

Abstract
Many individuals with abnormalities of mitochondrial respiratory chain complex III remain genetically undefined. Here, we report mutations (c.288G>T [p.Trp96Cys] and c.643C>T [p.Leu215Phe]) in CYC1, encoding the cytochrome c1 subunit of complex III, in two unrelated children presenting with recurrent episodes of ketoacidosis and insulin-responsive hyperglycemia. Cytochrome c1, the heme-containing component of complex III, mediates the transfer of electrons from the Rieske iron-sulfur protein to cytochrome c. Cytochrome c1 is present at reduced levels in the skeletal muscle and skin fibroblasts of affected individuals. Moreover, studies on yeast mutants and affected individuals’ fibroblasts have shown that exogenous expression of wild-type CYC1 rescues complex III activity, demonstrating the deleterious effect of each mutation on cytochrome c1 stability and complex III activity.
Main Text
Complex III (CIII) of the mitochondrial respiratory chain contains 11 subunits, one of which (cytochrome b) is encoded by mitochondrial DNA. CIII conveys electrons from ubiquinol to cytochrome c. It associates with complexes I (CI) and IV (CIV) to form the respirasome.1 Cytochrome c1 (Cyt c1), cytochrome b, and the Rieske protein are the three CIII redox components. The heme moiety of Cyt c1 is in the N-terminal domain, located in the intermembrane space, where it accepts electrons from the Rieske protein. Cyt c1 is anchored to the phospholipid bilayer by a single C-proximal transmembrane segment.2
Primary CIII deficiencies, although infrequent, have strikingly different clinical presentations. Mutations in mitochondrial CYTB or in the nuclear genes encoding CIII subunits or assembly factors elicit a wide range of tissue-specific defects in affected individuals; they include encephalopathy with renal involvement or lethal infantile hepatic failure (from BCS1L [MIM 124000] mutations in GRACILE syndrome [MIM 603358]);3,4 severe psychomotor retardation and extrapyramidal signs, dystonia, athetosis, and ataxia (from UQCRQ [MIM 615159] mutations);5 mitochondrial encephalopathy (from BCS1L, TTC19 [MIM 613814], CYTB [MIM 516020] mutations);6 pili torti and sensorineural hearing loss, known as Björnstad syndrome (from BCS1L mutations);7 optic neuropathy, exercise intolerance, and/or cardiomyopathy (from CYTB mutations);8 and transient episodes of hypoglycemia and lactic acidosis (from UQCRB [MIM 191330] and UQCRC2 [MIM 191329] mutations).9,10
We first carried out respiratory chain (RC) enzyme analysis of muscle, liver, and fibroblasts of two children (from unrelated consanguineous families) presenting with unexplained ketoacidosis and recurrent hyperlactacidemia. All investigations were carried out according to the recommendations of the relevant local ethical committees and with the informed consent of affected individuals and/or their families. The study revealed an isolated CIII deficiency in the two children (Table S1, available online).
The first child (P1; II-3, Lebanese family in Figure 1) is the son of first-cousin parents of Lebanese background. An older brother has autism and intellectual disability, and an older sister and a younger brother exhibit normal growth and development. The proband was born at 36 weeks of gestation (Table S2) after a pregnancy complicated by gestational diabetes, for which his mother needed insulin therapy. His birth weight was 2,300 g (10–50th percentile), his length was 46 cm (10–50th percentile), and his head circumference was 32 cm (50–90th percentile). He had normal early development. He first presented at 5 months of age with severe metabolic ketoacidosis (pH 7.04) after a 24 hr history of a febrile illness causing vomiting (Table S1). His initial blood lactate was 13 mmol/l (normal range = 0.7–2.0 mmol/l), and he also had hyperammonemia (260 μmol/l; normal range = 10–50 μmol/l). He was treated with fluid resuscitation, sodium benzoate, and arginine, and over the next few days his biochemistry corrected. He went on to have recurrent episodes of fulminant lactic acidemia with intercurrent illnesses; his lactate-to-pyruvate ratio was as high as 66, and his blood glucose was labile (it fluctuated from 3 to 30 mmol/l) and required insulin therapy. This prompted us to investigate RC enzyme in muscle, liver, and skin biopsies, which revealed a severe and consistent isolated CIII defect (the ratio of CIII to citrate synthase activity was 4%, 24,% and 25% of the mean control values in liver, muscle, and skin fibroblasts, respectively) (Table S1). At 34 months of age, he was exhibiting normal development and was assessed to have normal cardiac, ophthalmological, and audiological functions. His weight was 11.2 kg (first percentile), and his length was 86.5 cm (first percentile). His head circumference 6 months earlier was 48.0 cm (23rd percentile).
Figure 1.
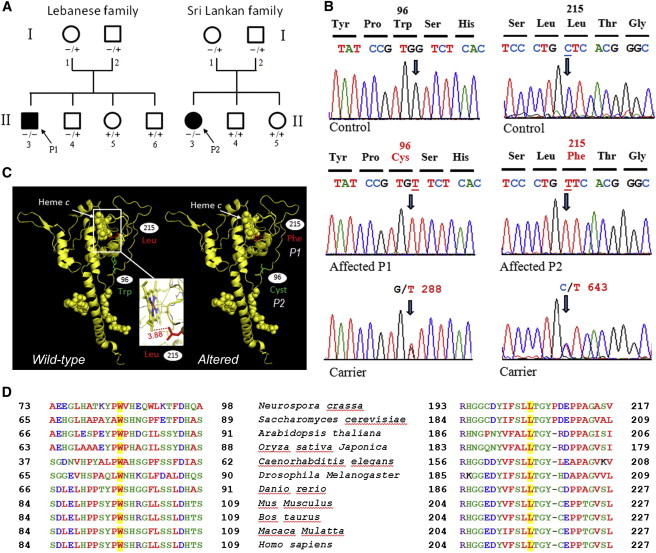
Gene Analysis in Families Affected by CYC1 Mutations
(A) Pedigrees of the Lebanese (with P1) and Sri Lankan (with P2) families. Affected individuals (dark symbols) harbor homozygous (−/−) mutations. Unaffected individuals are either heterozygous (−/+) or wild-type (+/+).
(B) Analysis of CYC1 genomic DNA. For P1, a single candidate disease-causing homozygous missense variant in CYC1 was identified by exome sequencing. For P2, who has defective CIII activity, a candidate-gene strategy resulted in sequencing cDNA obtained from total RNA of cultured fibroblasts and revealed a missense mutation (c.643C>T) in CYC1.
(C and D) Cyt c1 structures (C) and alignment (D).
The second affected child (P2; II-3, Sri Lankan family in Figure 1) is a girl (third child) born to first-cousin parents of Sri Lankan background. After an uneventful pregnancy and delivery, she presented with mild growth retardation and congenital left ptosis. She was admitted at 2.5 years of of age for acute vomiting with dehydration and progressive neurological deterioration, which led to a coma. A biological workup (Table S2) revealed hyperglycemic ketoacidosis with hyperlactatemia, liver failure, and hyperammonemia. The clinical condition rapidly improved with intravenous rehydration and insulin. During the following years, she exhibited normal psychomotor development, moderate failure to thrive, and numerous episodes of acute decompensation with ketoacidosis, hypoglycemia or hyperglycemia, and liver failure with hyperammonemia usually triggered by minor infections. A metabolic workup performed when the child was not decompensated disclosed mild permanent hyperlactacidemia (10 mmol/l; normal range = 1.0–3.75 mmol/l) with an increased lactate-to-pyruvate ratio (27 μmol/l; normal = 10–17 μmol/l), suggestive of a RC disorder. CIII deficiency was detected in muscle and was confirmed in cultured fibroblasts (Table S1). At the last follow-up examination, P2 was 18 years of age and had normal development and schooling. She still presents with rare episodes of metabolic decompensation with lactic acidosis.
For P1, who has isolated CIII deficiency, BCS1L, UQCRB, TTC19 (genes previously associated with CIII defects), and UQCR10 (MIM 610843; encoding a subunit of CIII) were sequenced, but no pathogenic mutations were identified. Whole-exome sequencing was subsequently performed. A library was first obtained from DNA (3 μg) fragmentation by sonication and ligation to SOLiDTM system sequencing adaptors and was next enriched for exomic sequences with the SeqCap EZ Human Exome Library v.2.0 exome-capture system (Nimblegen, Roche Diagnostics). After sequencing and alignment, >80% of the exome was covered at a depth of 20-fold or greater (average depth = 152-fold). Calling of single-nucleotide variants (SNVs) and indels was performed with LifeScope 2.5 (Life Technologies), and the resulting variant calls (>25,000 SNVs and >3,500 indels) were filtered with ANNOVAR 11 with in-house modifications. Genes were annotated with ENCODE Gencode v.11 (October 2011 freeze, GRCh37).Variants were first filtered against the 1000 Genomes database (2012 February release), whereby variants with a minor allele frequency greater than 0.5% were filtered out. Second, remaining variants were filtered against the dbSNP135 common database. The remaining variants were matched to a list of nuclear mitochondrial genes, and in silico prediction using Alamut v.2 (Interactive Biosoftware) identified variants of potential interest in 21 mitochondrial genes. Variants were filtered on the basis of recessive inheritance and a predicted role in CIII deficiency. A single candidate disease-causing homozygous missense variant in CYC1 (RefSeq accession number NM_001916.3) was identified: c.288G>T (p.Trp96Cys) (Figure 1). Bidirectional Sanger sequencing confirmed the variant in this individual and demonstrated segregation of the variant within the family (Figure 1). The mutation was absent in 81 unrelated Lebanese controls.
For the Sri Lankan girl (P2), who has low CIII activity in muscle and fibroblasts, the 11 genes coding for CIII subunits were sequenced (ABI Prism 3130XL; Applied Biosystems). DNA was extracted according to standard procedure, and mitochondrial DNA was examined by long-range PCR. Sequence analysis of CYTB and BCS1L was performed on gDNA. Sequence analysis of the nine nuclear genes (CYC1, UQCRFS1 [MIM 191327], UQCRC1 [MIM 191328], UQCRC2, UQCRH [MIM 613844], UQCRB, UQCR10, UQCRQ, and UQCR [MIM 609711]) encoding CIII subunits was performed on cDNA obtained from total RNA of cultured fibroblasts. A missense mutation (c.643C>T) resulting in a nonconservative substitution (p.Leu215Phe) within a highly conserved region of Cyt c1 was detected in CYC1 (Figure 1). This mutation was found to be heterozygous in each of her clinically unaffected parents and absent from her two unaffected siblings. This mutation is absent from dbSNP, 1000 Genomes databases, and 100 sequenced control alleles.
Immunoblot analysis of fibroblasts and skeletal-muscle samples from P1 and P2 showed a severe reduction in Cyt c1 levels (<10% of control levels), suggesting that the mutations make the Cyt c1 protein highly unstable (Figures 2A–2C and Figure S1). The reduction of Cyt c1 levels was accompanied by markedly reduced levels of assembly-dependent CIII subunits (core 2), especially in the muscle (Figures 2A and 2B). The severe depletion of Cyt c1, core 2 (P1), and core 1 (P2) subunits accounts for the loss of activity of the complex, which could hardly be assembled in the absence of these subunits. For P1, reduced levels of assembly-dependent subunits of CI (subunit NDUFB8) was also observed in fibroblasts only (Figure 2A), suggesting that CYC1 mutations might also affect the in vivo stability or assembly of other RC components. Accordingly, in P2 fibroblasts, the severe reduction of Cyt c1 and core 1 was accompanied by the absence of RC supercomplexes (Figure 2C).
Figure 2.
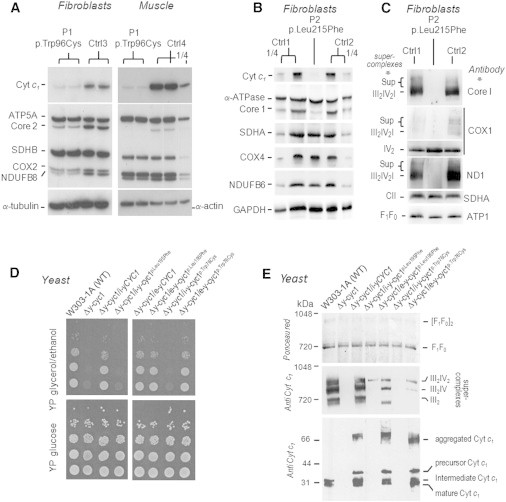
Analysis of Cyt c1 Mutant Cells and Rescue in Yeast Cells
(A–C) Immunoblots of affected individuals’ (P1 [with Cyt c1 p.Trp96Cys] and P2 [with Cyt c1 p.Leu215Phe]) RCs under denaturing (A and B) or native (C) conditions.
(A and B) Immunoblot of the levels of Cyt c1 oxidative-phosphoylation subunits in fibroblasts and skeletal muscle (A and B). Compared to those of the controls (Ctrl1–Ctrl4), levels of Cyt c1 and CIII were severely reduced in affected individual fibroblasts and skeletal muscle. Secondary reductions of assembly-dependent subunits of CI and CIV were also observed. SDS-PAGE and immunoblot were carried out on whole-cell lysate from fibroblasts (20 μg) or skeletal-muscle homogenate (5 μg). “¼” indicates that ¼ of the sample was loaded.
(C) Protein extract from fibroblasts (6% digitonin; 50 μg) was separated by blue-native PAGE on a 3.5%–12% polyacrylamide gel. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane and probed with a set of antibodies as indicated in the figure. The various supercomplexes and complexes and the ATP synthase (F1F0) are identified in the margins.
(D) Growth of wild-type and mutant yeast. Serial dilutions of the parental W303-1A strain, the Δy-cyc1 null mutant, and the null mutant harboring the wild-type (Δy-cyc1/i-yCYC1) or mutant genes in a single copy by integration at the URA3 locus (Δy-cyc1/i-y-cyc1) or in multiple copies in an episomal plasmid (Δy-cyc1/e-y-cyc1) were serially diluted and spotted on plates supplemented with glucose or glycerol plus ethanol and incubated at 30°C for 2 days.
(E) Immunoblot analysis of yeast Cyt c1. In the upper two panels, mitochondrial extracts (3% digitonin; 250 μg protein) were separated by blue-native PAGE on a 4%–13% polyacrylamide gel. Proteins were transferred to a PVDF membrane and stained with Ponceau red before being probed with a polyclonal antibody against yeast Cyt c1. The various supercomplexes and complexes and the ATP synthase (F1F0) are identified in the margins. In the lower panel, mitochondrial proteins (37 μg) were separated by SDS-PAGE on a 15% polyacrylamide gel and transferred to nitrocellulose and probed with a polyclonal antibody against yeast Cyt c1.
To confirm that the two CYC1 mutations were responsible for the observed clinical and biochemical phenotypes, we examined the effect of the mutations on mitochondrial function in Saccharomyces cerevisiae. The human mutations were engineered at orthologous positions c.228G>T (p.Leu195Phe) and c.585G>T (p.Trp76Cys) within the yeast gene (y-CYC1). The wild-type and mutant genes were either introduced in multiple copies on an episomal plasmid or integrated into the chromosomal DNA at the URA3 locus of a respiratory-deficient haploid strain carrying a null mutation in y-CYC1 (Δy-cyc1) Transformants confirmed to have a single copy of the mutant genes failed to grow on nonfermentable carbon sources (Figure 2D). In contrast, the growth defect of the Δy-cyc1 mutant was rescued in transformants harboring multiple copies of the mutant genes. As expected, the wild-type gene (either in single copy or multiple copies) restored wild-type growth on the respiratory substrates. The CIII-dependent activities of mitochondria from the different transformants were consistent with their growth phenotypes. Transformants with a single copy of the mutant genes had only 7% of the activity measured in the wild-type integrant (Table S3). The NADH cytochrome c reductase activity of CI and CIII increased to 52% and 33%, respectively, of that of the wild-type when the mutant genes were introduced on a multicopy plasmid.
Immunoblot analysis of yeast mitochondria revealed a near-complete absence of Cyt c1 and CIII in mutants with single copies of the p.Leu195Phe or p.Trp76Cys proteins (Figure 2E), consistent with results in affected individuals’ fibroblasts and muscles (Figures 2A–2C). Even though the levels of Cyt c1 in mutants overexpressing the genes were similar or even higher than those in the wild-type, immunochemically detectable CIII (as the dimer or as part of supercomplexes) was significantly reduced and correlated with their NADH cytochrome c reductase activities (Figure 2E and Table S3). Overexpression of the wild-type or mutant yeast gene led to some aggregation of Cyt c1, which was not completely dissociated in the sample buffer. Some overexpression of Cyt c1 was also evident in the strain with an integrated copy of the wild-type gene, resulting in aggregation as well. Presumably, when integrated at the URA3 locus, the mutant genes are also expressed above wild-type levels but are readily degraded as a result of their reduced stability. When expressed from a multicopy plasmid, however, the turnover rate is insufficient to clear all of the aggregated Cyt c1.
Data obtained with the recombinant lentiviral constructs containing either wild-type or mutant CYC1 cDNA of affected individual fibroblasts mimicked the results obtained with yeast. Moderate overexpression of wild-type, but not mutant, CYC1 (obtained with the use of a standard number of virus particles; multiplicity of infection [MOI] = 5) in the context of the endogenous c.643C>T mutant gene led to a partial rescue of CIII (see P2 in Figures S2A and S2B). Increased expression of CYC1 achieved with virus-particle levels of MOI = 120 allowed even mutant CYC1 to partially rescue CIII (see P1 in Figure S2C and S2D).
Together, the data obtained with cultured fibroblasts and yeast establish the deleterious effect of two CYC1 mutations for CIII assembly or stability in two individuals of different ethnic backgrounds; these mutations lead to quite a similar clinical presentation of ketoacidosis and lactic acidosis associated with insulin responsive hyperglycemia but with normal cognition. The mutations resulted in reduced levels of Cyt c1 and CIII and appeared to primarily affect the tertiary structure of Cyt c1 and thus render it more susceptible to proteolysis or alternatively less efficient to assembly with its partner subunits. Both circumstances would explain the low steady-state concentration of altered Cyt c1 in the single-copy yeast integrants and the partial rescue of CIII assembly when the genes are expressed from a multicopy plasmid. Analysis of the Cyt c1 crystal structure11 suggests that conformational changes due to the amino acid substitutions (Figure 1C) are most likely to occur in the extramembrane domain, whose function, though not clearly understood, plays a role in maintaining the structural and functional integrity of CIII.2 The tertiary-structure change might be a collapse of the long random coil containing the inward-facing tryptophan when the latter is replaced by the smaller cysteine or when a bulkier phenylalanine is substituted for the leucine, which is only 3.9Å away from a methyl group of the heme moiety. Unexpectedly, the redox activity of the altered proteins was retained in the transformants harboring multiple copies of the gene (Table S3). This was particularly surprising in the case of the p.Leu195Phe substitution (modeling the human p.Leu215Phe substitution), which is expected to alter the heme environment. These data suggest that the substitutions primarily affect the structural integrity of Cyt c1 and thereby reduce its concentration and its likelihood to assemble with other CIII subunits. The assembly defect can be compensated for by an increase in the pool of available altered Cyt c1, either in the yeast transformants harboring multiple copies of the gene or in fibroblasts with a high MOI. Under these conditions, the resultant CIII appears to be fully active in transferring electrons to cytochrome c.
It is interesting to note that even though the RC assays (Table S2) showed affected individuals to have mainly an isolated CIII deficiency, the immunoblot data for fibroblasts revealed a severe reduction of supercomplexes. Similarly, in the yeast complementation studies, there were reduced levels of CIII dimers and supercomplexes. Previous studies have suggested that it is essential to have a fully assembled CIII for the stability and activity of CI12 and that together, CI and CIII form a stable core respirasome to which CIV can also bind.1 These data collectively suggest that the CYC1 mutations affect the stability or assembly of the respirasome, most likely as a result of decreased CIII levels stemming from the depletion in Cyt c1, core 1, and core 2 subunits.
CIII deficiencies have highly variable clinical presentations, although they have common defects in insulin-signaling mechanisms and lactic acidosis (for example, UQCRB and UQCR29,10). Mitochondrial oxidative activity due to RC deficits can be considered one of the key determinants underlying hyperglycemia.13 Studies have suggested that CIII is one of the major sources of reactive oxygen species (ROS).14 Given that ROS has been shown to affect insulin signaling and action, this could explain the hyperglycemia phenotype in the affected inviduals.15
In conclusion, our studies show that two different CYC1 mutations cause ketoacidotic and lactic acidotic encephalopathy and insulin-responsive hyperglycemia. This clinical phenotype is the result of severe reductions in cytochrome c1, as well as marked reductions in other assembly-dependent CIII subunits. Rescue experiments confirm the CYC1 mutations to be the primary cause of CIII defects. The similarity of the clinical symptoms in the two affected individuals leads us to recommend mutation screening for CYC1 in young children presenting with recurrent lactic acidosis and hyperglycemia.
Acknowledgments
We thank Alison Compton and Justine Marum for Sequenom analysis and Wendy Salter for enzyme assays. This research was supported by Association contre les Maladies Mitochondriales, Action Rémy (A.S. and P.R.), Agence Nationale de la Recherche grants to P.R., National Institutes of Health grant HL02274 to A.T., Australian National Health and Medical Research Council (NHMRC) grant 1026891 to S.C. and J.C., an Australian Mitochondrial Disease Foundation (AMDF) PhD Scholarship to M.M., and AMDF funding support to M.D. and N.G.L. N.G.L. was supported by NHMRC Principal Research Fellowship 1002147 and project grant 1022707. D.R.T. was supported by NHMRC Principal Research Fellowship 1022896 and project grant 1023619. We also gratefully acknowledge donations to J.C. by the Crane and Perkins families.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
References
- 1.Schägger H., de Coo R., Bauer M.F., Hofmann S., Godinot C., Brandt U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004;279:36349–36353. doi: 10.1074/jbc.M404033200. [DOI] [PubMed] [Google Scholar]
- 2.Hunte C., Palsdottir H., Trumpower B.L. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 3.de Lonlay P., Valnot I., Barrientos A., Gorbatyuk M., Tzagoloff A., Taanman J.W., Benayoun E., Chrétien D., Kadhom N., Lombès A. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 2001;29:57–60. doi: 10.1038/ng706. [DOI] [PubMed] [Google Scholar]
- 4.Visapää I., Fellman V., Vesa J., Dasvarma A., Hutton J.L., Kumar V., Payne G.S., Makarow M., Van Coster R., Taylor R.W. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am. J. Hum. Genet. 2002;71:863–876. doi: 10.1086/342773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barel O., Shorer Z., Flusser H., Ofir R., Narkis G., Finer G., Shalev H., Nasasra A., Saada A., Birk O.S. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 2008;82:1211–1216. doi: 10.1016/j.ajhg.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghezzi D., Arzuffi P., Zordan M., Da Re C., Lamperti C., Benna C., D’Adamo P., Diodato D., Costa R., Mariotti C. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 2011;43:259–263. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- 7.Hinson J.T., Fantin V.R., Schönberger J., Breivik N., Siem G., McDonough B., Sharma P., Keogh I., Godinho R., Santos F. Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome. N. Engl. J. Med. 2007;356:809–819. doi: 10.1056/NEJMoa055262. [DOI] [PubMed] [Google Scholar]
- 8.Munnich A., Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- 9.Haut S., Brivet M., Touati G., Rustin P., Lebon S., Garcia-Cazorla A., Saudubray J.M., Boutron A., Legrand A., Slama A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum. Genet. 2003;113:118–122. doi: 10.1007/s00439-003-0946-0. [DOI] [PubMed] [Google Scholar]
- 10.Miyake N., Yano S., Sakai C., Hatakeyama H., Matsushima Y., Shiina M., Watanabe Y., Bartley J., Abdenur J.E., Wang R.Y. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum. Mutat. 2013;34:446–452. doi: 10.1002/humu.22257. [DOI] [PubMed] [Google Scholar]
- 11.Iwata S., Lee J.W., Okada K., Lee J.K., Iwata M., Rasmussen B., Link T.A., Ramaswamy S., Jap B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 12.Acín-Pérez R., Bayona-Bafaluy M.P., Fernández-Silva P., Moreno-Loshuertos R., Pérez-Martos A., Bruno C., Moraes C.T., Enríquez J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patti M.E., Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010;31:364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz F., Garcia S., Padgett K.R., Moraes C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012;21:5066–5077. doi: 10.1093/hmg/dds350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B.J., Skiba B., Ooms L.M. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.