

Abstract
The androgen receptor (AR) and its coregulators have important roles in the carcinogenesis of prostate cancer. p53 is an important tumour suppressor gene, and the absence of a fundamental p53 response may predispose to cancer. Transgelin, known as an ARA54-associated AR inhibitor, can suppress AR function in LNCaP cells. In addition to these effects, we aimed to elucidate the proapoptotic effects of the protein on LNCaP and its underlying mechanisms, especially the interaction between transgelin and p53. Cell counting, flow cytometric analysis and terminal deoxynucleotidyl transferase-dUTP nick-end labelling assays were applied to measure the proapoptotic effect of transgelin. Using western blotting of p53 and double immunofluorescence staining of p53 with transgelin, we show that transfection of transgelin results in increasing cytoplasmic translocation of p53 and upregulation of p53 expression. We also found an interaction between transgelin and p53 in vivo by mammalian two-hybrid and coimmunoprecipitation assays. The activation of the mitochondria-associated apoptosis pathway was observed in LNCaP cells after transfection with transgelin. These results are indicative of p53-mediated mitochondria-associated apoptotic effects of transgelin on LNCaP cells in addition to its known suppressive effects on the AR pathway.
Keywords: androgen receptor, apoptosis, mitochondria, p53, prostate cancer, transgelin
Introduction
Prostate cancer is the most common malignant disease in aging males, ranks top one of the cancer incidence and is the second leading cause of cancer-related death in USA 1. Despite the advances in early detection and diagnosis, prostate cancer morbidity remains high. Prostate cancer is highly resistant to cytotoxic therapies. The only curative approaches are radical prostatectomy or radiation therapy in cases in which the disease is organ confined. Surgical or chemical castration in combination with antiandrogens, known as maximal androgen ablation, has been widely used in the treatment of this disease 2. This treatment is initially effective, but with the progression to a hormone-refractory disease, it inevitably fails, in spite of consistent androgen receptor (AR) expression in the majority of patients in this androgen-independent stage 3, 4, 5, 6. The causes for the resistance of prostate cancer to most current treatment modalities are largely unknown. AR signalling and the mutual effect of AR and AR coregulators are under investigation, but how they contribute to the progression of prostate cancer is not fully understood.
AR belongs to the superfamily of nuclear receptors; in the presence of its ligands, it acts as a transcription factor 7, mediating signals that drive the development and differentiation of normal prostate epithelium 8. In tumours, several mechanisms have been involved in the unavoidable progression to androgen independency, by which tumour cells evolve to bypass the ligand-dependent regulation of AR. First, androgen ablation results in changes in ligand specificity and/or sensitivity followed by the activation of AR under low levels of androgens or antiandrogens 9, 10. In addition, ∼30% of androgen-independent tumours have AR gene amplification that leads to androgen hypersensitivity 11. Altered expression of AR coregulators may facilitate AR transactivation even at low levels of androgens 12. The proper function of AR requires coregulators for its optimal signalling. Several AR coregulators, including the cAMP response element-binding protein-binding protein, steroid receptor coactivator 1, ARA54, ARA67/PAT1, ARA70, hRad9 and phosphatase and tensin homologue deleted on chromosome 10 (PTEN) have been identified 3, 13, 14, 15, 16, 17, 18. ARA54 enhances AR transactivation in a ligand-dependent manner 17, 19, 20. It is also known that transgelin can inhibit ARA54-enhanced AR transactivation and prostate cancer growth 20.
In addition to the AR pathway facilitating the progression of the disease and the resistance to treatment, p53 is a regulator of genotoxic stress that has a variety of functions including cell cycle arrest and apoptosis, and it is involved in causing changes in metabolism and in triggering apoptosis after cell injury 21, 22. There is substantial evidence that TP53 mutations are associated with increased radioresistance and chemoresistance 23, 24. Interestingly, it is clear that TP53 mutations are less common in localized and organ-confined prostate tumours 25, but are significantly more frequent in metastatic and hormone-refractory tumours, reaching a mutation rate of ∼20%–25% 26. This indicates that the p53 function may already be attenuated or non-essential in local disease. Moreover, primary culture of human prostatic epithelial cells has shown a lack of p53 response to genotoxic stress 27. It has also recently been revealed that DNA damage agents cannot fully activate p53 after inhibition of AR or depletion of androgen, and that p53 downstream targets cannot be activated to induce apoptosis as a result 28. This evidence suggests a potential therapeutic approach to intervene in prostate cancer progression by activation of the p53 pathway.
As mentioned, transgelin functions as a suppressor by the inhibition of ARA54-enhanced AR transactivation. Transgelin, also known as SM22α, was isolated as a transformation-sensitive and shape change-sensitive actin-binding protein 29, which was lost in virally transformed cells 30, 31. Recently, Shields et al.32, 33 found that the Ras gene inactivated transgelin, which might represent an early event for tumour progression in breast and colon cancers. Here, we report that the suppressive effect of transgelin on the LNCaP cell line is not only caused by abolishing ARA54-enhanced AR function but also by inducing apoptosis through the p53 pathway.
Materials and methods
Cell culture, plasmid construction and transient transfection
The human prostate cell lines LNCaP, PC-3 cells and BJ cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The LNCaP cell line has a mutant but functional AR and wild-type p53, and it was established from a lymph node metastasis of a prostate cancer patient, whereas the PC-3 cell line is AR negative and p53-null. LNCaP and PC-3 cells were cultured in 35-mm culture dishes in RPMI 1640 medium and Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA), respectively, supplemented with 10% non-heat inactivated fetal bovine serum (FBS) (Invitrogen) in an incubator at 37°C with a humidified atmosphere containing 5% CO2. The BJ cell line is derived from human fibroblasts, and it is cultured in minimum essential medium, containing 2 mmol L−1 L-glutamine, 1 mmol L−1 sodium pyruvate, 0.1 mmol L−1 non-essential amino acids (Invitrogen) and 10% FBS. HCT116, harbouring wild-type p53, and HCT116-p53−/− were kindly provided by Dr Zhi-Ming Yang (Johns Hopkins University, Baltimore, MD, USA), grown in McCoy's 5A culture medium (Invitrogen) supplemented with 10% FBS. pVP16-transgelin, pG5-LUC and pRL-TK have been described previously 17, 20. After digestion of pVP16-transgelin using BamH I and Hind III (Fermentas, Glen Burnie, MD, USA), the product was subcloned into pcDNA 3.1 vector through the Bam I and Hind III sites. pM-53 and pVP16-T were purchased from CLONTECH (Clontech Laboratories, Madison, WI, USA). Transient transfections were performed using Superfect (QIAGEN, Germantown, MD, USA) according to the user manual. Briefly, before transfection, cells were seeded to 80% confluence on the day of transfection followed by introduction of plasmid and superfect reagent, and the total amount of transfected DNA per well was maintained as a constant by the addition of empty plasmid vectors. After incubation for 2–3 h, the medium was changed and cells were treated with R1881 (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) and incubated for the designated time points.
Cell viability assay
After transfection with pcDNA3.1-transgelin or vector for 16, 24, 48 or 72 h, cells were washed with phosphate-buffered saline (PBS) and trypsinized with 0.25% trypsin (Invitrogen). After mixing equal volumes of suspended cell solution and 0.2% trypan blue, cell viability was measured by auto T4 cell counter (Nexcelom Bioscience, Lawrence, MA, USA).
Terminal deoxynucleotidyl transferase-dUTP nick-end labelling (TUNEL) assay
TUNEL assay was performed according to the manufacturer's instructions using the TUNEL Alexa Fluor Imaging Assay (Invitrogen). Briefly, LNCaP cells were seeded on a 6-cm dish containing a coverslip. After treatment as indicated, cells were fixed with 4% paraformaldehyde in PBS followed by a permeabilization step with 0.25% Triton X-100. This was followed by the application of 100 μL terminal deoxynucleotidyl transferase reaction cocktail per coverslip at 37ºC for 1 h. After being washed with PBS twice, coverslips were incubated with 100 μL of the Click-iT reaction cocktail for 30 min at room temperature (RT). DNA was counterstained with Hoechst 33342 dye followed by mounting of coverslips and visualization with a fluorescence microscope.
Flow cytometric analysis
After treatment, LNCaP cells, both adherent and floating, were trypsinized, pelleted and washed with PBS. The cells were fixed with 75% ethanol at 4°C overnight. Samples were analyzed using a flow cytometer (Becton Dickinson FACScan, San Jose, CA, USA). Propidium iodide staining was applied to measure the percentage of cells in different phases of the cell cycle, and sub-G1 peaks were interpreted as representing the apoptotic cell population.
Cell fraction preparation
Nuclear and cytoplasmic lysates of LNCaP cells were separated according to the manufacturer's instructions, using the PARIS procedure (Applied Biosystems, Foster City, CA, USA). Briefly, cells were trypsinized, centrifuged and pelleted in PBS, followed by treatment with fractionation buffer. Finally, the samples were centrifuged for 5 min, and then the cytoplasmic and nuclear fractions were prepared.
Immunofluorescence staining
LNCaP cells were seeded on 8-mm round coverslips in RPMI 1640 medium containing 10% charcoal-stripped FBS (Sigma, St. Louis, MO, USA) 24 h before transfection. DNA was transfected with SuperFect. After 3 h of transfection, cells were treated with 10 nmol L−1 R1881 for 12, 24, 48 or 72 h. The cells were then fixed with 3.5% paraformaldehyde for 20 min on ice, followed by permeabilization with 0.5% NP-40/PBS for 5 min at room temperature (RT). Slides were washed and blocked with 2% bovine serum albumin in PBS for 15 min at RT. Then the cells were stained with 1:100 of mouse monoclonal anti-p53 antibody (Santa Cruz, San Diego, CA, USA) and with 1:50 of rabbit polyclonal antibody against transgelin (Santa Cruz) at 4°C overnight. After the primary antibody incubation, cells were washed and incubated with 1:100 Alexa-594-conjugated goat anti-rabbit IgG and Alexa-488-conjugated goat anti-mouse IgG (Invitrogen). All nuclei were stained with Hoechst 33342 or 33582 dye in 1:1 000 dilution (final concentration 2 μg mL−1 in PBS) (Invitrogen) for 1–2 min at RT. Coverslips were mounted and visualized with a fluorescence microscope (Carl Zeiss, Thornwood, NY, USA). The same methods were carried out for immunofluorescence staining of endogenous transgelin and p53 in BJ cells.
Mammalian two-hybrid assay
PC-3cells were transiently cotransfected with a GAL4-hybrid expression plasmid, a VP16-hybrid expression plasmid, the reporter plasmid pG5-LUC and the pRL-TK internal control plasmid. Cells were harvested for luciferase assay after 24 h. The dual-luciferase reporter system (Promega, Madison, WI, USA) was employed to measure luciferase activity. The activities of Renilla luciferase were used to normalize any variations in transfection efficiency.
Coimmunoprecipitation of transgelin and p53
Coimmunoprecipitation of transgelin and p53 was performed as described previously 34. Briefly, LNCaP cells cultured in RPMI-1640 supplemented with 10% non-heat inactivated FBS were lysed using buffer (1% Nonidet P-40, 10% glycerol, 135 mmol L−1 NaCl, 40 mmol L−1 Tris [pH 7.4], 1 mmol L−1 phenylmethylsulphonyl fluoride (PMSF), 1 mmol L−1 DTT (dithiothreitol) and 1 × protease inhibitor cocktail) (Sigma). After clarification and quantitation of lysate, 500 μg protein were incubated with 2 μg rabbit anti-transgelin antibody (Santa Cruz) or with normal rabbit IgG (Jackson Immunoresearch, West Grove, PA, USA) for 4 h at 4ºC with agitation, and 35 μL protein A/G plus agarose (GE Healthcare Life Sciences, Buckinghamshire, UK) was then added to each sample and incubated for 1 h. After washing thrice with radioimmunoprecipitation assay buffer, sample buffer was added to the beads, and the complex was boiled and separated on a 10% sodium dodecyl sulphate (SDS)-polyacrylamide gel. The separated proteins were transferred to a polyvinylidene difluoride membrane and then blotted with anti-transgelin and anti-p53 antibodies. The bands were visualized by ECL reagents (GE Healthcare Life Sciences). The immunoprecipitation was also carried out in the reverse order, meaning that mouse anti-p53 (Santa Cruz) or normal mouse IgG (Jackson Immunoresearch) were used to detect a p53 and transgelin complex. The same methods were applied to detect coimmunoprecipitation of endogenous transgelin with p53 in the BJ cell lysate.
SDS-polyacrylamide gel electrophoresis and Western blot analysis
LNCaP cells on 6-cm dishes were treated with 5 μg pcDNA3.1-transgelin for different time periods and the same dose of pcDNA3.1 was used as control. Cells were harvested and lysed with cell lysis buffer (1% Nonidet P-40, 10% glycerol, 135 mmol L−1 NaCl, 40 mmol L−1 Tris (pH 7.4), 1 mmol L−1 PMSF, 1 mmol L−1 DTT and 1 × protease inhibitor cocktail). Cell lysate was quantified using the bicinchoninic acid (BCA) method according to the manufacturer's instructions (Thermo Scientific, Barrington, IL, USA). A total of 30 μg of protein samples were separated on 10%–15% SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in Tris-buffered saline tween-20 buffer (150 mmol L−1 NaCl; 10 mmol L−1 Tris [pH 8.0] and 0.5% Tween 20) at RT for 1 h. Then, the membranes were incubated with 1:200 mouse anti-p53 (Santa Cruz), 1:200 mouse anti-transgelin (Santa Cruz), 1:200 rabbit anti-Bax (Santa Cruz), 1:200 mouse anti-Bcl-2 (Santa Cruz), 1:200 mouse anti-caspase-3 (Santa Cruz), 1:1 000 Rabbit anti-phospho-p53 Ser46 and 1:1 000 rabbit anti-Puma (Cell Signaling, Boston, MA, USA) for 2 h at RT or overnight at 4 ºC, followed by incubation with secondary antibodies for 1 h at RT. Blots were developed with ECL reagents. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) (US Biological, Swampscott, MA, USA) was used to normalize the quantity of the protein on the blot and as cytoplasmic protein marker. Lamin A (Sigma) was used as nuclear protein marker.
Statistical analysis
All the data were analyzed and expressed as the mean ± SD. Results were considered significant if P < 0.05 using the unpaired t-test.
Results
Transgelin inhibited the growth of LNCaP cells and induced apoptosis
We examined the effect of transient transfection with transgelin on LNCaP cell viability at different time points. Cell viability was inhibited by transgelin in a time-dependent pattern, and it reached a maximum at 72 h after transfection (Figure 1A). The TUNEL assay was used to detect the apoptotic cells, and the results showed that transgelin induces apoptosis of LNCaP cells (Figure 1B). In addition, to confirm the TUNEL assay results, we employed flow cytometric analysis, the results of which were consistent with those of the TUNEL assay. After transfection with transgelin, the population of apoptotic cells increased to reach a maximum at 72 h (37.79%) compared with that of vector-transfected cells (12.7%).
Figure 1.
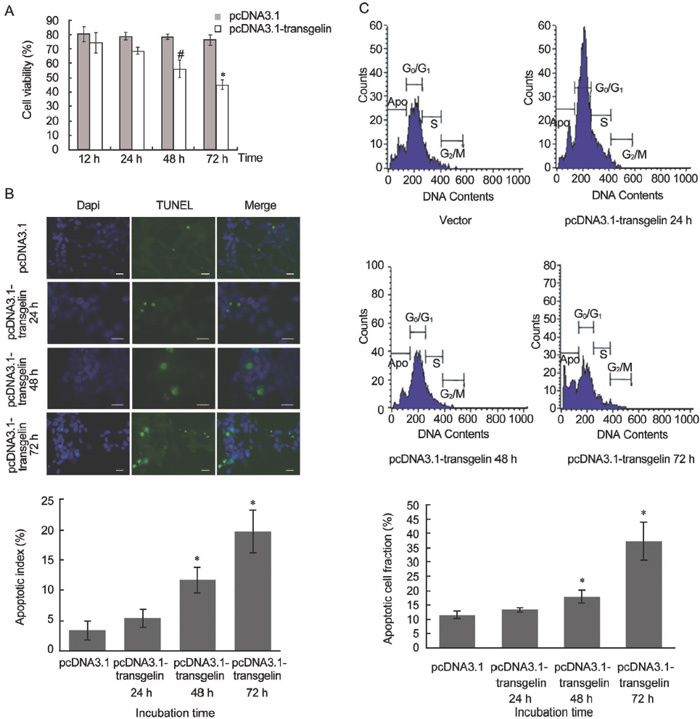
Effects of pcDNA3.1-transgelin on the LNCaP cell line. (A): Cells were transiently transfected with vector or pcDNA3.1-transgelin followed by incubation at the indicated time points. Cell viability was measured by auto cell T4 counter (#P < 0.05, *P < 0.01; compared with pcDNA3.1). (B): TUNEL assay was carried out for detecting apoptotic cells after transfection with pcDNA3.1-transgelin. Scale bars=20 μm. The graph below shows the apoptotic index, the percentage of TUNEL-positive cells out of 500 cells. All values represent the mean ± SD of three independent counting (*P < 0.01; compared with pcDNA3.1). (C): After transient transfection with vector or pcDNA3.1-transgelin, cells were harvested at the indicated time points and subjected to flow cytometric analysis. Apoptosis was measured using cell cycle analysis with propidium iodide and the percentage of hypodiploid cells (apoptotic cells) was calculated. The graph indicates the apoptotic population of cells of three independent experiments (*P < 0.01; compared with pcDNA3.1).
Transgelin upregulated the expression of p53 and resulted in an increase in cytoplasmic p53
Western blot was used to evaluate the expression of total p53 and nuclear and cytoplasmic p53. After transfection, p53 expression increased in a time-dependent manner, as did phospho-p53 Ser46 expression (Figure 2A). In addition, after transfection, accumulation and aggregation of cytoplasmic p53 was observed, and reached a maximum at 72 h (Figure 2B). However, nuclear p53 was not affected. GAPDH and Lamin A were used as cytoplasmic and nuclear protein markers, respectively (Figure 2B).
Figure 2.
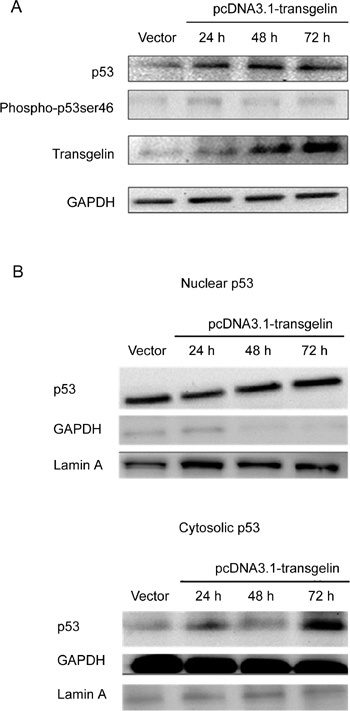
Effects of transfection with pcDNA3.1-transgelin on the expression of p53 in LNCaP cells. (A): Western blotting data reflected p53 and phospho-p53 Ser46 level in LNCaP cells after transfection with 5.0 μg vector or pcDNA3.1-transgelin. (B): Nuclear and cytosolic p53 levels were measured by western blotting using 30 μg fractions of each cell lysate.
Transgelin influenced the subcellular distribution of p53
To further confirm the western blot results showing an increasing accumulation of cytoplasmic p53, we used immunofluorescence staining methods to detect p53 distribution. p53 is an extensive tumour suppressor that mainly locates in the nucleus. Transfection with vector did not affect p53 distribution (Figure 3A). Transgelin, however, induced p53 translocation from the nucleus to the cytoplasm (Figure 3B).
Figure 3.
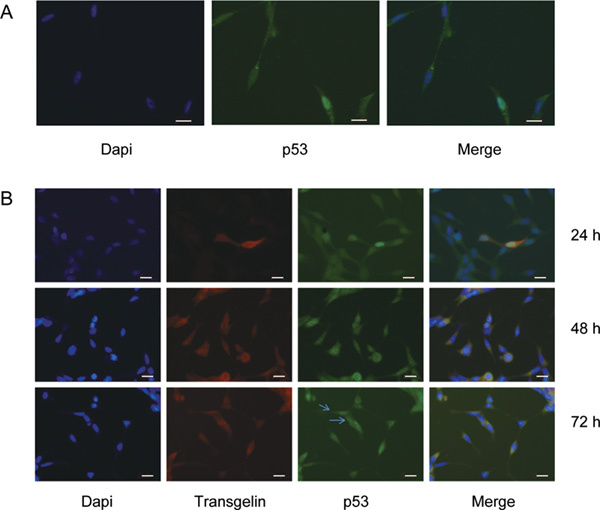
Transgelin affected the subcellular distribution of p53. (A): LNCaP cells were transfected with 5.0 μg pcDNA3.1. Immunofluorescence staining of p53 was applied to show the nuclear localization of p53. (B): Double immunofluorescence staining of transgelin and p53 was used to detect the translocation of p53 in LNCaP cells transfected with 5.0 μg pcDNA3.1-transgelin at the indicated time points. Arrowheads point out that p53 translocated to the cytoplasm. Scale bars=20 μm.
Transgelin interacted with p53 in vivo
To investigate the possible interaction of transgelin with p53, a mammalian two-hybrid assay was used. The results showed that transgelin interacted with p53 in PC-3 cells (Figure 4A). To confirm the interaction of transgelin with p53, we used the coimmunoprecipitation assay in LNCaP cells, which illustrated ectopic transgelin coimmunoprecipitated with p53 (Figure 4B and C). Transgelin is abundant in BJ cells, and immunofluorescence staining of BJ cells (passage 32) showed the location of endogenous transgelin. To further confirm the interaction of endogenous transgelin with p53, BJ cell lysates were used for immunoprecipitation assays, and the results showed that endogenous transgelin coimmunoprecipitated with p53 (Figure 4D).
Figure 4.
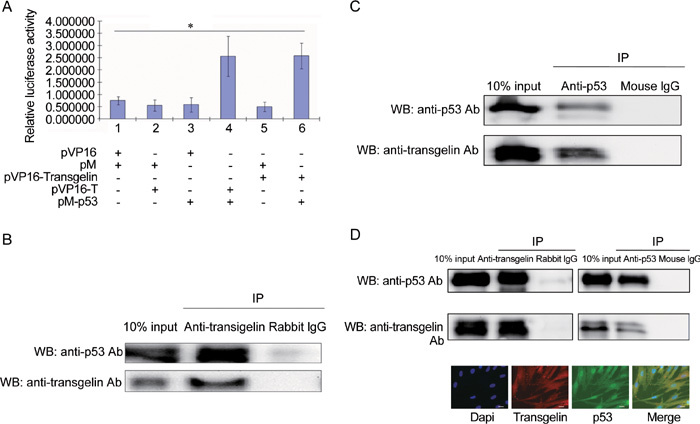
Interaction of transgelin with p53. (A): Mammalian two-hybrid assay. PC-3 cells cultured in 24-well plates were cotransfected with 0.5 μg pVP16, pM, pVP16-transgelin, pVP16-T and pM-p53 as indicated, together with 0.4 μg pG5-LUC reporter plasmid and 0.5 ng pRL-TK internal control plasmid. All values represent the mean ± SD of three independent experiments. The luciferase activity of the interaction between pM-p53 and pVP16-T was set as positive control (lane 4) and that of the interaction between pM and pVP16 was set as negative control (lane 1). Interaction between pVP16-transgelin and pM-p53 was evaluated by measuring relative luciferase activity (*P < 0.01). (B): Ectopic transgelin coimmunoprecipitated with p53. LNCaP cells were transfected with 4.0 μg pCDNA-transgelin, and harvested 24 h after transfection. A total of 2 μg anti-transgelin antibody or rabbit IgG was added to 500 μg of LNCaP cell lysate for 4 h at 4ºC to immunoprecipitate the transgelin and p53 complex. (C): p53 coimmunoprecipitated with ectopic transgelin. A total of 2 μg anti-p53 antibody or mouse IgG was added to 500 μg of LNCaP cell lysate for 4 h at 4ºC to immunoprecipitate transgelin and p53 complex. (D): Endogenous transgelin coimmunoprecipitated with p53. BJ cell lysate was incubated with anti-transgelin antibody or anti-p53 antibody to immunoprecipitate the transgelin and p53 complex, and rabbit IgG or mouse IgG was used as control, respectively. Immunofluorescence staining illustrated the localization of transgelin and p53 in BJ cells. Scale bars=20 μm. IP, immunoprecipitation; WB, Western blot.
Transgelin upregulated Bax expression, downregulated Bcl-2 expression and enhanced caspase-3 expression
The downstream target genes can be transactivated by p53, such as p53 upregulated modulator of apoptosis (Puma), to induce apoptosis. In addition to its transactivation activity, p53 also has biological activities that are cytosolic and transcription independent. Cytoplasmic p53 has effects on mitochondria, whereas the mitochondrial membrane constitutes the battleground on which proapoptotic factors (such as Bax and Bak) and anti-apoptotic factors (such as Bcl-2 and Bcl-XL) function. To determine whether the mitochondria-associated apoptosis pathway of LNCaP cells was activated by transgelin through p53, western blot was used to evaluate the expression of Puma, Bax, Bcl-2 and caspase-3. Results indicated upregulation of Puma and Bax, downregulation of Bcl-2 and induction of caspase-3 expression (Figure 5A). To further assure that the apoptotic effect of transgelin depends on p53, a transfection experiment on HCT116 and HCT116-p53−/− was carried out to evaluate Puma expression, and results indicated that upregulation of Puma from transgelin depends on p53. In the p53-null HCT116 cell line, the introduction of transgelin does not result in the upregulation of Puma, whereas transgelin upregulates Puma through p53 in p53-positive HCT116.
Figure 5.
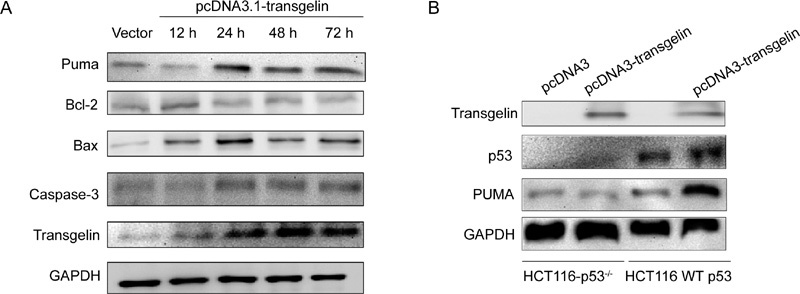
Effects of transgelin on intrinsic apoptotic pathway-related proteins. (A): Effect of transgelin on apoptosis-related genes in LNCaP cells. LNCaP cells were transfected with vector or transgelin followed by incubation for the indicated time periods, and cell lysate was analyzed for Western blotting to detect the expression of Puma, Bcl-2, Bax and caspase-3. GAPDH was used as internal control. (B): Function of transgelin depended on wild-type p53. HCT116 and HCT116-p53−/− cells were transfected with pcDNA3.1-transgelin or vector, and followed by harvest after 24 h. The cell lysate was analyzed by western blotting to evaluate p53 and Puma expression.
Discussion
Targeting apoptosis is a promising strategy for prostate cancer treatment. Bcl-2 and p53 represent two prime targets for such manipulation 35. The principal tumour suppressor protein, p53, acts as a nuclear transcription factor that transactivates genes involved in apoptosis, cell cycle regulation and numerous other processes 36. In addition, cytoplasmic p53 triggers apoptosis in a transcription-independent manner 21, 37. As we mentioned above, transgelin-induced p53 response in prostate cancer provides a potential therapeutic opportunity in addition to targeting the AR signalling pathway. Our study investigates the effect and mechanism of action of transgelin on p53 with respect to the induction of apoptosis in the human prostate cancer cell line LNCaP.
Transgelin, an actin-binding protein, has been recognized as a tumour suppressor 38 that specifically inhibits ARA54-enhanced AR transactivation with less severe side effects than those caused by systemic suppression of the AR signalling pathway, especially in many important organs. Transgelin has the advantage of specifically targeting the interaction between ARA54 and AR. In addition, because of the aberrant expression of ARA54 and the attenuated expression of transgelin in prostate cancer cells, this approach can specifically target prostate cancer cells with less effects on normal cells 20. Our study indicates that transgelin may induce LNCaP through its interaction with p53.
How prostate cancer cells survive after AR inhibition remains unknown. DNA damaging agents have been reported as unable to fully activate p53 in the absence of AR 28. Using transgelin as a possible gene therapy can avoid a paradoxical situation due to its dual effect on AR inhibition and p53 activation. Using transgelin in combination with chemotherapy or radiotherapy, the chances of prostate cancer cells developing chemoresistance or radioresistance would be reduced due to the activation of p53.
In this study, ectopic expression of transgelin results in an increasing translocation of p53 from the nucleus to the cytoplasm, leading to the accumulation of cytoplasmic p53 (as shown by immunofluorescence staining and western blot assay) and the induction of apoptosis in LNCaP cells (Figure 1B and C). In addition, transgelin interacts with p53 upregulating and stabilizing it (Figures 2A and 4). Phosphorylation of p53 at Ser46 is considered to be a primary determinant for the induction of apoptosis, by leading to selective transactivation of p53 target genes that have proapoptotic function 39. We found that the introduction of transgelin results in the upregulation of phospho-p53 Ser46, which can induce apoptosis. As a nuclear transcription factor, in addition to its transactivation-dependent effects in the nucleus, p53 also possesses biological activity that is located in the cytosol and is transcription independent, such as apoptosis induction through mitochondrial outer membrane permeabilization (MOMP) and inhibition of autophagy 21. The proapoptotic effects of cytoplasmic p53 are not dependent on transcription, in principle. However, the control of transcription by nuclear p53 decisively contributes to the function of cytoplasmic p53. Puma, one of the downstream target genes for p53, controls the sequestration of cytoplasmic p53 by anti-apoptotic proteins, releasing p53 to activate Bax. Our results indicate that transgelin results in p53 upregulation and cytoplasmic p53 accumulation, accompanied by an increased expression of Puma. The potentiation of the apoptotic response by cytoplasmic p53 may involve a direct interaction with members of Bcl-2 family of proteins 40, 41. The Bcl-2 family of proteins has an important function in regulating apoptosis. Some members of the family, such as Bcl-2 and Bcl-XL, suppress apoptosis by inhibiting MOMP, whereas others, such as Bax and Bid, somehow promote apoptosis through the conditional activation of MOMP. The balance between these two groups tunes up cell apoptosis.
In our study, transgelin induced the expression of p53 and Puma, affecting the subcellular distribution of p53. Consequently, accumulation of cytoplasmic p53 resulted in the upregulation of Bax and the downregulation of Bcl-2, which triggers caspase-3. Furthermore, the function of transgelin in the induction of apoptosis depends on p53. As indicated by the experiment on HCT116 and HCT116-p53−/− cells, transgelin cannot upregulate Puma expression in p53-null HCT116 cells compared with wild-type p53 HCT116 cells.
In conclusion, we show that the introduction of transgelin could induce apoptosis in the LNCaP cell line through p53. Transgelin induces not only p53 expression but also the translocation of p53 from the nucleus to the cytoplasm. In addition, transgelin interacts with p53 in vivo and triggers apoptosis by regulating the expression of Puma and controlling Bax and Bcl-2 expression in a p53-dependent pattern. These results suggest a therapeutic approach to battle prostate cancer through the activation of the p53 pathway and also provide insights into the function of transgelin.
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–96. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent EC, Hussain MH. The patient with hormone-refractory prostate cancer: determining who, when, and how to treat. Urology. 2003;62 Suppl 1:134–40. doi: 10.1016/j.urology.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Culig Z. Role of the androgen receptor axis in prostate cancer. Urology. 2003;62:21–6. doi: 10.1016/s0090-4295(03)00698-8. [DOI] [PubMed] [Google Scholar]
- Balk SP.Androgen receptor as a target in androgen-independent prostate cancer Urology 200260132–8.discussion 138–9. [DOI] [PubMed] [Google Scholar]
- Smith MR, Nelson JB.Future therapies in hormone-refractory prostate cancer Urology 2005659–16.discussion 17. [DOI] [PubMed] [Google Scholar]
- Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240:324–6. doi: 10.1126/science.3353726. [DOI] [PubMed] [Google Scholar]
- Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle. 2007;6:647–51. doi: 10.4161/cc.6.6.4028. [DOI] [PubMed] [Google Scholar]
- Haapala K, Hyytinen ER, Roiha M, Laurila M, Rantala I, et al. Androgen receptor alterations in prostate cancer relapsed during a combined androgen blockade by orchiectomy and bicalutamide. Lab Invest. 2001;81:1647–51. doi: 10.1038/labinvest.3780378. [DOI] [PubMed] [Google Scholar]
- Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–53. [PubMed] [Google Scholar]
- Wallen MJ, Linja M, Kaartinen K, Schleutker J, Visakorpi T. Androgen receptor gene mutations in hormone-refractory prostate cancer. J Pathol. 1999;189:559–63. doi: 10.1002/(SICI)1096-9896(199912)189:4<559::AID-PATH471>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633–48. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res. 2009;7:523–35. doi: 10.1158/1541-7786.MCR-08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto H, Rahman M, Takatera H, Kang HY, Yeh S, et al. A dominant-negative mutant of androgen receptor coregulator ARA54 inhibits androgen receptor-mediated prostate cancer growth. J Biol Chem. 2002;277:4609–17. doi: 10.1074/jbc.M108312200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang Y, Yeh S, Chang C. ARA67/PAT1 functions as a repressor to suppress androgen receptor transactivation. Mol Cell Biol. 2004;24:1044–57. doi: 10.1128/MCB.24.3.1044-1057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HY, Yeh S, Fujimoto N, Chang C. Cloning and characterization of human prostate coactivator ARA54, a novel protein that associates with the androgen receptor. J Biol Chem. 1999;274:8570–6. doi: 10.1074/jbc.274.13.8570. [DOI] [PubMed] [Google Scholar]
- Yang Z, Chang YJ, Miyamoto H, Ni J, Niu Y, et al. Transgelin functions as a suppressor via inhibition of ARA54-enhanced androgen receptor transactivation and prostate cancer cell growth. Mol Endocrinol. 2007;21:343–58. doi: 10.1210/me.2006-0104. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Kastan MB. Wild-type p53: tumours can't stand it. Cell. 2007;128:837–40. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Wang W, El-Deiry WS. Restoration of p53 to limit tumour growth. Curr Opin Oncol. 2008;20:90–6. doi: 10.1097/CCO.0b013e3282f31d6f. [DOI] [PubMed] [Google Scholar]
- Isaacs W, De Marzo A, Nelson WG. Focus on prostate cancer. Cancer Cell. 2002;2:113–6. doi: 10.1016/s1535-6108(02)00103-4. [DOI] [PubMed] [Google Scholar]
- Navone NM, Labate ME, Troncoso P, Pisters LL, Conti CJ, et al. p53 mutations in prostate cancer bone metastases suggest that selected p53 mutants in the primary site define foci with metastatic potential. J Urol. 1999;161:304–8. [PubMed] [Google Scholar]
- Girinsky T, Koumenis C, Graeber TG, Peehl DM, Giaccia AJ. Attenuated response of p53 and p21 in primary cultures of human prostatic epithelial cells exposed to DNA-damaging agents. Cancer Res. 1995;55:3726–31. [PubMed] [Google Scholar]
- Cohen MB, Rokhlin OW. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J Cell Biochem. 2009;106:363–71. doi: 10.1002/jcb.22022. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Heeley DH, Smillie LB. An abundant and novel protein of 22 kDa (SM22) is widely distributed in smooth muscles. Purification from bovine aorta. Biochem J. 1987;244:705–9. doi: 10.1042/bj2440705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi R, Kubota T, Hidaka H. Purification, characterization, and partial sequence analysis of a new 25-kDa actin-binding protein from bovine aorta: a SM22 homolog. Biochem Biophys Res Commun. 1994;198:1275–80. doi: 10.1006/bbrc.1994.1180. [DOI] [PubMed] [Google Scholar]
- Shapland C, Hsuan JJ, Totty NF, Lawson D. Purification and properties of transgelin: a transformation and shape change sensitive actin-gelling protein. J Cell Biol. 1993;121:1065–73. doi: 10.1083/jcb.121.5.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields JM, Rogers-Graham K, Der CJ. Loss of transgelin in breast and colon tumours and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J Biol Chem. 2002;277:9790–9. doi: 10.1074/jbc.M110086200. [DOI] [PubMed] [Google Scholar]
- Yeo M, Kim DK, Park HJ, Oh TY, Kim JH, et al. Loss of transgelin in repeated bouts of ulcerative colitis-induced colon carcinogenesis. Proteomics. 2006;6:1158–65. doi: 10.1002/pmic.200500390. [DOI] [PubMed] [Google Scholar]
- Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med. 2007;13:348–53. doi: 10.1038/nm1547. [DOI] [PubMed] [Google Scholar]
- Tang DG, Porter AT. Target to apoptosis: a hopeful weapon for prostate cancer. Prostate. 1997;32:284–93. doi: 10.1002/(sici)1097-0045(19970901)32:4<284::aid-pros9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Kakudo Y, Shibata H, Otsuka K, Kato S, Ishioka C. Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res. 2005;65:2108–14. doi: 10.1158/0008-5472.CAN-04-2935. [DOI] [PubMed] [Google Scholar]
- Assinder SJ, Stanton JA, Prasad PD. Transgelin: an actin-binding protein and tumour suppressor. Int J Biochem Cell Biol. 2009;41:482–6. doi: 10.1016/j.biocel.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–62. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–50. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]