

Abstract
Primary ciliary dyskinesia (PCD) is a ciliopathy characterized by airway disease, infertility, and laterality defects, often caused by dual loss of the inner dynein arms (IDAs) and outer dynein arms (ODAs), which power cilia and flagella beating. Using whole-exome and candidate-gene Sanger resequencing in PCD-affected families afflicted with combined IDA and ODA defects, we found that 6/38 (16%) carried biallelic mutations in the conserved zinc-finger gene BLU (ZMYND10). ZMYND10 mutations conferred dynein-arm loss seen at the ultrastructural and immunofluorescence level and complete cilia immotility, except in hypomorphic p.Val16Gly (c.47T>G) homozygote individuals, whose cilia retained a stiff and slowed beat. In mice, Zmynd10 mRNA is restricted to regions containing motile cilia. In a Drosophila model of PCD, Zmynd10 is exclusively expressed in cells with motile cilia: chordotonal sensory neurons and sperm. In these cells, P-element-mediated gene silencing caused IDA and ODA defects, proprioception deficits, and sterility due to immotile sperm. Drosophila Zmynd10 with an equivalent c.47T>G (p.Val16Gly) missense change rescued mutant male sterility less than the wild-type did. Tagged Drosophila ZMYND10 is localized primarily to the cytoplasm, and human ZMYND10 interacts with LRRC6, another cytoplasmically localized protein altered in PCD. Using a fly model of PCD, we conclude that ZMYND10 is a cytoplasmic protein required for IDA and ODA assembly and that its variants cause ciliary dysmotility and PCD with laterality defects.
Main Text
Motile cilia are present on various epithelial surfaces, including the respiratory airways, brain ependyma, and fallopian tubes, and are structurally similar to sperm flagella.1 Their core axoneme is composed of nine peripheral outer doublet microtubules surrounding a central-pair microtubule apparatus (9+2 arrangement), whereas motile embryonic node monocilia lack the central-pair apparatus (9+0 arrangement). Structures attached along the axoneme govern ciliary beating via a highly regulated and synchronous sliding between microtubules (inner-dynein-arm [IDA] and outer-dynein-arm [ODA] motor complexes) and regulate dynein activity (radial spokes and nexin-dynein regulatory complexes). Studies of ciliated organisms, including Chlamydomonas, Paramecium, Xenopus, Planaria, trypanosomes, and Drosophila,2–4 have helped to show that the axoneme is a superstructure facilitating both axoneme bending via the dynein motors’ ability to walk along the microtubules in a minus-ended fashion5 and signal communication between the central apparatus and dynein arms to regulate ciliary motility.
Primary ciliary dyskinesia (PCD [MIM 244400]) is a genetically heterogeneous autosomal-recessive disorder affecting 1 in 15,000–30,000 births and is caused by abnormal function of motile cilia and flagella.6–8 Abnormal motility is associated with axonemal ultrastructural defects, giving rise to symptoms including sinopulmonary disease, which is due to impaired mucociliary transport in the airways and which manifests with perinatal respiratory distress, chronic respiratory infections, rhinosinusitus, otitis media, and bronchiectasis.8 Subfertility can occur in both sexes, and in about half of affected individuals, situs abnormalities are linked to isomerism and, in some cases, congenital heart defects;9 hydrocephalus is a rare association. Mutations that cause PCD have been reported in 19 genes, all of which are associated with various ultrastructural defects, in addition to RPGR (MIM 312610), mutations in which might contribute to syndromic PCD.10 These 19 genes include those encoding axonemal proteins of the ODA or its docking complex (DNAH5, DNAH11, DNAI1, DNAI2, DNAL1, TXNDC3, and CCDC114),11–18 the radial spoke heads (RSPH4A and RSPH9),19 the central-pair apparatus (HYDIN),20 or the nexin-dynein regulatory complexes (CCDC164, CCDC39, and CCDC40).21–23
A distinct set of six proteins altered in PCD are either solely localized to the cytoplasm or found in both the cytoplasm and the axoneme: DNAAF1 (LRRC50), DNAAF2 (KTU), DNAAF3, CCDC103, HEATR2, and LRRC6.24–30 These are most likely involved in the cytoplasmic preassembly of dynein-arm complexes prior to their movement into the axoneme and/or in axonemal transport and attachment processes for the dynein-arm complexes. Variants in these six proteins are associated with a simultaneous ODA and IDA loss, a defect found in a significant proportion (24%–45%) of PCD cases (the variability is due in part to the difficulty in visualizing the IDA by transmission electron microscopy [TEM]).31–33 To determine the full spectrum of genetic defects causing IDA and ODA loss in individuals affected by PCD, we analyzed 38 unrelated PCD-affected families, representing a total of 60 affected individuals displaying reduced or absent IDA and ODAs in nasal biopsies. Signed and informed consent was obtained from all participants according to protocols approved by the institutional ethics review boards, and all cases were diagnosed with classic clinical PCD symptoms, including recurrent respiratory tract infections, chronic sinusitis, rhinitis, otitis media, bronchiectasis, and laterality and fertility defects.
We first used next-generation whole-exome sequencing (WES) to analyze an affected individual from 11 of these PCD-affected families by performing exome capture by in-solution hybridization followed by massively parallel sequencing. Approximately 3 μg of genomic DNA was sheared to a mean fragment size of 150 bp (Covaris), and the fragments were used for Illumina paired-end DNA library preparation and enrichment for target sequences (Agilent). Enriched DNA fragments were sequenced with 100 bp paired-end reads (HiSeq2000 platform, Illumina). Sequencing reads were aligned to the reference human genome with Novoalign (Novocraft Technologies) and the Burrows-Wheeler Aligner. Duplicate and multiple mapping reads were excluded, and the depth and breadth of sequence coverage was calculated with the use of custom scripts and the BedTools package.34 Single-nucleotide substitutions and small indels were identified with SAMtools and Pindel and were annotated with the ANNOVAR tool. Variant calling was performed with a previously published in-house pipeline.35 More than 5 Gb of sequence was generated per sample; >75% of the target exome was present at >20-fold coverage, and >95% was present at 5-fold coverage. We focused on homozygous and compound-heterozygous nonsynonymous or splice-site substitutions or indels that are either absent from or present with a frequency < 0.01 in the 1000 Genomes Project.36 We further filtered variants by removing any that are present at a frequency > 0.01 in 700 in-house non-PCD control exomes.
We found that out of a total of 23, 21, and 24 biallelic variants of interest fitting the filtering criteria, biallelic variants in ZMYND10 (MIM 607070; RefSeq accession number NM_015896.2) were shared by one affected individual from each of families UCL-88, UCL-142, and UCL-157, respectively. ZMYND10 is present in the Cildb ciliome database.37 It was also reported to have a likely role in ciliary motility because its expression is 14-fold higher in ciliated primary human airway epithelial cells upon stimulation of ciliogenesis by transfer to air-liquid interface culture38 and from expression profiling of bronchial biopsies from PCD cases.39 The exome-sequencing coverage in the three affected individuals is detailed in Table S1, available online, and their variant calling and filtering are summarized in Tables S2 and S3. All three cases are of North European descent, and two (UCL-142 II:1 from a genetic isolate and UCL-157 II:5 from a first-cousin consanguineous union) are homozygous for a ZMYND10 c.47T>G (p.Val16Gly) missense substitution, whereas the other (UCL-88 II:2) is compound-heterozygous for c.47T>G (p.Val16Gly) and a frameshift deletion (c.589_590del). The National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server reveals six carriers of c.47T>G (p.Val16Gly) in 4,300 European control exomes, corresponding to a low frequency of 0.000698 (rs138815960); this variant is absent from all 700 in-house control exomes.
Segregation analysis of the identified variants by Sanger sequencing in other available members of the three families confirmed the recessive inheritance of both variants (Figure 1A and Figure S1). Three more unrelated PCD cases carrying ZMYND10 variants were then identified from Sanger sequencing of ZMYND10 in a cohort of 27 additional individuals with ODA and IDA defects: North European origin individuals GVA-09 II:2 and UCL-233 II:1 are, respectively, homozygous for a missense variant (c.797T>C [p.Leu266Pro]) and compound-heterozygous for two missense substitutions (c.47T>G [p.Val16Gly] and c.116T>C [p.Leu39Pro]), whereas UCL-226 from a UK-Pakistani family arising from a first-cousin marriage is homozygous for a frameshift deletion (c.65delT [p.Phe22Serfs∗21]) (Figure 1A). The mutations in family GVA-09, first described in 2000 as showing linkage to ZMYND10 with a “potentially interesting” maximum nonparametric LOD score of 1.41,40 were also detected in a separate WES study performed on the two affected children and their unaffected sibling, II:3 (Table S4). Here, after filtering, ZMYND10 was the best scored option with reference to autosomal-recessive inheritance of variant homozygosity in this consanguineous pedigree (Table S5). Segregation analysis was performed in all three families and was again consistent with an autosomal-recessive disease-inheritance pattern (Figure 1A and Figure S1).
Figure 1.
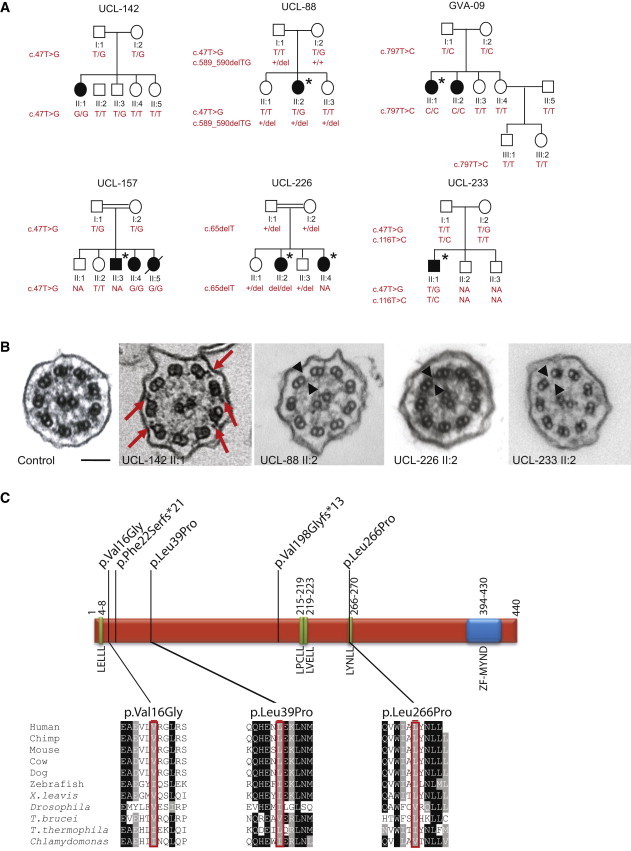
Family Segregation Analysis, Cilia Ultrastructure, and Localization of Variants within the Predicted ZMYND10
(A) Pedigree structure and segregation analysis of the six PCD-affected families harboring ZMYND10 mutations. Filled symbols indicate affected individuals, an asterisk indicates the laterality defect, and a slash indicates that an individual is deceased. Plus signs indicate a normal allele, and “NA” indicates that the result is not available.
(B) TEM shows that the nasal respiratory epithelial cell cilia of ZMYND10-mutant cases have either fewer (red arrows indicate remnant arms) or no (black arrowheads) IDAs and ODAs in comparison to those of the control. Scale bar represents 100 nm.
(C) Location of ZMYND10 variants. Green boxes indicate LxxLL motifs, the blue box represents the ZF-MYND domain, and the amino acid positions are shown. Conservation across species of residues affected by the three missense variants is shown with proteins (corresponding accession numbers are shown in parentheses) from the following species: H. sapiens (RefSeq NP_056980.2), P. troglodytes (RefSeq XP_516479.2), M. musculus (RefSeq NP_444483.2), B. taurus (RefSeq NP_001035638.1), C. lupus (RefSeq XP_533818.1), D. rerio (RefSeq NP_956691.1), X. leavis (RefSeq NP_001090272.2), D. melanogaster (RefSeq NP_648625.1), T. brucei (RefSeq XP_828897.1), T. thermophila (RefSeq XP_001026696.1), and C. reinhardtii (Phytozome accession Cre08.g358750.t1.3). The transcript annotations are based on the Augustus update u11.6 annotation of Joint Genome Institute assembly v.5.
The mutation summary and clinical information for the affected individuals carrying ZMYND10 mutations from all six families are shown in Table S6. The missense substitutions (p.Val16Gly, p.Leu39Pro, and p.Leu266Pro) were predicted to be “damaging” and “probably damaging” by SIFT and PolyPhen-2, respectively. Moreover, all three residues affected by missense variants are well conserved in ZMYND10 orthologs in other ciliated species (Figure 1C and Figure S2). Protein modeling shows that ZMYND10 contains four conserved classical LxxLL protein-binding motifs, in addition to its C-terminal MYND zinc-finger domain, implying that it requires interacting partners for its function. In proteins of this family, amino acid substitutions at any of the leucines within the LxxLL motif abrogate function.41 The first residue of the ZMYND10 LYNLL motif (at residues 266–270), which is the best conserved motif across vertebrates and also in the flagellate protozoan Trypanosoma brucei, is changed by the p.Leu266Pro missense variant in family GVA-09. This proline substitution might be damaging by creating a kink in the predicted coiled LxxLL motif, and this lends support to the functional significance of this motif (Figure 1C).
TEM of nasal biopsy respiratory cilia cross-sections (prepared as previously reported32) from individuals UCL-142 II:1, UCL-88 II:2, UCL-226 II:2, and UCL-233 II:2 detected in all cases a loss of IDAs and ODAs at the ultrastructural level (Figure 1B). In individual UCL-142 II:1 (with homozygous p.Val16Gly), we noticed an apparently intermediate phenotype in nasal respiratory cilia and variable retention of the IDAs and ODAs in different cross-sections. The absence of IDAs and ODAs in cilia axonemes was further confirmed in UCL-88 II:2 by high-resolution immunofluorescence staining for the ODA component DNAH5 and the IDA component DNALI1, respectively, which are now well-established diagnostic markers for PCD dynein-arm defects (Figure S3). An apparent accumulation of staining in the peribasal area of the affected individual’s cilia was noted, especially for DNAH5, indicating that these dynein-arm components might be retained in the cytoplasm, but not successfully assembled and/or transported to the axoneme.
To gain insights into the pathogenic nature of these ZMYND10 mutations, we performed high-speed video microscopic analysis of the affected individuals’ nasal cilia. Compared to controls, cases showing a complete absence of IDAs and ODAs (UCL-88 II:2, UCL-226 II:2, and UCL-233 II:2) had cilia that were almost completely static, consistent with other mutations associated with IDA and ODA defects (Movies S1 and S2).24–30 In contrast, a significant motility was retained by cilia from the c.47T>G (p.Val16Gly) homozygous individual UCL-142 II:1 in whom the dynein arms were partially retained. However, there was a slowed and stiff beating pattern that lacked the normal beat amplitude (a fully extended forward power stroke and backward recovery stroke) seen in controls (Movies S3 and S4); the reduced ciliary beat frequency had a median of 3.97 Hz (range = 2.92–5.24 Hz) compared to the normal range of 4–7 Hz at room temperature. Consistent with this, the IDAs and ODAs were also reported to be reduced, but not absent, in cilia in affected individuals from family UCL-157, which also carries homozygous c.47T>G (p.Val16Gly) mutations. In addition, cilia motility was recorded as variable in family UCL-157: cilia from affected individual II:4 were almost static, whereas II:5 had more normal motility (Table S6).
ZYMND10 orthologs are found widely in ciliated eukaryotes, and notably, no ortholog exists in Caenorhabditis elegans, which has only immotile cilia. There is, however, an ortholog in both Ostreococcus, a green alga that lacks cilia but retains IDA genes, and Physcomitrella, a moss with motile flagellated sperm. The mouse ortholog (Zmynd10) is enriched in the testes and is 1 of 99 genes whose expression pattern is correlated with tissues rich in ciliated cells.42 In humans, alternative splicing leads to two ZMYND10 isoforms differing across 35 centrally positioned amino acids; these are expressed in the testes and lungs (we note that the five sequence variants identified here would equally affect both transcripts).43 ZMYND10 (also known as BLU) is within a tumor-suppressor gene cluster in chromosomal region 3p21.3 and has been found to be inactivated in common human cancers, including lung, nasopharyngeal, and ovarian cancers, but no susceptibility variants have been identified as of yet.44
To investigate the ciliary role of ZMYND10, we determined the tissue distribution of Zmynd10 expression in mouse embryos at embryonic day 18.5 by in situ hybridization as previously described.19 This showed specific expression in the ciliated epithelial layer associated with nasal and lung epithelium (Figures 2A and 2B and Figure S4). This restricted expression in regions where motile cilia are located replicates the localization pattern of other proteins that cause PCD when deficient.11,19 For functional evaluation of ZMYND10, we turned to a Drosophila model using flies maintained on standard media at 25°C and a w1118 strain as a wild-type control. In Drosophila, sensory neurons and sperm are the only cells that bear cilia or flagella. We recently noted that the Drosophila ortholog of ZMYND10, CG11253 (referred to here as Zmynd10), is highly expressed in the transcriptome of developing chordotonal (Ch) sensory neurons,45 which are proprioceptors with motile mechanosensory cilia.46 In situ hybridization of whole embryos by standard methods with the use of a digoxygenin-labeled RNA probe3 confirmed that during embryogenesis Zmynd10 mRNA was restricted to developing Ch neurons and was notably absent from other ciliated sensory neurons (Figure 2C and Figures S5A–S5D). This is mirrored by the expression of a Zmynd10-mVenus fusion gene, which was constructed from the entire Zmynd10, including 1.5 kb of upstream flanking sequence (Figure S5I). The Zmynd10-mVenus fusion gene was expressed in embryonic Ch neurons (Figure 2D). In the pupal antenna, the fusion gene was also expressed exclusively in the Ch neurons of Johnston’s organ; these cells are auditory receptors (Figure 2E). Unlike other Drosophila sensory cilia, Ch neuron cilia have the hallmarks of motility: the proximal portion of their 9+0 axoneme bears dynein arms (Figure 3H), and Ch ciliary movement is important for auditory transduction.48,49 Moreover, genetic perturbation of dynein-arm components causes Drosophila to be deaf50 and also perturbs proprioception.3 In adults, Zmynd10 is specifically expressed in the testes.51 Thus, Zmynd10 expression correlates exclusively with the development of cells with flagella or motile cilia.
Figure 2.
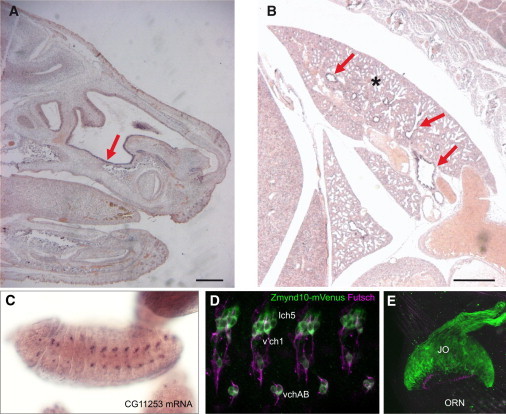
Restricted Expression of ZMYND10 within Motile Ciliated Tissues
(A and B) Expression of Zmynd10 mRNA in embryonic day 18.5 mouse embryos by whole-mount in situ hybridization reveals specific expression in the nasal epithelium (A, arrow) and the bronchi (B, arrows) of the lung (indicated by an asterisk). Scale bars represent 0.5 mm.
(C) Expression of Zmynd10 (CG11253) in Drosophila embryos by whole-mount in situ hybridization reveals expression restricted to differentiating Ch neurons.
(D) The Drosophila Zmynd10-mVenus fusion gene (green) is expressed in Ch neurons (lch5, v’ch5, and vchAB), but not other ciliated sensory neurons (marked by FUTSCH, magenta) in the embryo.
(E) In the pupal antenna, the Zmynd10-mVenus fusion gene is strongly expressed in the Ch neurons of Johnston’s Organ (“JO”), but not the olfactory receptor neurons (“ORNs”).
Figure 3.
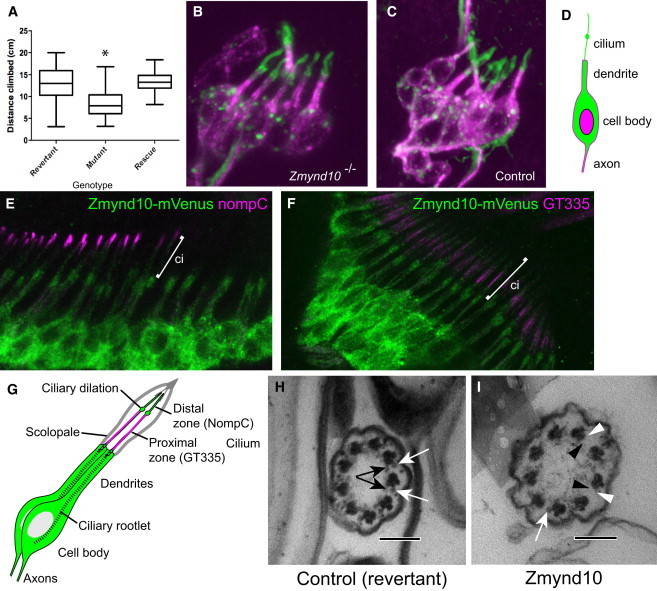
Zmynd10-Mutant Flies Have Sensory Defects and Loss of the Axonemal Dynein Arms
(A) A box and whisker plot of climbing assay for proprioceptive defects. Twenty 2- to 7-day-old flies were placed in a measuring cylinder, and then after a 1 min recovery period, they were banged to the bottom of the cylinder, and the height climbed in 10 s was then recorded. Homozygous mutant Zmynd10EY10886 flies performed significantly less well in this assay than did the “wild-type” revertants (in which the P element had been lost by excision) (p < 0.0001). The Zmynd10-mVenus fusion transgene completely restored the climbing behavior of homozygous mutant flies (“rescue”). Significance was determined by a Kruskal-Wallis test (H = 35.48; n = 44, 42, and 28; p < 0.0001) followed by a Dunn’s test.
(B and C) The lch5 neuron cluster from an embryonic abdominal segment stained with FUTSCH (magenta) and anti-HRP, which detects the cilium at this stage (green). In mutant embryos, the cilia are present and appear grossly normal in length.
(D) Schematic of embryonic Ch neuron.
(E and F) In Ch neurons in pupal antennae, Zmynd10-mVenus expression (green) is largely cytoplasmic relative to nompC and GT335 (distal and proximal ciliary markers, magenta).
(G) Schematic of pupal Ch neurons.
(H and I) TEM of adult antennal Ch neuron cilia. A control with ODA and IDA complexes (white and black arrows, respectively) is indicated in (H). A Zmynd10 mutant, showing loss of some ODA and IDA complexes (white and black arrowheads, respectively) is shown in (I). Primary antibodies used were mAb-22C10 (1:200), RbAb-HRP (1:500), RbAb-GFP (1:500) (Molecular Probes), mAb-NompC (1:100),47 and GT335 (Sigma, 1:200). Secondary antibodies were from Molecular Probes. Scale bars represent 100 nm.
Transcriptional regulation of Zmynd10 supports a role in ciliary motility. The transcription factors Fd3F and Rfx were recently shown to coregulate genes for ciliary motility in Ch neurons (including axonemal dynein genes).3 Indeed, Fd3F is related to vertebrate Foxj1, which is strongly linked to the differentiation of cells bearing motile cilia.52–54 Zmynd10 expression in Ch neurons was greatly reduced in Rfx49 and fd3F1 mutant embryos3,55 (Figures S5E–S5G). The region immediately upstream of Zmynd10 (which supported Ch-neuron-specific expression of the Zmynd10-mVenus fusion gene) contains conserved binding motifs for these factors (Figure S5I). In summary, expression of Zmynd10 is confined to the only Drosophila cells bearing a motile cilium or flagellum, and its expression is dependent on the transcription factors that regulate motile cilia.
Fly stock p{EPgy2}CG11253EY10886 (obtained from the Bloomington Stock Center [Indiana Univerisity]) contains a P element inserted in the last intron of Zmynd10 (Figure S5I). In situ hybridization showed that Zmynd10 mRNA appeared to be strongly reduced or absent in embryos homozygous for this P element insertion, indicating a strong loss-of-function mutation (Figure S5H). Homozygous Zmynd10EY10886 flies are viable and have no visible morphological defects. However, a climbing assay revealed that they are uncoordinated (Figure 3A). This phenotype was not observed in revertant flies in which the P element had been excised from the Zmynd10 locus, and coordinated locomotion was completely restored by the introduction of the Zmynd10-mVenus fusion gene (Figure 3A). Together with expression-pattern data, this suggests that Zmynd10EY10886 homozygotes have defective proprioception as a result of malfunctioning Ch neurons. Indeed, antennal Ch neurons are also defective, given that Zmynd10-mutant flies have recently been reported to be deaf.50
Ch neuron structure was examined by immunofluorescence in embryos, larvae, and pupal antennae. No gross morphological defects in Ch neurons or their terminal cilia were observed with morphology markers (Figures 3B–3D). Compartmentalization of the Ch neuron cilium also appeared normal in that it had correctly localized markers of the proximal motile zone (GT335, anti-polyglutamylated tubulin) and distal sensory zone (the TRPN channel, NOMPC)56 (Figure S6 and data not shown). This suggests that there are no general defects in ciliogenesis, compartmentalization of the cilium, or intraflagellar transport (IFT). Ch cilium ultrastructure was examined by TEM of Johnston’s organ in the adult antenna by Electron Microscopy Research Services, Newcastle University Medical School, as described previously.3 This revealed the presence of a normal 9+0 cilium on each Ch neuron dendrite. However, the proximal axonemal zone showed a reduction in observable dynein arms (Figures 3H and 3I). Both ODAs and IDAs were reduced, and IDAs were possibly affected more strongly (Table S7). Localization of the protein encoded by Zmynd10-mVenus was most strongly observed in the Ch neuron cell bodies, but less reached the inner dendritic segment (Figures 3E–3G). A low level was also observed in the cilium, although this did not extend to the tip.
Homozygous Zmynd10-mutant males were infertile given that they produced no offspring when crossed to wild-type females (n = 60). In mutant males, the testes appeared normal and contained developing sperm bundles (Figures 4A and 4B). TEM showed that the sperm bundles generally consisted of 64 sperm (data not shown), suggesting that spermatocyte divisions and sperm differentiation were largely unaffected. However, motile sperm were never observed in testis dissections; indeed, the seminal vesicles were completely devoid of sperm, suggesting that the sperm were not transferred from the testes to the seminal vesicles (Figures 4A and 4B). This phenotype is similar to that of mutants of Dic61B, a testis-specific dynein intermediate chain homolog.57
Figure 4.
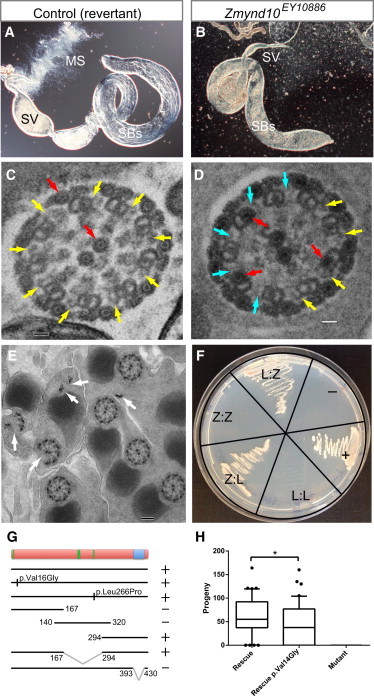
Zmynd10-Mutant Male Flies Lack Motile Sperm
(A) In a dissected wild-type adult (5-day-old) testis, sperm bundles (“SBs”) can be seen, the mature sperm are transferred to the seminal vesicle (“SV”), and motile sperm (“MS”) can be seen emerging from this structure.
(B) In a Zmynd10-mutant fly, sperm bundles are still observed in the testis, but the seminal vesicle is empty and no motile sperm are seen emerging. There is also some disruption of sperm-bundle coiling at the proximal end of the testis, which might be a secondary effect of sperm immotility.
(C–E) TEM transverse sections of sperm bundles in a testis.
(C) In a control fly, yellow arrows point to dynein arms. Also visible (red arrows) are the inner pair and outer accessory microtubules, which have an electron-dense core (the luminal filament). Scale bar represents 20 nm.
(D) In the mutant, there is loss of dynein arms (cyan arrows) and ectopic luminal filaments in the A microtubule of the doublets (red arrows). Scale bar represents 20 nm.
(E) In the mutant, some flagella have fragmented axonemes (arrows). Scale bar represents 100 nm.
(F) Yeast two-hybrid assay of human ZMYND10 (“Z”) and LRRC6 (“L”). In each sector, the Bait:Prey combinations are indicated. Also present are positive (p53) and negative (Lamin) controls.
(G) A summary of yeast two-hybrid results of the interaction between LRRC6 and mutated or truncated forms of ZMYND10 shows that interaction (+) is not affected by the PCD-linked protein variants but relies on the presence of the MYND domain. Variants were introduced by site-directed mutagenesis with the use of the Strataclone Quickchange 2 kit (Stratagene).
(H) A box and whisker blot shows the progeny produced by single male flies. Zmynd10-mutant males (“mutant”) were infertile, but fertility was restored with the rescue transgene (“rescue”). A two-tailed Mann-Whitney test shows that restoration of fertility was significantly less with a rescue transgene containing the p.Val14Gly missense variant (“rescue V14G”) (U = 586; n = 40, 40; p = 0.0385). For fertility analysis, 40 2- to 5-day-old males were crossed individually to wild-type females. After 2 days of prelaying, flies were transferred to new vials and were allowed to lay eggs for 3 days. Progeny from the latter were counted.
TEM showed sperm flagella with a partial loss of dynein arms (Figures 4C and 4D), and 12% (n = 384) showed axoneme splitting, whereby one or more doublet complexes became detached from the rest of the axoneme (Figure 4E). This might suggest impaired nexin or radial-spoke connections. An additional phenotype was the presence of ectopic luminal filaments (electron-dense cores) within the “A” microtubule of some doublets (Figure 4D). Interestingly, these phenotypes have also been reported to be associated with mutations in the axonemal β2-tubulin gene B2t6. Like Zmynd10-mutant males, B2t6-mutant males generate largely intact but immotile sperm with missing ODAs.58
Recently, mutations linked to PCD were reported in LRRC6 (MIM 614930), whose Drosophila ortholog is touch-insensitive larval B (tilB).29,30The tilB-mutant phenotype, developmental expression pattern, and protein subcellular localization all closely resemble that of mutant Zmynd10.59,60 These similarities suggest that their two encoded proteins might function together, and indeed, a Zmynd10 interaction was reported in a large-scale interaction mapping of the Drosophila proteome.61 Given these links, we investigated whether human ZMYND10 and LRRC6 interact. Human ZMYND10 and LRRC6 cDNAs were cloned into pGADT7 activation-domain and pGBKT7 DNA-binding-domain vectors for use in a Matchmaker yeast two-hybrid assay according to the manufacturer’s protocols (Clontech). A clear interaction between these proteins was revealed (Figure 4F). In the same assay, the p.Val16Gly and p.Leu266Pro variants of ZMYND10 still showed interaction with LRRC6 (Figure 4G). Furthermore, deletion analysis of ZMYND10 showed that the interaction strictly required the MYND zinc-finger domain (Figure 4G). To further confirm the interaction between the two proteins, we performed a GST pull-down assay on Myc-tagged in-vitro-translated LRRC6 by using either GST or GST-ZMYND10. After electrophoresis of the washed GST samples alongside a sample of the Myc-tagged LRR6 (input), the immunoblot was probed with Myc antibody and showed specific binding of the GST-ZMYND10 only (Figure S7).
We used the fly to model the putative hypomorphic effects of the p.Val16Gly missense variant that correlated with retained ciliary beating activity in affected individuals. We found that fertility and sperm motility in mutant male flies were completely rescued by the introduction of the wild-type Zmynd10-mVenus fusion gene. When the PCD-associated variant, p.Val16Gly, was engineered into the fusion gene (p.Val14Gly in the Drosophila protein) by site-directed mutagenesis with the StrataClone QuikChange 2 kit (Stratagene), fertility was restored, but not as fully as for the wild-type protein (Figure 4H).
In this study, we have shown that recessive loss-of-function mutations in ZMYND10 underlie PCD with abnormal axonemal ODA and IDA assembly. This study expands our current understanding of the genetic heterogeneity underlying PCD, given that ZMYND10 is the seventh gene associated with disease arising from dual IDA and ODA defects and static cilia; in this study, its mutations caused 16% (6/38) of the cases of IDA and ODA defects. We identified two predicted frameshift and three missense variants in ZMYND10; notably, one missense change (p.Leu266Pro) affects the first leucine residue of one of the protein’s key predicted protein-interaction domains, a conserved LxxLL sequence motif. Being found present on six disease chromosomes in four out of six families affected by ZYMND10-associated PCD in this study, the c.47T>G (p.Val16Gly) missense variant could possibly represent a common European-founder-effect mutation, and we investigated this further by deriving haplotype data from SNPs surrounding the variant in the three affected individuals who carry the variant and for whom exome data are available (Table S1). A shared 1.6 Mb haplotype spanning the c.47T>G (p.Val16Gly) variant could be derived, which seems likely to indicate a common ancestral mutation event, but the uninformative nature of the SNP markers within this region prevented firm conclusions (Table S8). These findings can inform future developments in both genetic testing and gene-based therapeutic strategies for PCD and will thus be useful for improved diagnosis, counseling, and carrier testing in families affected by PCD.
In affected individuals, we noted a difference in the ciliary dysmotility conferred between different ZMYND10 mutations, given that the homozygous inheritance of the c.47T>G (p.Val16Gly) missense variant was found to correlate with retention of cilia motility. The equivalent p.Val16Gly-containing protein was still able to rescue the male infertility phenotype of mutant flies but did so less completely than the wild-type protein, supporting the idea of some retention of function. Despite the fact that we found a cellular genotype-phenotype correlation for PCD, suggesting that c.47T>G (p.Val16Gly) is a functionally more “mild” allele than a complete null, the clinical significance is unclear: affected individuals carrying the homozygous c.47T>G (p.Val16Gly) mutation still exhibit ineffective mucociliary clearance and classic disease symptoms.
The presence of the LxxLL motifs in the conserved ZMYND10 is intriguing. This motif was first identified in diverse transcriptional coactivators recruited to nuclear receptors in the presence of an agonist, including histone acetyltransferases and histone lysine demethylases (p160s, p300 [also known as CBP], and KDM1A).62 Several proteins with LxxLL motifs have been well studied structurally and functionally. Our PCD-associated p.Leu266Pro variant is of interest because it represents an amino acid substitution associated with a human disease in an LxxLL motif. Potentially, ZMYND10 could interact with transcription factors and related coregulator complexes in the cytoplasm and thereby influence the expression of other proteins that are known to cause PCD when deficient. Consistent with this, ZMYND10 has been shown to interact with the TSC22D4 (also known as THG1) corepressor and RNA-processing enzyme.63–65 However, there is no evidence supporting a direct transcriptional regulatory role for ZYMND10 as of yet. The alternative possibility based on our findings, including the protein interaction revealed between ZMYND10 and LRRC6, is that ZMYND10 plays a direct role in the assembly of the IDAs and ODAs into the axoneme by interacting with the essential family of cytoplasmic dynein-arm preassembly factors that have already been associated with the etiology of PCD. In this case, our study might have identified a mechanism whereby the LxxLL motifs common in transcriptional regulatory complexes play a role in the formation of non-transcription-related complexes, as has been observed for the highly specialized LD motif (LDxLL) found in the paxillin superfamily.66 Further studies are therefore warranted to provide insight into the role of the LxxLL motifs identified in ZYMND10 and their potential role in dynein-arm assembly.
We have shown here that Drosophila can be used as a suitable model organism for PCD in that it possesses two unique systems available for motile axonemal and ciliogeneis characterization and genetic manipulation: the Ch sensory neurons and the male reproductive system. The heritable mutations mimicking human PCD can be investigated in flies with the use of relatively simple neurological and fertility assays, providing an in vivo platform for modeling PCD-related gene function both for understanding the underlying cellular basis and for testing the ability of PCD-associated variants to rescue the fly mutations. This model will thus be widely applicable to advancing our understanding of the genetic and cellular basis of PCD.
Using this model organism, we determined that Zmynd10 has a highly restricted expression pattern and function confined to motile-ciliated cells and sperm. The developmental expression of Zmynd10 in relation to cilium formation in Ch neurons showed that Zmynd10 expression in developing neurons precedes expression of dynein genes and completion of neuronal terminal differentiation and cilium outgrowth by at least several hours (Figures S5A–S5C),3 which suggests that Zmynd10 plays a developmental role in dynein-arm synthesis, assembly, or transport. In flies, we were also able to localize ZMYND10 primarily as a cytoplasmic component of cells containing motile cilia. Its mainly cytoplasmic localization is reminiscent of that of three other proteins linked to ODA and IDA loss in PCD: DNAAF1, DNAAF2, and DNAAF3, which are regarded as dynein-arm assembly factors.24,26,27 Our data showing that human ZMYND10 interacts with the reported cytoplasmic dynein assembly factor LRRC6 further supports a role for ZMYND10 in the dynein-arm assembly process that causes PCD when deficient.
Acknowledgments
We would like to thank all the primary ciliary dyskinesia (PCD)-affected families for their participation in the study, Fiona Copeland, and the UK PCD Family Support Group. We wish to acknowledge the contribution of Björn Afzelius, who originally ascertained the GVA-09 family. We thank Jeanette Dankert-Roelse, Maggie Meeks, Celia D. Delozier, and R. Mark Gardiner for family recruitment and their past involvement in the project. We thank Paul Griffin, Sarah Ollosson, Mellisa Dixon, Patricia Goggin, Claire Jackson, and Maria Philipsen for light and electron microscopy. We acknowledge technical assistance from Panagiotis Maghsoudlou, Corinne Gehrig, and Anne Vannier. The pBID-UASC-GV plasmid was a gift from B. McCabe. S.E.A. is supported by a Gebert Foundation grant and grants ERC 249968 and SNF 144082. J.-L.B. is supported by grants from the Swiss National Science Foundation (#32003B_135709). J.-L.B., L.B., and H.M.M. are supported by grants from the Milena Carvajal Pro-Kartagener Foundation of Geneva. P.M. is supported by a grant from the Bodossakis Foundation. M.S. is supported by an Action Medical Research UK Clinical Training Fellowship (RTF-1411). A.O., S.P., E.M.K.C., and H.M.M. are supported by Action Medical Research awards GN1773 and GN2101 and Newlife Foundation for Disabled Children UK award 10-11/15. P.I.z.L. and A.P.J. are supported by the Medical Research Council of Great Britain (MR/K018558/10). D.J.M. and G.G. are supported by studentships from the Biotechnology and Biological Sciences Research Council and the Medical Research Council, respectively.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
FlyBase, www.flybase.org
NHLBI Exome Variant Server/Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
References
- 1.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 2.Bower R., Tritschler D., Vanderwaal K., Perrone C.A., Mueller J., Fox L., Sale W.S., Porter M.E. The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol. Biol. Cell. 2013;24:1134–1152. doi: 10.1091/mbc.E12-11-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newton F.G., zur Lage P.I., Karak S., Moore D.J., Göpfert M.C., Jarman A.P. Forkhead transcription factor Fd3F cooperates with Rfx to regulate a gene expression program for mechanosensory cilia specialization. Dev. Cell. 2012;22:1221–1233. doi: 10.1016/j.devcel.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broadhead R., Dawe H.R., Farr H., Griffiths S., Hart S.R., Portman N., Shaw M.K., Ginger M.L., Gaskell S.J., McKean P.G., Gull K. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 5.Lindemann C.B., Lesich K.A. Flagellar and ciliary beating: the proven and the possible. J. Cell Sci. 2010;123:519–528. doi: 10.1242/jcs.051326. [DOI] [PubMed] [Google Scholar]
- 6.Afzelius B.A. Genetics and pulmonary medicine. 6. Immotile cilia syndrome: past, present, and prospects for the future. Thorax. 1998;53:894–897. doi: 10.1136/thx.53.10.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbato A., Frischer T., Kuehni C.E., Snijders D., Azevedo I., Baktai G., Bartoloni L., Eber E., Escribano A., Haarman E. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 2009;34:1264–1276. doi: 10.1183/09031936.00176608. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 10.Bukowy-Bieryłło Z., Ziętkiewicz E., Loges N.T., Wittmer M., Geremek M., Olbrich H., Fliegauf M., Voelkel K., Rutkiewicz E., Rutland J. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 2013;48:352–363. doi: 10.1002/ppul.22632. [DOI] [PubMed] [Google Scholar]
- 11.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 12.Bartoloni L., Blouin J.L., Pan Y., Gehrig C., Maiti A.K., Scamuffa N., Rossier C., Jorissen M., Armengot M., Meeks M. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pennarun G., Escudier E., Chapelin C., Bridoux A.M., Cacheux V., Roger G., Clément A., Goossens M., Amselem S., Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duriez B., Duquesnoy P., Escudier E., Bridoux A.M., Escalier D., Rayet I., Marcos E., Vojtek A.M., Bercher J.F., Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onoufriadis A., Paff T., Antony D., Shoemark A., Micha D., Kuyt B., Schmidts M., Petridi S., Dankert-Roelse J.E., Haarman E.G., UK10K Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:88–98. doi: 10.1016/j.ajhg.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knowles M.R., Leigh M.W., Ostrowski L.E., Huang L., Carson J.L., Hazucha M.J., Yin W., Berg J.S., Davis S.D., Dell S.D., Genetic Disorders of Mucociliary Clearance Consortium Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:99–106. doi: 10.1016/j.ajhg.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W.S., Loges N.T., Pennekamp P., Lindberg S., Stenram U. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horani A., Druley T.E., Zariwala M.A., Patel A.C., Levinson B.T., Van Arendonk L.G., Thornton K.C., Giacalone J.C., Albee A.J., Wilson K.S. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:685–693. doi: 10.1016/j.ajhg.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.F., Siffroi J.P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papon J.F., Coste A., Roudot-Thoraval F., Boucherat M., Roger G., Tamalet A., Vojtek A.M., Amselem S., Escudier E. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2010;35:1057–1063. doi: 10.1183/09031936.00046209. [DOI] [PubMed] [Google Scholar]
- 32.Shoemark A., Dixon M., Corrin B., Dewar A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J. Clin. Pathol. 2012;65:267–271. doi: 10.1136/jclinpath-2011-200415. [DOI] [PubMed] [Google Scholar]
- 33.Chilvers M.A., Rutman A., O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones W.D., Dafou D., McEntagart M., Woollard W.J., Elmslie F.V., Holder-Espinasse M., Irving M., Saggar A.K., Smithson S., Trembath R.C. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 2012;91:358–364. doi: 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnaiz O., Malinowska A., Klotz C., Sperling L., Dadlez M., Koll F., Cohen J. Cildb: a knowledgebase for centrosomes and cilia. Database (Oxford) 2009;2009:bap022. doi: 10.1093/database/bap022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross A.J., Dailey L.A., Brighton L.E., Devlin R.B. Transcriptional profiling of mucociliary differentiation in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2007;37:169–185. doi: 10.1165/rcmb.2006-0466OC. [DOI] [PubMed] [Google Scholar]
- 39.Geremek M., Bruinenberg M., Ziętkiewicz E., Pogorzelski A., Witt M., Wijmenga C. Gene expression studies in cells from primary ciliary dyskinesia patients identify 208 potential ciliary genes. Hum. Genet. 2011;129:283–293. doi: 10.1007/s00439-010-0922-4. [DOI] [PubMed] [Google Scholar]
- 40.Blouin J.L., Meeks M., Radhakrishna U., Sainsbury A., Gehring C., Saïl G.D., Bartoloni L., Dombi V., O’Rawe A., Walne A. Primary ciliary dyskinesia: a genome-wide linkage analysis reveals extensive locus heterogeneity. Eur. J. Hum. Genet. 2000;8:109–118. doi: 10.1038/sj.ejhg.5200429. [DOI] [PubMed] [Google Scholar]
- 41.McInerney E.M., Rose D.W., Flynn S.E., Westin S., Mullen T.M., Krones A., Inostroza J., Torchia J., Nolte R.T., Assa-Munt N. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McClintock T.S., Glasser C.E., Bose S.C., Bergman D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics. 2008;32:198–206. doi: 10.1152/physiolgenomics.00128.2007. [DOI] [PubMed] [Google Scholar]
- 43.Agathanggelou A., Dallol A., Zöchbauer-Müller S., Morrissey C., Honorio S., Hesson L., Martinsson T., Fong K.M., Kuo M.J., Yuen P.W. Epigenetic inactivation of the candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene. 2003;22:1580–1588. doi: 10.1038/sj.onc.1206243. [DOI] [PubMed] [Google Scholar]
- 44.Chang J.W., Hsu H.S., Ni H.J., Chuang C.T., Hsiung C.H., Huang T.H., Wang Y.C. Distinct epigenetic domains separated by a CTCF bound insulator between the tandem genes, BLU and RASSF1A. PLoS ONE. 2010;5:e12847. doi: 10.1371/journal.pone.0012847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cachero S., Simpson T.I., Zur Lage P.I., Ma L., Newton F.G., Holohan E.E., Armstrong J.D., Jarman A.P. The gene regulatory cascade linking proneural specification with differentiation in Drosophila sensory neurons. PLoS Biol. 2011;9:e1000568. doi: 10.1371/journal.pbio.1000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jarman A.P. Studies of mechanosensation using the fly. Hum. Mol. Genet. 2002;11:1215–1218. doi: 10.1093/hmg/11.10.1215. [DOI] [PubMed] [Google Scholar]
- 47.Liang X., Madrid J., Saleh H.S., Howard J. NOMPC, a member of the TRP channel family, localizes to the tubular body and distal cilium of Drosophila campaniform and chordotonal receptor cells. Cytoskeleton (Hoboken) 2011;68:1–7. doi: 10.1002/cm.20493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Göpfert M.C., Humphris A.D., Albert J.T., Robert D., Hendrich O. Power gain exhibited by motile mechanosensory neurons in Drosophila ears. Proc. Natl. Acad. Sci. USA. 2005;102:325–330. doi: 10.1073/pnas.0405741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Göpfert M.C., Robert D. Motion generation by Drosophila mechanosensory neurons. Proc. Natl. Acad. Sci. USA. 2003;100:5514–5519. doi: 10.1073/pnas.0737564100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Senthilan P.R., Piepenbrock D., Ovezmyradov G., Nadrowski B., Bechstedt S., Pauls S., Winkler M., Möbius W., Howard J., Göpfert M.C. Drosophila auditory organ genes and genetic hearing defects. Cell. 2012;150:1042–1054. doi: 10.1016/j.cell.2012.06.043. [DOI] [PubMed] [Google Scholar]
- 51.Marygold S.J., Leyland P.C., Seal R.L., Goodman J.L., Thurmond J., Strelets V.B., Wilson R.J., FlyBase consortium FlyBase: improvements to the bibliography. Nucleic Acids Res. 2013;41(Database issue):D751–D757. doi: 10.1093/nar/gks1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X., Ng C.P., Habacher H., Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 53.Jacquet B.V., Salinas-Mondragon R., Liang H., Therit B., Buie J.D., Dykstra M., Campbell K., Ostrowski L.E., Brody S.L., Ghashghaei H.T. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development. 2009;136:4021–4031. doi: 10.1242/dev.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stubbs J.L., Oishi I., Izpisúa Belmonte J.C., Kintner C. The forkhead protein Foxj1 specifies node-like cilia in Xenopus and zebrafish embryos. Nat. Genet. 2008;40:1454–1460. doi: 10.1038/ng.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dubruille R., Laurençon A., Vandaele C., Shishido E., Coulon-Bublex M., Swoboda P., Couble P., Kernan M., Durand B. Drosophila regulatory factor X is necessary for ciliated sensory neuron differentiation. Development. 2002;129:5487–5498. doi: 10.1242/dev.00148. [DOI] [PubMed] [Google Scholar]
- 56.Lee J., Moon S., Cha Y., Chung Y.D. Drosophila TRPN(=NOMPC) channel localizes to the distal end of mechanosensory cilia. PLoS ONE. 2010;5:e11012. doi: 10.1371/journal.pone.0011012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fatima R. Drosophila Dynein intermediate chain gene, Dic61B, is required for spermatogenesis. PLoS ONE. 2011;6:e27822. doi: 10.1371/journal.pone.0027822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raff E.C., Hoyle H.D., Popodi E.M., Turner F.R. Axoneme beta-tubulin sequence determines attachment of outer dynein arms. Curr. Biol. 2008;18:911–914. doi: 10.1016/j.cub.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kavlie R.G., Kernan M.J., Eberl D.F. Hearing in Drosophila requires TilB, a conserved protein associated with ciliary motility. Genetics. 2010;185:177–188. doi: 10.1534/genetics.110.114009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eberl D.F., Hardy R.W., Kernan M.J. Genetically similar transduction mechanisms for touch and hearing in Drosophila. J. Neurosci. 2000;20:5981–5988. doi: 10.1523/JNEUROSCI.20-16-05981.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., Godwin B., Vitols E. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 62.Chen W., Roeder R.G. Mediator-dependent nuclear receptor function. Semin. Cell Dev. Biol. 2011;22:749–758. doi: 10.1016/j.semcdb.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 64.Hyde S.J., Eckenroth B.E., Smith B.A., Eberley W.A., Heintz N.H., Jackman J.E., Doublié S. tRNA(His) guanylyltransferase (THG1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc. Natl. Acad. Sci. USA. 2010;107:20305–20310. doi: 10.1073/pnas.1010436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kester H.A., Blanchetot C., den Hertog J., van der Saag P.T., van der Burg B. Transforming growth factor-beta-stimulated clone-22 is a member of a family of leucine zipper proteins that can homo- and heterodimerize and has transcriptional repressor activity. J. Biol. Chem. 1999;274:27439–27447. doi: 10.1074/jbc.274.39.27439. [DOI] [PubMed] [Google Scholar]
- 66.Brown M.C., Curtis M.S., Turner C.E. Paxillin LD motifs may define a new family of protein recognition domains. Nat. Struct. Biol. 1998;5:677–678. doi: 10.1038/1370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.