Colocalize with Centriolar Proteins in Rat Trachea
(A) Coimmunofluorescence of ZMYND10 (Sigma) with acetylated tubulin (Sigma).
(B–D) Coimmunofluorescence of ZMYND10 (Abnova) with SAS6 (spindle assembly abnormal protein 6) (Sigma) (B), PCM1 (pericentriolar material 1) (Cell Signaling Technology) (C), and LRRC6 (Novus) (D).
(E–G) Coimmunofluorescence of LRRC6 (Sigma) with acetylated tubulin (E), SAS6 (F), and PCM1 (G). PCM1 is a component of centriolar satellites, which are electron-dense granules scattered around centrosomes. PCM1 also localizes to fibrous granules of ciliogenic cells, but not to deuterosomes.
Scale bars represent 5 μm in (A)–(G).
When examining LRRC6 localization (Figures 4D–4G), we found that LRRC6 colocalized with ZMYND10 (Figure 4D) to the cytoplasmic puncta, but not to the axonemal domain that is marked by acetylated tubulin (Figure 4E). Like ZMYND10, LRRC6 fully colocalized to cytoplasmic puncta with SAS6 (Figure 4F) and PCM1 (Figure 4G). We concluded that ZMYND10 and LRRC6 are in a shared protein complex with SAS6 and PCM1 and have localization of cytoplasmic punta in respiratory epithelial cells, strongly suggesting a role of this protein complex in motile cilia.
Because we found ZMYND10 and LRRC6 to be in a complex and Horani et al. showed that LRRC6 is involved in transcriptional regulation of DNAI1 and DNAH7,24 we investigated whether ZMYND10 also regulates transcription of some dynein proteins. We examined the expression of DNAH5 and DNALI1 because we showed that DNAH5 (Figures 2 and Figure S3) and DNALI1 (Figure S4) were not present in ZMYND10-mutant ciliary axonemes from respiratory epithelia. In HTEpCs transfected with ZMYND10-specific shRNA, the amount of DNAH5 and DNALI1 mRNA was significantly lower than that in control cells (Figure S12). These results suggest that the protein complex including ZMYND10 and LRRC6 is involved in transcriptional regulation of some dynein components.
It was shown that LRRC6 interacts with disheveled (DVL)30 and that loss of lrrc6 function in the ciliopathy zebrafish model seahorse increases canonical Wnt signaling.31 We confirmed here by coimmunoprecipitation that LRRC6 interacts with all three disheveled proteins (DVL1, DVL2, and DVL3) (Figures S13A and S13B) and found that the four proximal LRRC6 truncating alterations (p.Gln188∗, p.Lys200Glufs∗3, p.Trp210Cysfs∗12, and p.Thr237Lysfs∗7) abrogated interaction with DVL3 but that the most distal truncating alteration (p.Ala298Profs∗2) and the two alterations resulting from missense mutations did not, indicating that the LRRC6 fragment distal of Ala298 is dispensable for interaction with DVL3.
Increased canonical Wnt signaling has been implicated in ciliopathies of both sensory cilia32 and motile cilia, an effect that is mediated by DVL. We therefore tested whether overexpression of WT constructs of ZMYND10 and LRRC6 decreases canonical Wnt signaling by inhibiting catenin-induced activation of a TCF-dependent reporter gene in the TOPFlash system. We found that whereas all the truncated forms of LRRC6 failed to inhibit TCF reporter activity, two defective proteins resulting from missense mutations (c.259T>C [p.Cys87Arg] and c.436G>C [p.Asp146His]) did not act any differently than the WT. WT or truncated (p.Phe101Serfs∗38, p.Gln38∗, and p.Gln366∗) forms of ZMYND10 did not have any effect on TCF reporter activity (Figure S13C). Because of the divergent behavior of ZMYND10 and LRRC6 mutations in this assay, an upregulation on canonical Wnt signaling appears to be concomitant in LRRC6 mutations, but it does not appear to represent a shared pathogenic pathway for the generation of the PCD phenotype.
In summary, we identified ZMYND10 mutations as causing PCD. ZMYND10 takes part in a protein complex with LRRC6, localizes to cytoplasmic puncta in respiratory epithelial cells, and regulates transcription of dynein proteins, strongly suggesting that this protein complex plays a role in motile cilia. Identification of genes involved in the cytoplasmic, nonaxonemal components of motile cilia will help in further elucidating the molecular mechanisms involved in dynein-arm assembly, and it will be important because PCD caused by mutations in these genes might be particularly amenable to pharmacologic modification.
Acknowledgments
We are grateful to all individuals with primary ciliary dyskinesia (PCD) and family members for their participation, as well as Michele Manion, who founded the United States PCD Foundation. We thank the German support group “Kartagener Syndrom und Primaere Ciliaere Dyskinesie e.V.” This research was supported by grants from the National Institutes of Health (NIH) to F.H. (DK068306 and DK090917), to W.Z. (DK091405), to I.A.D. (DK053093 and DK070263), to D.S.W (EY013408), and to Z.S. (DK092808-01A1). This work was supported in part by NIH grants UL1 TR000083 and UL1 TR000154 from the National Center for Advancing Translational Sciences. M.K. is supported by the project “Studies of nucleic acids and proteins—from basic to applied research,” sponsored by the International PhD Projects Programme of Foundation for Polish Science. The project is cofinanced by the European Union Regional Development Fund. D.A.M. was supported by the Department of Pathology, Faculty of Medicine, Kuwait University. M.A.Z., M.W.L., S.D.D., M.R., T.W.F., S.D.S., J.E.P., K.N.O., and M.R.K. are supported by NIH research grant 5 U54 HL096458-06, funded by the Office of the Director, and supported by the Office of Rare Diseases Research and the National Heart, Lung, and Blood Institute (NHLBI). M.A.Z. and M.R.K. are supported by NIH NHLBI grant 5 R01HL071798. H.O. is supported by the Deutsche Forschungsgemeinschaft (DFG Om 6/4 and Om 6/5, GRK1104, and SFB592), IZKF Münster, the Cell Dynamics and Disease graduate school, and project SYSCILIA from the European Community. Additional acknowledgements are provided in the Supplemental Data.
Contributor Information
Michael R. Knowles, Email: knowles@med.unc.edu.
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Supplemental Data
This movie shows rhythmic beating of cilia bundles in the lumen of the pronephric tubule. It was acquired at 240 frames per second (fps) and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows distended tubule lumen and an irregular and paralyzed cilia beat. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows regular cilia waves at the periphery of the placode. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows near-complete cilia paralysis. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.
Web Resources
The URLs for data presented herein are as follows:
GeneReviews, Zariwala, M.A., Knowles, M.R., and Leigh, M.W. (1993). Primary Ciliary Dyskinesia, http://www.ncbi.nlm.nih.gov/pubmed/20301301
HomozygosityMapper, http://www.homozygositymapper.org/HomozygosityMapper/
Human Gene Mutation Database, http://www.biobase-international.com/product/hgmd
Human Protein Atlas, http://www.proteinatlas.org/
Mutation Taster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Leigh M.W., Pittman J.E., Carson J.L., Ferkol T.W., Dell S.D., Davis S.D., Knowles M.R., Zariwala M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noone P.G., Leigh M.W., Sannuti A., Minnix S.L., Carson J.L., Hazucha M., Zariwala M.A., Knowles M.R. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 3.Zariwala M.A., Knowles M.R., Omran H. Genetic defects in ciliary structure and function. Annu. Rev. Physiol. 2007;69:423–450. doi: 10.1146/annurev.physiol.69.040705.141301. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 5.Schwabe G.C., Hoffmann K., Loges N.T., Birker D., Rossier C., de Santi M.M., Olbrich H., Fliegauf M., Failly M., Liebers U. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008;29:289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- 6.Knowles M.R., Leigh M.W., Carson J.L., Davis S.D., Dell S.D., Ferkol T.W., Olivier K.N., Sagel S.D., Rosenfeld M., Burns K.A., Genetic Disorders of Mucociliary Clearance Consortium Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax. 2012;67:433–441. doi: 10.1136/thoraxjnl-2011-200301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W.S., Loges N.T., Pennekamp P., Lindberg S., Stenram U. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otto E.A., Hurd T.W., Airik R., Chaki M., Zhou W., Stoetzel C., Patil S.B., Levy S., Ghosh A.K., Murga-Zamalloa C.A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W., Otto E.A., Cluckey A., Airik R., Hurd T.W., Chaki M., Diaz K., Lach F.P., Bennett G.R., Gee H.Y. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaki M., Airik R., Ghosh A.K., Giles R.H., Chen R., Slaats G.G., Wang H., Hurd T.W., Zhou W., Cluckey A. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 16.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 17.McClintock T.S., Glasser C.E., Bose S.C., Bergman D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics. 2008;32:198–206. doi: 10.1152/physiolgenomics.00128.2007. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X., Liu H., Li B., Huang P., Shao J., He Z. Tumor suppressor BLU inhibits proliferation of nasopharyngeal carcinoma cells by regulation of cell cycle, c-Jun N-terminal kinase and the cyclin D1 promoter. BMC Cancer. 2012;12:267. doi: 10.1186/1471-2407-12-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Werner M.E., Hwang P., Huisman F., Taborek P., Yu C.C., Mitchell B.J. Actin and microtubules drive differential aspects of planar cell polarity in multiciliated cells. J. Cell Biol. 2011;195:19–26. doi: 10.1083/jcb.201106110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.F., Siffroi J.P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horani A., Ferkol T.W., Shoseyov D., Wasserman M.G., Oren Y.S., Kerem B., Amirav I., Cohen-Cymberknoh M., Dutcher S.K., Brody S.L. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS ONE. 2013;8:e59436. doi: 10.1371/journal.pone.0059436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettencourt-Dias M., Hildebrandt F., Pellman D., Woods G., Godinho S.A. Centrosomes and cilia in human disease. Trends Genet. 2011;27:307–315. doi: 10.1016/j.tig.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagiwara H., Ohwada N., Takata K. Cell biology of normal and abnormal ciliogenesis in the ciliated epithelium. Int. Rev. Cytol. 2004;234:101–141. doi: 10.1016/S0074-7696(04)34003-9. [DOI] [PubMed] [Google Scholar]
- 27.Sorokin S.P. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J. Cell Sci. 1968;3:207–230. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- 28.Kubo A., Sasaki H., Yuba-Kubo A., Tsukita S., Shiina N. Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 1999;147:969–980. doi: 10.1083/jcb.147.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albee A.J., Kwan A.L., Lin H., Granas D., Stormo G.D., Dutcher S.K. Identification of Cilia Genes That Affect Cell-Cycle Progression Using Whole-Genome Transcriptome Analysis in Chlamydomonas reinhardtti. G3 (Bethesda) 2013;3:979–991. doi: 10.1534/g3.113.006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serluca F.C., Xu B., Okabe N., Baker K., Lin S.Y., Sullivan-Brown J., Konieczkowski D.J., Jaffe K.M., Bradner J.M., Fishman M.C., Burdine R.D. Mutations in zebrafish leucine-rich repeat-containing six-like affect cilia motility and result in pronephric cysts, but have variable effects on left-right patterning. Development. 2009;136:1621–1631. doi: 10.1242/dev.020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kishimoto N., Cao Y., Park A., Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev. Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Germino G.G. Linking cilia to Wnts. Nat. Genet. 2005;37:455–457. doi: 10.1038/ng0505-455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This movie shows rhythmic beating of cilia bundles in the lumen of the pronephric tubule. It was acquired at 240 frames per second (fps) and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows distended tubule lumen and an irregular and paralyzed cilia beat. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows regular cilia waves at the periphery of the placode. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.
This movie shows near-complete cilia paralysis. It was acquired at 240 fps and is displayed at 15 fps. The actual duration of the movie is 250 ms.