

Abstract
BACKGROUND
We investigate the influence of oligomeric forms of Aß and the influence of duration of exposure on the development of tau phosphorylation.
METHODS
Aβ oligomers were injected intracranially either acutely into 5 mo old rTg4510 mice and tissue collected 3 days later or chronically into 3 mo old mice and tissue collected 2 mo later. Several forms of phosphorylated tau, glycogen synthase kinase 3 (GSK3) activation, microglial and astrocyte activation were measured.
RESULTS
Acute injections of Aß oligomers had no effect on phospho-tau epitopes, but did result in elevation of phosphorylated/activated GSK3. Chronic infusion of Aß oligomers into the right hippocampus resulted in a 3-4 fold elevations of several phospho-tau isoforms with no changes in total tau levels. A significant elevation of pGSK3 accompanied these changes. Microglial staining with CD68 paralleled the increase in tau phosphorylation, however CD45 staining was unaffected by Aß. Control experiments revealed that the infusion of Aß from the minipumps was largely complete by 10 d after implantation. Thus, the elevation of phospho-tau 2 mo after implantation implies that the changes are quite persistent.
CONCLUSION
Soluble Aβ1-42 oligomers have long-lasting effects on tau phosphorylation in rTg4510 model, possibly due to elevations in GSK3. These data suggest that even brief elevations of Aß production, may have enduring impact on the risk for tauopathy.
Keywords: β-amyloid, phospho-tau, glycogen synthase kinase 3 α/β (GSK3), microglia, inflammation, microosmotic pump
INTRODUCTION
In Alzheimer’s Disease (AD) and other tauopathy disorders, tau is involved in a series of pathological events, including hyperphosphorylation, aggregation into straight and paired helical filaments [1], and finally, formation of neurofibrillary tangles (NFT). The consequence of this process is disruption of microtubule assembly [2] and axonal transport [3, 4], which ultimately may lead to neurodegeneration.
Transgenic mouse models expressing human tau mutations develop pathology that mimics frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) disorder, and have improved understanding of tau-mediated neurodegeneration [5, 6]. The inducible rTg4510 mouse model used here has demonstrated progression of tau pathology with age, formation of NFT, cognitive impairments and neuronal loss [7, 8]. Understanding the mechanisms of cross-talk between tau and β-amyloid has moved this area to the center of research efforts. It is still unclear in the literature whether hAPP/β-amyloid (Aβ) pathology is responsible for tau pathology in AD; reports from studies of transgenic mouse models suggest that manipulation of Aβ levels drives changes in the tau pathology [9-11]. Notably, the role of AD brain-derived and synthetically prepared Aβ oligomers in causing early synaptic toxicity [12, 13], LTP deficits [14, 15] and cognitive dysfunction [16] supports a direct role of Aβ1-42 derived oligomers in the early pathology of disease. However, other reports suggest that reduction of endogenous tau levels protects against excitotoxicity in vitro [17] and prevents Aβ-related behavioral deficits in transgenic mice expressing hAPP, potentially via tyrosine kinase Fyn activation [18, 19]. Moreover, a dendritic role of tau in conferring Fyn-mediated Aβ toxicity was suggested as a possible mechanistic explanation on the Aβ, tau and Fyn triad [20].
In the present study, we examined the effect of acute or chronically infused synthetic oligomeric Aβ1-42 on tau phosphorylation (p-tau) in 5-month-old rTg4510 mice. The questions we wished to address concerned a) the dwell time of oligomeric Aß after administration intracranially, b) the impact of the Aß oligomers on tau metabolism and its persistence in the early stages of tauopathy and c) the relationship of these changes to kinase activity and microglial activation. Our findings are consistent with the accumulating body of evidence that Aß oligomers exacerbate tau pathology in vivo.
MATERIALS AND METHODS
Production and SDS-PAGE/Immunoblotting of oligomers
Oligomers of Aβ1-42 were prepared as previously described [21]. For initial characterization Aβ (1–42) peptide obtained from Biosource, was dissolved in ice-cold HFIP, thoroughly mixed. Aliquots were taken, the HFIP was completely evaporated, and samples were frozen at −80°C until use. For oligomer preparations the dried Aβ was first dissolved in 100% DMSO to 5 mM Aβ, then further diluted 50-fold with F12 medium (PromoCell) supplemented with glutamine, 146mg/L (PromoCell, GmbH, Germany) to obtain a 100 μM Aβ solution. Samples were incubated for 24h at 4°C and then centrifuged at 14000g for 10 min at 4°C to remove large aggregates. The size distribution of the oligomers in the supernatant was examined by atomic force microscopy (AFM).
The 100 μM of Aβ1-42 sample prepared as described above was incubated for 30 days inside a microosmotic pump (Alzet #1004, Durect Corp. Cupertino, CA) at 37°C in PBS (pH 7.4). Due to the properties of the osmotic pump, we were able to collect aliquots of oligomers pumped from the reservoir at various time points. Aliquots were diluted 10-fold, and applied to SDS-PAGE as described previously [22]. Monoclonal mouse antibody 6E10 against β-amyloid was used (Convance, Dedham, MA; 1:1000 dilution). Immunostained proteins were visualized using chemiluminescence, in accordance with the manufacturer’s recommendations (ECL, SignaGen Laboratories, Gaithersburg, MD).
Oligomeric Solution Physical Characterization
Atomic force microscopy (AFM, XE-100 PSIA Inc., Santa Clara, CA) was used to determine the morphology of the species present in the Aβ1-42 oligomeric solution. Silicon nitride with aluminum coating (Tap300Al-G, Budget Sensors, Bulgaria) cantilevers with 300 kHz frequency and 40 N/m force constant were used in this study. The images were collected at a scan rate of 1Hz/min and gain 1. Five microliters of the oligomeric solution were deposited on freshly cleaved mica at a concentration of 25 μM, incubated for 5 minutes, rinsed with Millipore water, and dried with ultrapure nitrogen (NI UHP300, Airgas, Tampa FL). The samples were imaged using tapping mode to preserve their integrity.
Mice
The rTg4510 mice originate from the mouse model of tauopathy described elsewhere [8]. Both rTg4510 mice and the littermate control mice that do not express tau were used between 3 and 5 months of age. Animals were housed and treated according to institutional and National Institutes of Health standards.
Stereotaxic Intracranial injections and Infusion of Aβ1-42 oligomers
Acute injections of 100 μM oligomers or scramble Aβ1-42 into cortex and hippocampus of 5 month old rTg4510 and age-matched nontransgenic (ntg) littermates were performed using the convection-enhanced delivery (CED) method previously described [23]). The coordinates of injection were as follows: hippocampus, anteroposterior, − 2.7 mm; lateral −2.7 mm, vertical −3.0 mm from bregma; cortex, anteroposterior, + 1.7 mm; lateral − 2.2 mm, vertical −3.0 mm from bregma. Nontransgenic mice injected with oligomers were sacrificed after 1 hr after intracranial injections to visualize injected material, while rTg4510 injected with oligomers or scrambled Aβ1-42 survived for 3 days. The chronic infusion of human Aβ1-42 oligomers or saline into 3 month old rTg4510 and nontransgenic mice was performed as previously described [24]. Briefly, a cannula (Plastics One, Roanoke, VA) was stereotactically implanted into the right hippocampus (coordinates from bregma: −2.7 mm anteroposterior; −2.7 mm lateral; 3.0 mm vertical), and an osmotic pump (Alzet #1004, Durect Corp. Cupertino, CA) was attached and implanted subcutaneously near the scapula. Pumps contained either oligomeric Aβ1-42 (Biosource, Camarillo, CA) in vehicle (F12 medium) or saline alone. The infusion rate was 0.11 μl/h (67 ng/h Aβ1-42). Pumps remained in place for Aβ delivery for 28 days, and mice were sacrificed 1 month after the termination of the infusion (2 months after pump placement).
Immunohistochemistry/ Immunofluorescence
Immunohistochemistry was performed on free-floating sections collected from brain tissue and prepared as described in detail previously [25]. Sections were incubated with primary antibodies (Table 1) overnight at 4°C, washed and incubated with either goat anti-rabbit IgG Alexa 594, streptavidin-Alexa 488 (Invitrogen, US) for immunoflourescence or with the appropriate biotinylated secondary antibody for immunohistochemistry (VectorLabs, Burlingame, CA) for 2h at room temperature. For immunohistochemistry, sections were incubated for 1 hr in avidin-biotin complex (Vectastain Elite, Vector Laboratories, Inc., Burlingame CA) and washed. Color development was performed using 3,3′-diaminobenzidine (DAB; Sigma, St. Louis, MO) enhanced with nickelous ammonium sulfate (J. T. Baker Chemical Company, Phillipsburg, NJ). Staining was analyzed with Digital Scanning Microscope (Mirax) and quantified by Zeiss Image HIS segmentation analyzing program (Zeiss). For immunofluorescence purpose, sections were mounted on slides and imaged using a Zeiss AxioVision Imager Z1 microscope, and processed using AxioVision 4.8 image software (Gottingen, Germany).
Table 1.
Antibodies used for immunohistochemistry and immunofluorescence
| Marker | Antibody | Recognition status | Host Species | Dilution | Vendor |
|---|---|---|---|---|---|
| TAU | |||||
| S202/T205 | AT8 | PHF tau - hyperphosphorylated | mouse | 1:10 000 | ThermoScientific |
| S199/202 | Ser199/202 | PHF tau - hyperphosphorylated | rabbit | 1:100 000 | Anaspec |
| S396 | Ser396 | PHF tau - hyperphosphorylated | rabbit | 1:30 000 | Anaspec |
| S356 | Ser356 | PHF tau - hyperphosphorylated | rabbit | 1:10 000 | Anaspec |
| total tau | H-150 | rabbit | 1: 3 000 | SantaCruz Biotechnology | |
|
| |||||
| Microglia/Astrocyte | |||||
| CD45 | activated microglial | rat | 1:30 00 | Serotec | |
| CD11B | general microglia | rat | 1:1 000 | Serotec | |
| CD68 | phagocytic microglia | rabbit | 1:3 000 | Serotec | |
| GFAP | activated astrocyte | rabbit | 1:10 000 | Dako | |
|
| |||||
| Anyloid beta | |||||
| polyclonal Aβ | Aβ 40/42 | rabbit | 1:10 000 | Gift from Dr. P. Gottschall | |
|
| |||||
| Kinase | |||||
| GSK3 α/β pTyr 216+279 |
GSK3 | activated GSK α/β | rabbit | 1:3 000 | Abcam |
Abbreviations: CD, cluster of differentiation; GFAP, glial fibrillary acidic protein; GSK 3, glycogen synthase kinase; S, serine; T, threonine; Tyr, tyrosine; p, phosphorylated.
Statistics
Statistical analysis were performed using Student t-test or one-way ANOVA followed by Fisher’s LSD post hoc means comparison test using Stat View software version 5.0 (SAS Institute Inc, Cary NC). Graphs were generated using GraphPad Prism 4.0 (La Jolla, CA).
RESULTS
Aβ1-42 oligomer characterization
Based on results suggesting a link between amyloid and tau pathology [10, 26, 27], we administered oligomers of Aβ1-42 into rTg4510 mice. Oligomeric species were prepared using defined conditions previously described [21]. Western blotting and atomic force microscopy were used to determine the secondary structure of Aβ1-42 derived oligomers. Figure 1A, shows the bands identified on the SDS-PAGE and western blot. The preparation contains a mixture of low (17-14kDa, LMW) and high molecular weight oligomers (HMW, 38-180 kda) as well as the monomer. We exploited atomic force microscopy to investigate the physical features of the oligomers after 24 h incubation at 37°C. As shown in Fig 1B, most of the sample after 24h incubation contained globular structures with a mean diameter of 22 nm and height of 2.8 nm.
Figure 1. Biophysical characterization of oligomer preparations.
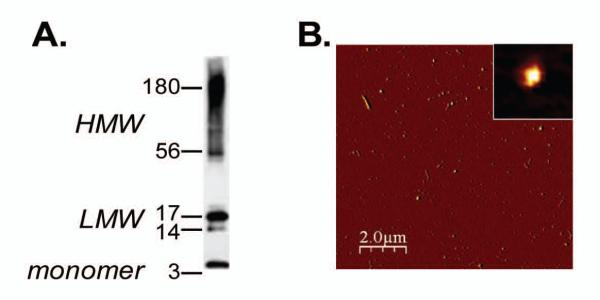
A. 100· M Aβ42-derived oligomers were prepared as described in Material and Method session and subjected to SDSPAGE and western blot using the 6E10 antibody. Monomers, band at 4.5 kDa, low molecular weight species (LMW) ranging from 14-17 kDa, and high molecular weight species (HMW) between 38-180 kDa are visible. B. Atomic force microscopy image of the 100· M stock concentration of oligomers incubated for 24 h at 37°C. Globular structure of oligomers was observed in a 10 × 10 μm x-y scan size. (The scale bar represents 2 μm; the maximum height in the z-axis is 10 nm). The insets represent higher magnification of an oligomer with a diameter of 22 nm and a height of 2.78 nm.
Aβ1-42 oligomers after injection into the mouse CNS
To monitor the distribution of Aß oligomers after injection into mouse brain, we performed acute injections into the anterior cortex and hippocampus unilaterally and monitored the dispersion of the injected material using immunohistochemistry. When tissues were collected one hour after the intra-cerebral injection, we found extensive diffusion of Aß from the sites of injection (arrowheads, Fig 2A) with some distribution into the striatum after cortical injection and to the entorhinal cortex after hippocampal injection. There was no immunostaining visible in the uninjected hemisphere (left, Fig 2B), suggesting that the injected material did not cross themidline. However, in tissue collected 3 days after the acute injection, we were unable to detect Aß oligomers in any brain region (Fig 2B). Hence we concluded that oligomeric Aβ1-42 was quickly cleared/degraded from the animal’s brain. Thus, for protracted exposure of the brain to oligomeric Aß, it would seem that a more sustained infusion would be required.
Figure 2. Detection of Aβ42 in the brain after acute intra-cerebral injection of oligomers.
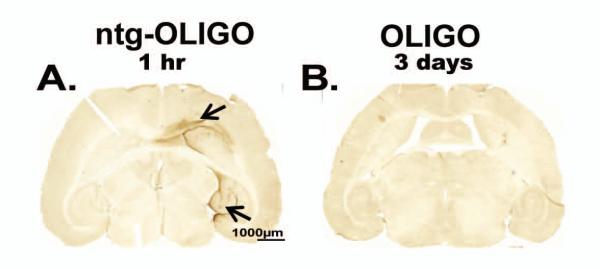
100μM Aβ42-derived oligomers (OLIGO) were injected in the cortex and hippocampus of nontransgenic (ntg) mice for 1 hr (A, arrows indicating the injection sites, n=3). 100μM OLIGO was injected in brain regions of rTg4510 mice for 3 days (B n=5). Tissue was stained for Aβ immunoreactivity. The scale bar is 1000μm for both panels.
Pathological p-tau was not altered by acute administration of Aβ1-42 oligomers
We subsequently performed immunohistochemical studies and image analyses to determine the levels of phosphorylated tau (p-tau) in rTg4510 mice injected with oligomeric Aß1-42 compared with scrambled Aβ using murine littermates (Fig. 3). Multiple phophorylated tau epitopes (S202/T205, S396, S199/S202) as well as total tau were evaluated. The total tau antibody recognizes an epitope in the N terminal of human tau independent of phosphorylation P-tau staining in the anterior cortex (3A, B, E, F, I, J), and CA1 field of hippocampus (3C, D, G, H, K, L), were readily detected by immunohistochemistry in rTg4510 mice. However, injection of oligomers did not alter the levels of tau phosphorylation in either region measured with any marker when compared to mice injected with the scrambled Aß peptide as a control for the injection procedure (Fig. 3Q-S). Similar results were observed when comparing p-tau levels in injected vs. uninjected hemispheres (data not shown). Acute injections of Aβ1-42 oligomers did not produce striking changes in total tau staining (Fig. 3M-P), and this was reflected in image analysis measurements of the immunostaining (Fig. 3T).
Figure 3. Acute injection of oligomers did not affect p-tau levels.

P-S202/T205, S396, S199/S202 tau and total tau immunoreactivity in the ipsilateral frontal cortex (CX) and CA1 field of hippocampus (Fig. 3A-P) of rTg4510 mice injected with Scrambled Aβ (n=5) or OLIGO (n=5) are shown. The levels of total tau or p-tau were undetectable in the ntg mice injected with OLIGO preparations and hence were not shown. Staining density was analysed as percent of stained area for each region and represented as ratio of ipsilateral to contralateral side (Fig. 3 Q-T). Data from frontal cortex are presented in the left side of each panel Q-T, while data from hippocampus are presented on the right side. There were no statistically significant changes in staining for any tau marker. Statistical comparisons by Student t-test were performed using StatView software. The scale bar represents 200μm for all panels.
Acute Aβ1-42 oligomers injection was associated with alternations in GSK3 activity
Given the direct effect of GSK3 on tau pathology [28] we evaluated whether acute oligomer injections affect GSK3 activity in this model. Staining for phosphorylated GSK-3 α/β kinase at pY279/ pY216 increased after acute injections of oligomers in the brain of rTg4510 mice compared with mice injected with scrambled Aß in anterior cerebral cortex (Fig. 4A, C) and the CA1 area of the hippocampus (Fig. 4B, D). Quantification revealed that oligomeric Aß1-42 injection significantly induced phosphorylated GSK-3 α/β levels compare to scrambled Aβ1-42group in both frontal cortex (Fig. 4E, * p < 0.05) and hippocampus (Fig. 4F, ** p < 0.01) of rTg4510 mice.
Figure 4. GSK3 activation occurred following acute injection of oligomers but not scramble Aβ.

Phosphorylated GSK3 α/β immunoreactivity in the frontal cortex (CX) and CA1 (HPC) of rTg4510 scramble Aβ (Sc Aβ, A, B) or OLIGO (C, D) is presented. Staining density was analysed as percent of stained area is represented as ratio of the ipsilateral to the contralateral side for each region after treatment (E, F). Scale bar is 200μm. * p< 0.05; ** p< 0.01, n=5.
Acute injection of Aβ1-42 oligomers did not exacerbate the microgliosis present in rTg4510 mice
Acute injections of oligomeric Aß did not modify the activation state of microglia, assessed by either CD45 (Fig. 5) or CD68 immunostaining (Fig. 6). Although the data presented here compare the hemispheres injected with oligomeric Aβ and scrambled Aß peptide, similar findings were true in comparisons between the oligomeric Aβ-injected hemisphere and the contralateral hemisphere (not shown).
Figure 5. CD45 immunoreactivity was prominent in rTg4510 mouse brain and it did not alter following acute injections of oligomers.
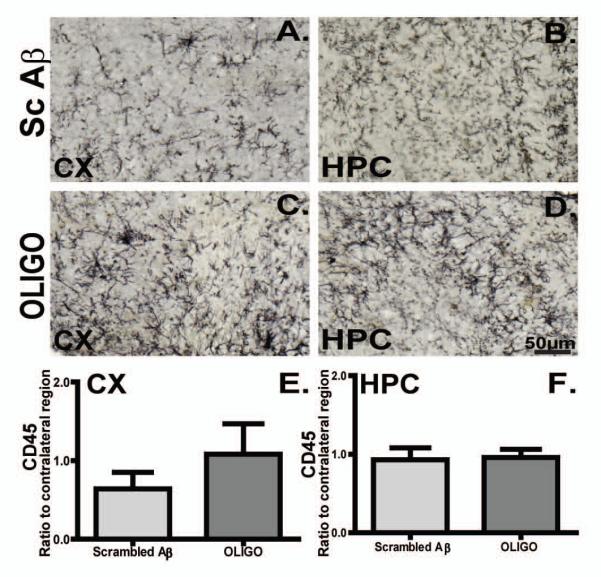
Micrographs represent CD45 immunoreactivity in the frontal cortex (CX) and hippocampus (HPC) of rTg4510 mice following Sc Aβ (A, B, n=5) or OLIGO injections (C, D, n=5). Staining density was analysed as percent of stained area for each region and graphed as ratio of ipsilateral to the contralateral side (E, F). The scale bar represents 50μm.
Figure 6. CD68 immunoreactivity present in the transgenic mice did not alter following acute oligomer injections.

Micrographs representing CD68 immunoreactivity in rTg4510 mice regions injected with Sc Aβ (A, B, n=5), or OLIGO (C, D, n=5). Staining density was analysed as percent of stained area for each region and expressed as ratio to the contralateral side (E, F). The scale bar represents 50μm.
Chronic infusion of Aβ1-42 oligomers in rTg4510 mice enhances phosphorylation of tau
Although acute administration of Aß oligomers had no impact on tau phosphorylation, it is less clear what the impact of longer exposures would be. We infused a solution containing 100 μM Aβ1-42 oligomers or saline (as a surgical control) into the right hippocampus of 3-month-old rTg4510 mice using 28 day osmotic minipumps and indwelling intracranial cannulae. We also infused oligomeric Aß into nontransgenic animals, lacking human tau. We then collected tissue 60 days after pump implantation, to ascertain if there were any enduring effects of the Aß oligomers on histopathology in the rTg4510 mice.
We evaluated several phosphorylated tau epitopes (Table 1) in the CA1 field of hippocampus and entorhinal cortex. P-tau S199/202 was localized to neuronal cell bodies of the pyramidal cells of CA1 (Fig. 7C, E) as well as to neurons in entorhinal cortex (Fig. 7D, F) in Tg4510 mice. Infusion of oligomers into hippocampus increased p-tau S199/202 in this region when compared to saline-treated rTg4510 animals (Fig 7C, E, G; p< 0.01). A trend toward increased p-tau S199/202 level was observed in entorhinal cortex, but did not reach significance (Fig. 7D, F, H). Injection of nontransgenic mice with oligomers did not result in phosphorylation of S199/202 epitopes of endogenous mouse tau (Fig. 7A, B; not quantified due to absence of signal).
Figure 7. Chronic infusion of oligomers increased immunostaining for pS199/202 tau.

P-S199/202 immunoreactivity in the CA1 region of hippocampus (CA1, depicted images are taken 700 μm from the center of cannula; C, E) or entorhinal cortex (EC, 1600 μm from cannula’s center; D, F) of rTg4510 mice infused with saline (C-D) or OLIGO (E-F) into right hippocampus are shown in all figures from this point unless otherwise stated. Ntg mice infused with OLIGO preparations are used as controls (A-B). Staining density was analysed as percent of stained area for each region (G-H). The scale bar represents 200μm for all panels. Student t-test; ** p< 0.01, n=6.
Similar results were observed for p-tau S202/T205 staining. In saline-treated rTg4510 mice, p-tau S202/T205 was localized to neuronal soma of pyramidal and cortical neurons (Fig. 8A and inset, B). Interestingly, p-tau at the S202/T205 epitope in oligomer-treated mice displayed somatodendritic localization in pyramidal CA1 neurons (Fig. 8C and insets). Oligomers dramatically increased the levels of p-tau S202/T205 in the right hippocampus of rTg4510 mice compared to the saline-treated group (Fig. 8E, p< 0.01). In contrast, infusion of oligomeric Aβ did not significantly induce p-tau S202/T205 in the entorhinal cortex areas compare to saline infused animals (Fig. 8F).
Figure 8. Infusion of oligomers increased immunostaining for pS202/T205 tau.

Micrographs of pS202/T205 immunoreactivity in the CA1 and EC of rTg4510 mice infused with saline (A, B) or OLIGO (C, D) are presented. Insets in panels A and B represent higher magnification images of p-tau localization in the cell body and dendrites of neurons. Staining density was analysed as percent of stained area for each region (E, F). Scale bars represent 200 μm in each panel, and 50 μm in the insets. Student t-test; ** p< 0.01, n=6.
Based on the evidence of the involvement of tau phosphorylated at S396 in modifying microtubule binding and promoting neuronal degeneration [29], we evaluated the pS396 levels as an indicator of tau pathology. Significantly increased levels of pS396 tau were observed in the right hippocampus of oligomer vs. saline treated rTg4510 groups (Fig. 9C, E, G; p< 0.05). Furthermore, Aß oligomer infusion did not induce significant changes in p-tau levels in entorhinal cortex (Fig. 9D, F, H).
Figure 9. Infusion of oligomers increased pS396 tau levels.

P-S396 immunoreactivity in the CA1 (HPC; A, C, E) and entorhinal cortex (EC; B, D, E) of ntg mice infused with OLIGO (A, B), or rTg4510 mice infusion with saline (C, D) or OLIGO (E, F). Staining density in rTg4510 mice was analysed as percent of stained area for each region (G, H). Scale bar is 200μm. Student t-test; * p< 0.05, n=6.
We investigated total tau expression using an antibody which recognizes an N terminal domain epitope of human tau irrespective of phosphorylation status (Fig. 10). We were unable to detect tau in the nontransgenic mice with this antibody (Fig. 10 A, B). Total tau was localized in the neuron cell body and oligomer delivery did not significantly affect total tau levels in rTg4510 mice compared to the saline-treated cohort (Fig. 10C-H). Similarly, staining for another PHF- tau epitope, pS356, demonstrated p-tau somatic localization in neurons, but no significant effect of oligomers was observed between groups (data not shown).
Figure 10. Total tau levels remain unchanged following chronic infusion of oligomer.

Total tau immunoreactivity in CA1 and entorhinal cortex of rTg4510 mice infused with saline (C-D) or OLIGO (E-F) is shown. Endogenous tau was not detectable in nontransgenic (ntg) mice (A-B). Staining density was measured as percent of stained area for each region (G, H), but was unaffected by treatment. Scale bar represents 200μm, Student t-test, n=6.
GSK 3 is elevated in the rTg4510 model and in response to Aβ1-42 oligomers
In rTg4510 mice, Aß oligomer infusion augmented phosphorylated GSK3 immunoreactivity in the hippocampus compared to the saline-treated group (Fig. 11A, C, E and G; p<0.05). In spite increased levels of phosphorylated GSK-3 in rTg4510 mice compared to the nontrangenic group in entorhinal cortex (Fig. 11B, D), the difference between saline-injected and oligomer-injected rTg4510 mice did not achieve statistical significance. Notice the neuronal localization of phosphorylated/activated GSK3 in Fig 11.
Figure 11. Activation of GSK3 increased following chronic infusion.

Phosphorylated GSK3 α/β immunoreactivity in the CA1 and entorhinal cortex (EC) of rTg4510 or nontransgenic mice (A, B) following infusion of saline (C, D) or OLIGO (E, F) into right hippocampus. Staining density was analysed as percent of stained area for each region of rTg4510 (G, H). Scale bar represents 200μm. * p< 0.05; n=6.
Finally, we looked at the relationship between activated GSK3 and phosphorylated tau in rTg4510 mice. We used immunofluorescence to co-localize S202/T205 p-tau (Fig. 12A, E) and pGSK-3 (Fig 12B, E) in the tissue from oligomer-injected rTg4510 mice. Immunofluorescent staining revealed a considerable degree of co-localization of GSK3 with AT-8 p-tau in the CA1 (Fig. 12A-C), and entorhinal cortex (Fig. 12D-F). Both markers appeared to be located to the perinuclear area, as shown by the merged images with nuclear DAPI staining (Fig. 12C and F).
Figure 12. Phosphorylated tau co-localized with activated GSK3 in rTg4510 mouse brain.
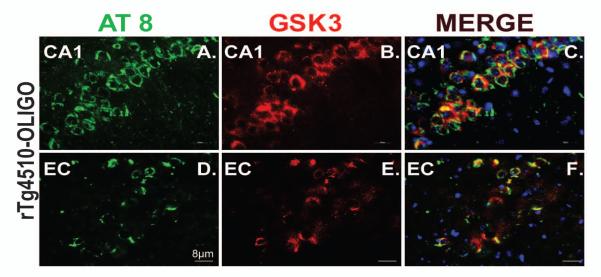
Tau at pS202/T 205 (AT8; green) is co-localized with phosphorylated GSK3 α/β (red) in the neuronal cell bodies of CA1 (A-C) and entorhinal cortex (D-F) in rTg4510 mice infused with OLIGO. DAPI was used to stain nuclei (blue). The scale bar represents 8 μm for all panels.
CD68 positive microglia but not CD45 microglia was induced following chronic Aβ1-42 oligomers infusion
Intracerebral injection of β-amyloid aggregates and derived oligomers has been linked with increased microgliosis [24]. We stained for CD45 and CD68 to monitor microglial activation. Consistent with our group’s previous observations [30], we report that at 5 months of age, the presence of tau pathology in rTg4510 mice is accompanied by increased CD45 immunoreactivity when compared with ntg mice (Fig. 13A, B). After oligomer infusion, the microglial morphology tended towards a more activated phenotype, with enlarged somata and thickened processes (Fig. 13E) compared to saline injected mice (Fig 13C). However, statistically significant differences in immunostained area were observed in hippocampus in comparison to the nontransgenic mice (Fig 13A and G). This suggests that the CD45expression of activated microglia in rTg4510 mice is tau pathology-related and independent of oligomer administration. These results were in agreement with the acute effects of oligomers on CD45 activated microglia (Fig. 5).
Figure 13. CD45 immunoreactivity is prominent in rTg4510 mouse brain and independent of oligomer infusion.

Micrographs represent CD45 immunoreactivity in the CA1 region of hippocampus (HPC) and entorhinal cortex (EC) of OLIGO-infused ntg mice (A, B), rTg4510 mice following saline (C, D) or OLIGO infusion (E, F). Staining density was analysed as percent of stained area for each region (G, H). The scale bar represents 50μm. Statistical comparisons by one-way ANOVA and Fisher’s PLSD were performed using StatView software. * p< 0.05, n=6.
We did not observe activation of CD68, phagocytic microglia in ntg-oligomer infused animals (Fig. 14A, B). Moreover, rTg4510 infused with saline displayed modest amounts of CD68 immunoreactivity (Fig. 14C, D). Chronic infusion of oligomers induced activation of CD68 phagocytic microglia (Fig. 14E, F). Image analysis showed that Aß oligomer infusion significantly increased CD68 expression in the hippocampus (Fig. 14G; p< 0.05) and entorhinal cortex (Fig. 14H; p< 0.05) compared to both ntg and rTg4510-saline infused animals. We also examined another microglial marker, CD11b and an astrocyte marker, glial fibrillary acidic protein (GFAP), by immunofluorescence. We observed an increase in CD11b+ microglia in the hippocampus of rTg45140 animals chronically infused with oligomers (red, Supplement Fig. 1C) compared to the saline controls (supplement Fig. 1A), with less effect in the entorhinal cortex (Supplement Fig. 1B vs. D). However, no changes of astrocytosis were observed as shown by GFAP immunohistochemistry analysis (green, supplement fig 1E-H). Merging of the CD11b and GFAP images indicate little overlap of these two distinct populations of cells (microglia and astrocytes).
Figure 14. CD68 immunoreactivity is increased in oligomer infused rTg4510 mouse.

Micrographs representing CD68 immunoreactivity in the CA1 region of hippocampus (HPC) and entorhinal cortex (EC) of ntg mice infused with OLIGO (A, B), and rTg4510 mice following saline (C, D) or OLIGO infusion (E, F) are shown. Staining density was analysed as percent of stained area for each region (G, H). The scale bar represents 50μm. CD68 immunostaining was elevated in OLIGO-treated rTg4510 mice compared with either nontransgenic mice treated with OLIGO (ntg-OLIGO) or rTg4510 mice treated with saline. Statistical comparisons by one-way ANOVA and Fisher’s PLSD were performed using StatView software. * p< 0.05, n=6.
Stability of Oligomeric Aß within the minipump
To monitor structural changes in the injected sample, the same oligomer preparation used for the intra-cranial injections was loaded into an osmotic pump, kept in physiological conditions (isotonic buffer at 37°C), and samples were collected after 1, 10 or 27 days of incubation. Interestingly, after one day of incubation within the pump, the monomeric species decreased precipitously and a cluster of LMW material appeared, primarily migrating at a molecular weight consistent with trimers (Supplement Fig 2). HMW oligomers (38 to 180 kDa bands) were present at all sampling time, but decreased during the 27 day incubation period. Large aggregates were also visible in the top of the gel and continued to be present up to 10 days (data not shown). Western blot data suggested that oligomers kept ex vivo but inside the osmotic pump contain a mixture of oligomer species. At day 27 the solution exiting the pump contained little Aβ protein. It is possible that this hydrophobic protein adhered to the inside of the osmotic pump or aggregated and precipitated in a manner that prevented additional release from the pump.
DISCUSSION
Several lines of evidence suggest that Aβ pathology precedes and can potentiate tau pathology. First, it is generally regarded that the initial pathology to develop in Alzheimer’s patients is Aß deposition, and this is followed with several years delay by tau pathology [31]; (but see [32] for a different perspective). Second, mutations in tau do not recapitulate the pathological or behavioral correlates of AD [33], while mutations affecting the amyloid precursor protein or its processing do reproduce most features associated with sporadic AD [34]. Moreover in AD the brain regions developing tau pathology are, at least sequentially, distinct from those brain regions affected by other taupoathies. This leads to the suggestion that one consequence of amyloid deposition in AD may be to precipitate tau pathology.
Similar evidence is accumulating in animal models that Aß can exacerbate tau pathology. Geula C et al [35] observed that intracranial injections of fibrillar, but not soluble Aß could initiate tau pathology in aged marmosets (this was prior to attempts to isolate oligomeric forms of Aß). Forebrain tau pathology has been shown to be enhanced by Aβ administration in tau transgenic mouse models [10, 27]. More recently, injection of aged APP brain extract into the brain of young B6/P301L mice was also shown to increase tau pathology [9]. Immunotherapeutic approaches or genetic modifications which reduce β-amyloid accumulation slow the development of tau pathology in models exhibiting both pathologies [36, 37]. Furthermore, studies suggest a postsynaptic role for tau in conferring β-amyloid effects on cognitive function [18] and dendritic toxicity [20, 38].
In this experiment, we examined the impact of the acute vs. chronic administration of Aβ1-42 oligomers in tau phosphorylation. First, we investigated the composition of the Aβ1-42 oligomers that were used for in vivo infusion. The Aß oligomer sample initially contained a mixture of low molecular weight forms, stable, globular structures, and Aß oligomers are cleared and/or degraded from brain parenchyma within days (Fig 2), consistent with earlier work using nonfibrillar forms of Aß [39, 40]. Study of the material exiting from the pumps found that larger aggregates predominated as the material incubated at 37° in physiological buffer inside the osmotic minipump. This observation is in agreement with data published previously showing metastable structures of Aβ with increased temperature [21]. This led to either precipitation of the aggregates or adsorption of the material onto the lining of the pump chamber or polyethylene tubing. In either case, these results indicate that the infusion of Aß oligomers was largely complete by 10 days after pump implantation. Had we not included this in vitro control condition in the study, we would not have discovered this early termination of Aß infusion.
For these experiments, we selected the age of 5 mo in the rTG4510 model because this is a time when some tau pathology is evident, but is not maximal [8, 41]. Acute injections of Aβ oligomers did not alter the p-tau levels at any of the epitopes investigated in vivo at the 3 day post-injection interval (Fig. 3). We selected this interval as one in which we have previously observed effects of intra-cranially administered agents [42-45]. It is unclear whether the lack of effect indicates that none had yet developed, or that the clearance of the Aß led to rapid reversal of p-tau changes that might have occurred. This is in contrast to the recent evidence on an acute effect of Aβ oligomers on tau phosphorylation in cultured hippocampal neurons [46]. Whether this represent true differences between in vitro and in vivo models, or is a product of reduced magnitude of effect in vivo is not clear at this stage. However, a key observation in the mice injected acutely with Aß oligomers was a significant increase in the active form of GSK3, a kinase capable of phosphorylating multiple sites on tau [47, 48].
Unlike the acute treatment, the chronic infusion of Aβ oligomers increased phosphorylation of tau in vivo. Staining for p-tau using several antibodies (pS199/S202, pS202/Th205and pS396) were elevated 3-4 fold in the injected hippocampus (Fig. 7, 9) compared to the saline-treated rTg4510 group. Although the adjacent entorhinal cortex indicated a trend towards an increase, this was not statistically significant. No differences were observed contralateral to the side of injection. The pS396 epitope is suggested to be involved in microtubule destabilization and PHF formation in AD brain [29]. However, not all forms of tau were elevated. Total tau and the pS356 specific antibody were unaffected by the Aß oligomer treatment. It is important to note that these effects are enduring, as the infusion of Aβ oligomers probably ceased 6 weeks prior to tissue collection. This observation implies that even transient stimulation of tau pathology can have long-lasting impact on the rate of tauopathy development, and at some stage, these changes are irreversible (consistent with suppression of the transgene with doxycycline [8]).
Perhaps most intriguing was the persistence of the GSK3 activation induced by the injection of Aß oligomers into the hippocampus. As in the acute study, the phosphorylation and activation of this enzyme persists after the injected Aß has been cleared from the brain (Fig 11). Moreover, there is a large degree of overlap between cells expressing elevated pS202/T205 and GSK3, implying that this elevated kinase expression may be responsible for the increased phosphorylation of tau. During both brain development and in neurodegenerative diseases, activated GSK-3 is associated with hyperphosphorylated tau and the two markers are co-localized within neurons [49]. Consistent with our findings, in vitro [50] and in vivo studies [51], report β-amyloid oligomer-induced tau pathology through active GSK3 α/β. To our knowledge, this is the first report that the activation of GSK3α/β kinase by Aβ oligomers persists beyond the period of direct exposure to the Aß. This maybe one mechanism by which short term elevations of Aß, such as following head injury [52], can increase risk for subsequent tauopathy.
We have previously demonstrated the role of potent inflammatory stimulus such as LPS, on exacerbating tau pathology [30]. Furthermore, we reported that increases in several markers of microglial activation were age and pathological tau accumulation dependent in rTg4510 mice [30]. Our data show two important findings 1) rTg4510 mice exhibit microglial activation (measured with CD68) relative to nontransgenic animals independent of acute exposure to the oligomers and 2) chronic infusion of Aβ 1-42 oligomer exacerbates microglia activation. A straightforward interpretation of these data is that the p-tau pathology resulted in microglial over expression of CD68. Interestingly, our data demonstrate that CD45-immunoreactive, microglia were not affected by either acute or chronic administration of oligomers in the brain. This is quite distinct from observations in mice with amyloid deposits [25]. Given the suggestion that CD45-high immune-positive cells are derived from infiltrating monocytes [53], this would suggest that tau pathology does not lead to significant recruitment of blood derived cells.
In conclusion, we demonstrate evidence of the involvement of Aβ -derived oligomers in modifying the level of hyperphosphorylated tau. The neuropathological symptoms of AD are multifactorial and many kinases and molecular mechanism contribute to the disease, however we demonstrate here persistently increased phosphorylation/activation of GSK-3 α/β kinase in both acutely and chronically Aβ oligomers -treated rTg4510 mice. These data would suggest that GSK3 inhibitors may be effective in diminishing some of the effects of Aß oligomers on exacerbation of tau pathology.
Supplementary Material
Supplement Figure 1. Microglial but not astrocytic activation was increased by chronic oligomer infusion in rTg4510 mouse brain. Activated microglia are visualized by CD11b immunofluorescence stain of tissue from saline and OLIGO infused rTg451 mice (A, C), Higher magnification of CD11b positive microglia is shown in panels B and D. Astrocytes are visualized by immunofluorescence stain using GFAP antibody (green) in saline (E) and OLIGO (G) infused rTg4510 mice. Higher magnification of glia markers were presented in panels F and H. DAPI is used to stain nuclei (blue) and is represented by panels I-L. Panels M-P depict the merging the three fluorescent channels. The scale bar represents 10 μm in B, D, F, H, J, L, N and P and 20 μm in A, C, E, G, I, K, M and Q.
Supplement Figure 2. Stability of oligomeric Aß within the minipump decreases after 10 days. 100μM Aβ42-derived oligomers were incubated within the osmotic pump in PBS at pH 7.4 and 37°C. Aliquots were collected 1, 10 or 27 days and subjected to SDS-PAGE and western blot using the 6E10 antibody. Densitometry analysis of the monomer band at 4.5 kDa, low molecular weight species (LMW) ranging from 6-17 kDa, and high molecular weight species (HMW) between 38-180 kDa, were plotted.
Acknowledgment
This work was supported by the following NIH grants: AG15490 and NS 76308.
Footnotes
Authors have no actual or potential conflicts of interest.
Disclosure Statement: Dave Morgan has had consultancies and/or contracts with the following companies; AstraZeneca, Baxter, Bristol-Myers-Squibb, Chiesi, Eisai, Elan, Ethicon, Forest, Lilly, Lundbeck, Merck, NeuroImmune, NiCox, Pfizer, Rinat, Wyeth. None of these activities pertain to the material presented within this manuscript.
REFERENCES
- 1.Trojanowski JQ, Schmidt ML, Shin RW, Bramblett GT, Rao D, Lee VM. Altered tau and neurofilament proteins in neuro-degenerative diseases: diagnostic implications for Alzheimer’s disease and Lewy body dementias. Brain Pathol. 1993;3(1):45–54. doi: 10.1111/j.1750-3639.1993.tb00725.x. [DOI] [PubMed] [Google Scholar]
- 2.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113(5):501–11. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E. Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci. 2007;27(37):9916–27. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87(2):440–51. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 6.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 7.Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25(46):10637–47. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, et al. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007;171(6):2012–20. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 11.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281(3):1599–604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 12.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 13.O’Nuallain B, Klyubin I, Mc Donald JM, Foster JS, Welzel A, Barry A, Dykoski RK, Cleary JP, et al. A monoclonal antibody against synthetic Abeta dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J Neurochem. 119(1):189–201. doi: 10.1111/j.1471-4159.2011.07389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 31(20):7259–63. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100(18):10417–22. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta - amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99(9):6364–9. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 31(2):700–11. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 20.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 142(3):387–97. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 21.Stine WB, Jr., Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278(13):11612–22. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 22.Selenica ML, Wang X, Ostergaard-Pedersen L, Westlind-Danielsson A, Grubb A. Cystatin C reduces the in vitro formation of soluble Abeta1-42 oligomers and protofibrils. Scand J Clin Lab Invest. 2007;67(2):179–90. doi: 10.1080/00365510601009738. [DOI] [PubMed] [Google Scholar]
- 23.Carty N, Lee D, Dickey C, Ceballos-Diaz C, Jansen-West K, Golde TE, Gordon MN, Morgan D, et al. Convection-enhanced delivery and systemic mannitol increase gene product distribution of AAV vectors 5, 8, and 9 and increase gene product in the adult mouse brain. J Neurosci Methods. 194(1):144–53. doi: 10.1016/j.jneumeth.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craft JM, Watterson DM, Van Eldik LJ. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia. 2006;53(5):484–90. doi: 10.1002/glia.20306. [DOI] [PubMed] [Google Scholar]
- 25.Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, et al. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173(2):183–95. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- 26.LaFerla FM, Oddo S. Alzheimer’s disease: Abeta, tau and synaptic dysfunction. Trends Mol Med. 2005;11(4):170–6. doi: 10.1016/j.molmed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 28.Selenica ML, Jensen HS, Larsen AK, Pedersen ML, Helboe L, Leist M, Lotharius J. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br J Pharmacol. 2007;152(6):959–79. doi: 10.1038/sj.bjp.0707471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10(6):1089–99. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 30.Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, et al. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kantarci K, Boeve BF, Wszolek ZK, Rademakers R, Whitwell JL, Baker MC, Senjem ML, Samikoglu AR, et al. MRS in presymptomatic MAPT mutation carriers: a potential biomarker for tau-mediated pathology. Neurology. 75(9):771–8. doi: 10.1212/WNL.0b013e3181f073c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 70(11):960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 33.Hutton M. Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology. 2001;56(11 Suppl 4):S21–5. doi: 10.1212/wnl.56.suppl_4.s21. [DOI] [PubMed] [Google Scholar]
- 34.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 35.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4(7):827–31. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 36.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-beta vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer’s disease. J Neurosci. 2009;29(25):7957–65. doi: 10.1523/JNEUROSCI.1339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 68(6):1067–81. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, et al. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18(6):2161–73. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holcomb LA, Gordon MN, Benkovic SA, Morgan DG. A beta and perlecan in rat brain: glial activation, gradual clearance and limited neurotoxicity. Mech Ageing Dev. 2000;112(2):135–52. doi: 10.1016/s0047-6374(99)00086-x. [DOI] [PubMed] [Google Scholar]
- 41.Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, Lebson L, Lee D, et al. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol. 2009;174(1):228–38. doi: 10.2353/ajpath.2009.080764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carty NC, Wilcock DM, Rosenthal A, Grimm J, Pons J, Ronan V, Gottschall PE, Gordon MN, et al. Intracranial administration of deglycosylated C-terminal-specific anti-Abeta antibody efficiently clears amyloid plaques without activating microglia in amyloid-depositing transgenic mice. J Neuroinflammation. 2006;3:11. doi: 10.1186/1742-2094-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22(6):1007–12. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 44.Herber DL, Mercer M, Roth LM, Symmonds K, Maloney J, Wilson N, Freeman MJ, Morgan D, et al. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2(2):222–31. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- 45.Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23(9):3745–51. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 30(36):11938–50. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3alpha exhibits beta-catenin and tau directed kinase activities that are modulated by Wnt. Eur J Neurosci. 2006;24(12):3387–92. doi: 10.1111/j.1460-9568.2006.05243.x. [DOI] [PubMed] [Google Scholar]
- 48.Engel T, Lucas JJ, Gomez-Ramos P, Moran MA, Avila J, Hernandez F. Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging. 2006;27(9):1258–68. doi: 10.1016/j.neurobiolaging.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 49.Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33(1):43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- 50.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29(9):1334–47. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, et al. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res. 2006;83(3):374–84. doi: 10.1002/jnr.20734. [DOI] [PubMed] [Google Scholar]
- 52.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57(4):419–25. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1. Microglial but not astrocytic activation was increased by chronic oligomer infusion in rTg4510 mouse brain. Activated microglia are visualized by CD11b immunofluorescence stain of tissue from saline and OLIGO infused rTg451 mice (A, C), Higher magnification of CD11b positive microglia is shown in panels B and D. Astrocytes are visualized by immunofluorescence stain using GFAP antibody (green) in saline (E) and OLIGO (G) infused rTg4510 mice. Higher magnification of glia markers were presented in panels F and H. DAPI is used to stain nuclei (blue) and is represented by panels I-L. Panels M-P depict the merging the three fluorescent channels. The scale bar represents 10 μm in B, D, F, H, J, L, N and P and 20 μm in A, C, E, G, I, K, M and Q.
Supplement Figure 2. Stability of oligomeric Aß within the minipump decreases after 10 days. 100μM Aβ42-derived oligomers were incubated within the osmotic pump in PBS at pH 7.4 and 37°C. Aliquots were collected 1, 10 or 27 days and subjected to SDS-PAGE and western blot using the 6E10 antibody. Densitometry analysis of the monomer band at 4.5 kDa, low molecular weight species (LMW) ranging from 6-17 kDa, and high molecular weight species (HMW) between 38-180 kDa, were plotted.