

Poor semen quality is an attribute of the human condition that we share with our closest primate ancestors and may be part of our evolutionary inheritance. This characteristic may also reflect the lack of selection pressure on male fertility in advanced societies that have gone through the demographic transition. In the future, such trends may be further exacerbated by the widespread use of ART, which will impair the elimination of low fecundity genotypes from the human gene pool. Environmental pollutants may also be influencing male fertility, however, neither the chemical identity of these factors nor their mechanism-of-action are fully understood.
Over 20 years ago, Carlsen et al.1 published a highly cited paper in the British Medical Journal which purported to detect a significant decline in sperm counts over a period of 60 years (1934–1996). This was followed shortly afterwards by the Skakkebaek and Sharpe hypothesis,2 published in Lancet, which argued that the increasing incidence of reproductive abnormalities in the human male may be related to increased oestrogen exposure in utero, citing maternal exposure to diethylstilbestrol (DES) during pregnancy as evidence for such a link. These classic papers have now become inextricably linked in the global consciousness and have led to a public misconception that male fertility is declining as a consequence of fetal exposure to environmental estrogens.3 In the 20 years that have elapsed since these landmark papers were published, a great deal of data has appeared that seem to support and refute their validity in equal measure, resulting in a state of confusion. The European Science Foundation4 has recently published a review of the current status of male reproductive health. Using this summary as a guide we are now in an excellent position to dissect some of the key arguments and formulate questions for future consideration.
Falling sperm counts
One of the major questions raised by the Carlsen paper1 is whether there has truly been a global decline in sperm counts in recent decades. There are obvious difficulties in establishing the truth of this situation from a global perspective because of differences between studies in terms of location, donor selection criteria, analytical methods, age distribution, ejaculation frequency, socio-economic background and racial composition, independent of any differences in environmental or lifestyle exposures that might have influenced testicular development and function. There have been a number of studies lending support to the falling sperm count hypothesis but others that have failed to observe any perceptible temporal change.5 We can only conclude with confidence that sperm counts vary significantly between locations for a variety of reasons and are particularly low in Denmark compared with countries such as Finland, although even the latter has recently seen a decline in semen quality.6 However, the data do not seem to indicate a consistent global change in sperm numbers. In the end, we now know what we have always known—that sperm numbers are extremely variable, even within a single individual, and represent an arcane and inherently inaccurate way of assessing male reproductive function.
Moreover, this entire debate has been dominated by the public perception that falling sperm counts equate with falling fertility, which is as unhelpful as it is unsubstantiated. Irrespective of whether there are secular trends, we should just acknowledge that male infertility is an extremely common condition affecting around one in 20 of the male population7 and instead of counting spermatozoa, focus on trying to determine the underlying aetiology of this condition, and specifically, whether it is likely to be environmentally or genetically induced.
Importance of the demographic transition
It is possible to mount an argument that the developed world is witnessing a rising tide of male infertility for reasons that have nothing to do with sperm counts but everything to do with affluence. As societies pass through the demographic transition they experience a fall in family size in concert with a fall in perinatal mortality. In societies experiencing a high level of poverty (Victorian London or modern-day Uganda, Niger, Afghanistan), the fertility rate (number of children born per women) is typically above five. Such large numbers of children are necessary in order to ensure that one or two progeny survive to reproductive age and pass the parents' genes onto the next generation. Under such circumstances we are constantly selecting for high fecundity genes and genetic causes of infertility will be extremely rare. However, in developed societies, particularly those where assisted conception services are readily available, there are no selection pressures on fertility any more, and as an inevitable consequence, the incidence of genetic infertility will rise. Furthermore, the rapid growth of the assisted conception industry and our unprecedented ability to solve male-factor infertility by using intracytoplasmic sperm injection has only added fuel to the fire. The increased use of such aggressive assisted conception technologies to treat male infertility will only result in more poor fecundity genes being passed to the offspring. As an inevitable consequence, the more we turn to assisted reproductive technologies to solve infertility problems in one generation, the more we are going to need it in the next. If such changes in the distribution of high-fecundity genes are in fact occurring, they will be manifest on a time scale that covers successive generations (hundreds of years) rather that the decades measured in current epidemiological studies.
If genetic causes of infertility are on the rise because of a lack of selection pressure, what genes are likely to be involved? Deletions on the Y-chromosome are clearly part of the answer and arise spontaneously in the germ line of patients, possibly as a result of aberrant intrachromosomal recombination events involving repetitive palindromic sequences on the long arm of the Y. The AZFb and AZFc deletions occur between such palindromes, whereas the AZFa deletion is the result of intrachromosomal recombination events between two repetitive specific HERV15 (Human Endogenous Retroviral #15) sequence blocks.8 These mutations are thought to arise spontaneously in the germ line of the fathers or are created by aberrant repair of DNA-damaged spermatozoa in the oocyte, for reasons that are still unresolved.9,10 In addition to Y-chromosome deletions, genetic factors are also involved in other types of male infertility including congenital hypogonadotrophic hypogonadism and congenital absence of the vas deferens. Overall, such major genetic lesions are thought to account for around 15% of male infertility.11 This may be an underestimate of the genetic contribution to male infertility, however, because the latter is unlikely to be due to a single genetic defect but rather to suboptimal patterns of expression within an intricate network of genes, the coordinated action of which is required to achieve the complex task of differentiating a fully functional spermatozoon from a spermatogonium. A minor defect anywhere in this complex system is likely to result in infertility, as demonstrated by the large number of genetically manipulated mice that ultimately exhibit a male infertility phenotype.12
Evolutionary origins
The poor semen quality characteristic of human males may also be a consequence of their evolutionary origins. The polymorphism typical of human spermatozoa is also seen in gorillas and is thought to be the result of a mating system involving precoital competition between males rather than the postcoital competition typical of promiscuous species such as the chimpanzee.13 In the case of the gorilla, precoital competition leads to large silver-backed alpha males corralling a harem of females with whom they will have exclusive mating rights. Under these circumstances there is little selection pressure on semen quality and the latter is poor as a consequence. By contrast, when female chimpanzees are in estrus, they are mated by multiple males, creating intense competition between the spermatozoa of different individuals within the female reproductive tract, to be the first to reach and fertilize the egg.14 Under these circumstances, there is intense selection pressure on the relative performance of the spermatozoa from each individual and the evolutionary result is the production of high quality ejaculates characterized by large numbers of morphologically normal, vigorously motile spermatozoa.15 Since most human societies have opted for a monogamous mating system, sperm competition will be minimal and the quality of the ejaculate will be correspondingly poor.16 Thus, a lack of evolutionary selection pressure may again be part of the complex mixture of factors generating the relatively high incidence of male infertility in our species.
Environmental factors
Even if the poor semen quality seen in our species is related to a lack of evolutionary selection pressure, such a mechanism would not be expected to account for any rapid changes in sperm numbers as suggested by Carlsen et al.1 For disruptive effects that are occurring over a matter of two or three decades, we have to look, not to our evolutionary history, but to possible environmental toxicants as the cause célèbre. The best evidence that such toxicants might be having an impact on male reproduction comes not from sperm counts but from records depicting the incidence of testicular cancer. Although this is a rare condition, testicular cancer seems to be increasing in incidence, at least in developed countries, and is now regarded as the most common cancer affecting young men. Figure 1 illustrates the changes that have occurred in testicular cancer rates for New South Wales (NSW) over the past 40 years. The linear increase in the incidence of this cancer contrasts sharply with the appearance of cancers of the female reproductive tract in the same NSW population over the same period of time. In women, the incidence of reproductive tract cancers has either remained constant or, in the case of cervical cancer, decreased due to improved surveillance and treatment. The increased incidence of testicular cancer is not an indirect reflection of any change in life expectancy because this is a disease of young men with a peak incidence at around 30 years of age. Furthermore, the upward trend is not reflected by any reported changes in the efficiency with which the disease is diagnosed. Rather, testicular cancer does seem to be showing a genuine increase in incidence in the last 30 years in most industrialized countries in North America, Europe and Oceania.17 Notwithstanding significant differences in absolute incidence between countries, this increase is occurring at a rate that would be consistent with an environmental, rather than a genetic, cause. The inflection point for the recent rise in testicular cancer levels coincides with the end of World War II, which, in turn, is linked to a massive expansion in the chemicals industry.18 As a result, our environment has been contaminated with a range of novel chemicals over the past half century that are unfamiliar to our cytochrome P450 detoxification systems. A variety of these chemical structures have been suggested to influence male reproduction including alkylphenols, polychlorinated biphenyls, polyvinyl chlorides, phthalates and cigarette smoke. These observations raise a number of questions concerning the relative potency of these various pollutants and their mechanisms of action.
Figure 1.
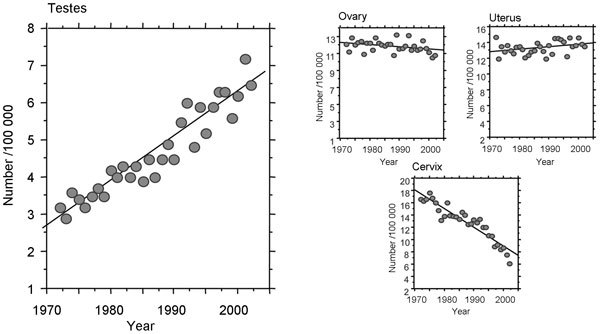
Age-standardized incidence of reproductive tract cancers in NSW. The changing incidence of testicular, ovarian, uterine and cervical cancer in NSW are illustrated. (Cancer Institute of NSW). NSW, New South Wales.
Endocrine disruptors, testicular cancer and infertility
In principle, reproductive toxicants could act on the adult individual, during fetal development when the male reproductive tract is being formed, or on the gametes whose union created the individual in the first instance. The ‘estrogen hypothesis' was constructed around an action during pregnancy and specifically during the primordial germ cell-spermatogonial stem cell transition, which is the point-of-origin for carcinoma in situ.19 In addition, environmental estrogens acting during pregnancy were proposed to be responsible for a spectrum of additional, related male reproductive tract disorders including hypospadias and cryptorchidism, known collectively as the testicular dysgenesis syndrome (TDS).20
There is good evidence to suggest that environmental estrogens are contaminating our waterways and having an impact on the sexual differentiation of aquatic organisms.21 Furthermore, the evidence that such compounds are affecting human reproductive health is less compelling. If we take DES administration to pregnant women as the extreme case, then there can be no doubt that in utero exposure to such powerful estrogens can disrupt embryonic development leading, notoriously, to an increased incidence of vaginal clear cell adenocarcinoma.22,23 In keeping with the environmental estrogen hypothesis, a meta-analysis of such patients has also detected a significantly increased risk of testicular cancer, hypospadias and cryptorchidism in the male offspring of women who were exposed to DES during pregnancy.24 However, DES is an extremely powerful estrogen that was administered directly to women during pregnancy. Environmental exposures involve estrogens with potencies that are much less than DES (1000–1 000 000 times lower that of estradiol 17β in the case of alkylphenols) and occur at a fraction of the concentration achieved with this directly-administered compound. Notwithstanding the extremely high level of DES exposure achieved clinically, there is no evidence to suggest that the male offspring of DES-treated women suffer from any loss of fertility.25 Thus, while acknowledging that DES can induce developmental defects in men and can even influence semen quality,26,27 it remains debatable whether the low concentrations of environmental estrogens to which human are exposed during pregnancy could out-compete the high levels of endogenous estrogen pervading the reproductive system at this time of the reproductive cycle.
In order to widen the scope of the toxicants-are-responsible concept, the ‘environmental estrogens' hypothesis has been extended to ‘endocrine disruptors' in order to encompass the action of anti-androgenic compounds, such as phthalate esters. These compounds are clearly capable of inducing developmental defects in animal models.28 Furthermore, a relationship between maternal exposure to phthalates and the sexual differentiation of male offspring has been observed by Swan et al. and weak associations observed with semen quality.28,29 These associations are not consistently observed however, and the picture has been confused by inaccurate linkages being created between developmental abnormalities within the male reproductive tract and the aetiology of infertility.30 That we are being exposed to a wide range of potential toxicants at different stages of reproductive life is incontrovertible Furthermore, the idea that some of these toxicants might have an impact upon male reproduction seems likely, particularly when acting in combination.31 The task that now lies before us is to determine which compounds, acting via what mechanisms, are responsible for which male reproductive pathologies. Given the complex array of environmental factors involved, the multiple points in the reproductive cycle when they might operate, the confounding influence of genetic and epigenetic factors, and the diverse spectrum of conditions potentially influenced by such exposures, this task will not be an easy one. In this context a key question that will shape future research in this area is whether the aetiology of developmental defects covered by TDS, extends to male infertility.
Genetics, environmental factors and assisted conception
Whether there is a secular downward trend in human semen quality is a question that cannot be resolved easily or rapidly. Above, I have argued that a gradual loss of sperm function is inevitable given the current lack of selection pressure on high fecundity genes. The extent to which such projected reductions in semen quality could lead to a loss of fertility would, under normal circumstances, be counterbalanced by the imperative to procreate. However, if the uptake of assisted conception therapy continues to increase at its present rate, then such a balance of forces will be compromised and a general decline in male fertility becomes predictable. It is always possible that such genetically induced trends will be exacerbated by environmental factors but they are not necessarily dependent on them.
On the other hand, most of the available evidence does point to environmental factors being involved in the widespread increase in testicular cancer, as well as the other developmental abnormalities that constitute TDS.4,32 However, the precise identities of the environmental factors contributing to this syndrome are still not understood, nor is their underlying mechanism of action. There is also a familial, genetic component to testicular cancer and, as yet, we do not understand how the genotype of an individual might interact with environmental toxicants to perturb primordial germ cell differentiation and fuel the generation of testicular cancer. Furthermore, whether such environmental factors are relevant to the aetiology of male infertility is not understood. The primary notion that a spectrum of pathological conditions, from testicular cancer to male infertility have a common developmental origin, is certainly seductive. However, despite the publicity this concept has received, there are few data to support its validity. From a mechanistic viewpoint there is very little to associate the high rates of male infertility seen in developed countries (∼1 case in 20 males), with relatively rare developmental pathologies such as testicular cancer (4–8 cases per 100 000 males) or hypospadias (100–200 in 100 000 male births).
It is certainly possible that male infertility has an environmental component but the identity of the toxic agents and their modus operandi may be different from those of the environmental factors driving developmental diseases such as testicular cancer. In cases of male infertility the current consensus is that a range of environmental factors (including small molecular mass organic compounds, heavy metals, heat, radio-frequency electromagnetic radiation, smoking, diet etc.), acting on a suboptimal genetic background, converge on the spermatogenic process to generate poor quality spermatozoa that readily default to an apoptotic state characterized by high levels of oxidative stress.33 There is little evidence to suggest an endocrine or even a developmental origin for male infertility or to suggest that it may be a facet of TDS. Of course, the small number of patients exhibiting testicular cancer may have poor semen quality but this should not be confused with the large number of males who have no evidence of testicular cancer, or any other aspect of TDS, and yet experience infertility to varying degrees. While we cannot rule out a link between the environmental factors causing TDS and the environmental contribution to male infertility, for the time being, it may be better to keep an open mind and continue to acquire data on all possible associations between environmental toxicants and the reproductive process. We shall then have to listen to the message delivered by these data and, in their wake, develop the next generation of hypotheses to explain how environmental factors impinge on male reproductive health.
The author declares that there are no competing financial interests.
References
- Carlsen E, Giwercman A, Keiding N, Skakkebæk NE. Evidence for decreasing quality of semen during past 50 years. BMJ. 1992;305:609–13. doi: 10.1136/bmj.305.6854.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe RM, Skakkebaek NE. Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract. Lancet. 1993;341:1392–5. doi: 10.1016/0140-6736(93)90953-e. [DOI] [PubMed] [Google Scholar]
- te Velde ER, Bonde JP.Misconceptions about falling sperm counts and fertility in Europe. Asian J Androle-pub ahead of print 3 December 2012; doi: 10.1038/aja.2012.122. [DOI] [PMC free article] [PubMed]
- ESF Science Policy Briefing 40; 2010. Male reproductive health—its impacts in relation to general wellbeing and low European fertility rates. http://www.esf.com
- Merzenich H, Zeeb H, Blettner M. Decreasing sperm quality: a global problem. BMC Public Health. 2010;10:24. doi: 10.1186/1471-2458-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordkap L, Joensen UN, Blomberg Jensen M, Jørgensen N. Regional differences and temporal trends in male reproductive health disorders: semen quality may be a sensitive marker of environmental exposures. Mol Cell Endocrinol. 2012;355:221–30. doi: 10.1016/j.mce.2011.05.048. [DOI] [PubMed] [Google Scholar]
- McLachlan RI, de Kretser DM. Male infertility: the case for continued research. Med J Aust. 2001;174:116–7. doi: 10.5694/j.1326-5377.2001.tb143180.x. [DOI] [PubMed] [Google Scholar]
- Krausz C, Degl'Innocenti S. Y chromosome and male infertility: update. Front Biosci. 2006;11:3049–61. doi: 10.2741/2032. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Krausz CG. Oxidative stress, DNA damage and the Y chromosome. Reproduction. 2001;122:497–506. doi: 10.1530/rep.0.1220497. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Koopman P, Lewis SE. Seeds of concern. Nature. 2004;432:48–52. doi: 10.1038/432048a. [DOI] [PubMed] [Google Scholar]
- Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25:271–85. doi: 10.1016/j.beem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Jamsai D, O'Bryan MK. Genome-wide ENU mutagenesis for the discovery of novel male fertility regulators. Syst Biol Reprod Med. 2010;56:246–59. doi: 10.3109/19396361003706424. [DOI] [PubMed] [Google Scholar]
- Seuanez HN, Carothers AD, Martin DE, Short RV. Morphological abnormalities in spermatozoa of man and great apes. Nature. 1977;270:345–7. doi: 10.1038/270345a0. [DOI] [PubMed] [Google Scholar]
- Anderson MJ, Dixson AF. Sperm competition: motility and the midpiece in primates. Nature. 2002;416:496. doi: 10.1038/416496a. [DOI] [PubMed] [Google Scholar]
- Tourmente M, Gomendio M, Roldan ER. Sperm competition and the evolution of sperm design in mammals. BMC Evol Biol. 2011;11:12. doi: 10.1186/1471-2148-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MJ, Chapman SJ, Videan EN, Evans E, Fritz J, et al. Functional evidence for differences in sperm competition in humans and chimpanzees. Am J Phys Anthropol. 2007;134:274–80. doi: 10.1002/ajpa.20674. [DOI] [PubMed] [Google Scholar]
- Huyghe E, Matsuda T, Thonneau P. Increasing incidence of testicular cancer worldwide: a review. J Urol. 2003;170:5–11. doi: 10.1097/01.ju.0000053866.68623.da. [DOI] [PubMed] [Google Scholar]
- Richiardi L, Bellocco R, Adami HO, Torrang A, Barlow L, et al. Testicular cancer incidence in eight northern European countries: secular and recent trends. Cancer Epidemiol Biomarkers Prev. 2004;13:2157–66. [PubMed] [Google Scholar]
- Looijenga LH. Human testicular (non)seminomatous germ cell tumours: the clinical implications of recent pathobiological insights. J Pathol. 2009;218:146–62. doi: 10.1002/path.2522. [DOI] [PubMed] [Google Scholar]
- Toppari J, Virtanen HE, Main KM, Skakkebaek NE. Cryptorchidism and hypospadias as a sign of testicular dysgenesis syndrome (TDS): environmental connection. Birth Defects Res A Clin Mol Teratol. 2010;88:910–9. doi: 10.1002/bdra.20707. [DOI] [PubMed] [Google Scholar]
- Jobling S, Williams R, Johnson A, Taylor A, Gross-Sorokin M, et al. Predicted exposures to steroid estrogens in U.K. rivers correlate with widespread sexual disruption in wild fish populations. Environ Health Perspect. 2006;114 Suppl 1:32–9. doi: 10.1289/ehp.8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston GP, Gooch JW, Breslin WJ, Shuey DL, Nikiforov AI, et al. Environmental estrogens and reproductive health: a discussion of the human and environmental data. Reprod Toxicol. 1997;11:465–81. doi: 10.1016/s0890-6238(97)00014-2. [DOI] [PubMed] [Google Scholar]
- Harris RM, Waring RH. Diethylstilboestrol—a long-term legacy. Maturitas. 2012;72:108–12. doi: 10.1016/j.maturitas.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Martin OV, Shialis T, Lester JN, Scrimshaw MD, Boobis AR, et al. Testicular dysgenesis syndrome and the estrogen hypothesis: a quantitative meta-analysis. Environ Health Perspect. 2008;116:149–57. doi: 10.1289/ehp.10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Baird DD, Weinberg CR, Hornsby PP, Herbst AL. Fertility in men exposed prenatally to diethylstilbestrol. N Engl J Med. 1995;332:1411–6. doi: 10.1056/NEJM199505253322104. [DOI] [PubMed] [Google Scholar]
- Stenchever MA, Williamson RA, Leonard J, Karp LE, Ley B, et al. Possible relationship between in utero diethylstilbestrol exposure and male fertility. Am J Obstet Gynecol. 1981;140:186–93. doi: 10.1016/0002-9378(81)90107-1. [DOI] [PubMed] [Google Scholar]
- Whitehead ED, Leiter E. Genital abnormalities and abnormal semen analyses in male patients exposed to diethylstilbestrol in utero. J Urol. 1981;125:47–50. doi: 10.1016/s0022-5347(17)54895-8. [DOI] [PubMed] [Google Scholar]
- Grady R, Sathyanarayana S. An update on phthalates and male reproductive development and function. Curr Urol Rep. 2012;13:307–10. doi: 10.1007/s11934-012-0261-1. [DOI] [PubMed] [Google Scholar]
- Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005;113:1056–61. doi: 10.1289/ehp.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini G, Del Vecchio A, Massaro M, Verrotti A, de Felice C. Phthalate exposure and male infertility. Toxicology. 2006;226:90–8. doi: 10.1016/j.tox.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Christiansen S, Kortenkamp A, Axelstad M, Boberg J, Scholze M, et al. Mixtures of endocrine disrupting contaminants modelled on human high end exposures: an exploratory study in rats. Int J Androl. 2012;35:303–16. doi: 10.1111/j.1365-2605.2011.01242.x. [DOI] [PubMed] [Google Scholar]
- Mathur PP, D'Cruz SC. The effect of environmental contaminants on testicular function. Asian J Androl. 2011;13:585–91. doi: 10.1038/aja.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken RJ, De Iuliis GN, Gibb Z, Baker MA. The Simmet lecture: new horizons on an old landscape–oxidative stress, DNA damage and apoptosis in the male germ line. Reprod Domest Anim. 2012;47 Suppl 4:7–14. doi: 10.1111/j.1439-0531.2012.02049.x. [DOI] [PubMed] [Google Scholar]