

Abstract
Desulfotomaculum kuznetsovii is a moderately thermophilic member of the polyphyletic spore-forming genus Desulfotomaculum in the family Peptococcaceae. This species is of interest because it originates from deep subsurface thermal mineral water at a depth of about 3,000 m. D. kuznetsovii is a rather versatile bacterium as it can grow with a large variety of organic substrates, including short-chain and long-chain fatty acids, which are degraded completely to carbon dioxide coupled to the reduction of sulfate. It can grow methylotrophically with methanol and sulfate and autotrophically with H2 + CO2 and sulfate. For growth it does not require any vitamins. Here, we describe the features of D. kuznetsovii together with the genome sequence and annotation. The chromosome has 3,601,386 bp organized in one contig. A total of 3,567 candidate protein-encoding genes and 58 RNA genes were identified. Genes of the acetyl-CoA pathway, possibly involved in heterotrophic growth with acetate and methanol, and in CO2 fixation during autotrophic growth are present. Genomic comparison revealed that D. kuznetsovii shows a high similarity with Pelotomaculum thermopropionicum. Genes involved in propionate metabolism of these two strains show a strong similarity. However, main differences are found in genes involved in the electron acceptor metabolism.
Keywords: Thermophilic spore-forming anaerobes, sulfate reduction, autotrophic, methylotrophic, Peptococcaceae, Clostridiales
Introduction
Desulfotomaculum kuznetsovii strain 17T (VKM B-1805; DSM 6115) is a moderately thermophilic sulfate-reducing bacterium isolated from deep subsurface thermal mineral water [1]. It grows with a wide range of substrates, including organic acids, such as long-chain fatty acids, short-chain fatty acids (butyrate, propionate, acetate), lactate, pyruvate, fumarate and succinate as well as ethanol and methanol. These substrates are degraded to CO2 coupled to sulfate reduction. The strain is also able to grow autotrophically with H2/CO2 and sulfate and to ferment pyruvate and fumarate. For growth, D. kuznetsovii has no vitamin requirement.
Desulfotomaculum is a genus of Gram-positive, spore-forming anaerobes that is phylogenetically and physiologically very diverse. The genus is poorly studied physiologically, while its members are known to play an important role in the carbon and sulfur cycle in a variety of often adverse environments. The genus is divided phylogenetically into different sub-groups [2,3]. To get a thorough understanding of the evolutionary relationship of the different Desulfotomaculum sub-groups and the physiology of the individual species, it is important to have genome sequence information. Here, we present a summary of the features of D. kuznetsovii strain 17T, together with the description of the complete genomic sequencing and annotation. Moreover, we describe a physiological and genomic comparison of D. kuznetsovii strain 17T and Pelotomaculum thermopropionicum strain SIT, because phylogenetically P. thermopropionicum is the closest related organism with validly published name that has a completely sequenced genome. However, the two strains have different physiological traits. For example, P. thermopropionicum is not able to grow by sulfate reduction, but is able to grow in syntrophy with methanogens. D. kuznetsovii lacks this ability. By comparing the genomes of the two bacteria we were able to identify the main similarities and differences.
Classification and features
D. kuznetsovii is a member of the phylum Firmicutes. Phylogenetic analysis of the 16S rRNA genes of D. kuznetsovii shows that it clusters in Desulfotomaculum cluster 1. This cluster not only contains Desulfotomaculum species, but also members of the genera Sporotomaculum, Cryptanaerobacter and Pelotomaculum. D. kuznetsovii is part of sub-group 1c together with D. solfataricum, D. luciae, D. thermosubterraneum, D. salinum, D. australicum, and D. thermocisternum, while Pelotomaculum species belong to sub-group 1h (Figure 1) [2].
Figure 1.

Neighbor joining tree based on 16S rRNA sequences showing the phylogenetic affiliation of Desulfotomaculum and related species divided in the subgroups of Desulfotomaculum cluster 1. D. kuznetsovii is printed in bold type. The sequences of different Thermotogales were used as outgroup, but were pruned from the tree. Closed circles represent bootstrap values between 75 and 100%. The scale bar represents 10% sequences difference.
D. kuznetsovii cells are rod-shaped (1.0-1.4 x 3.5-5 μm) with rounded ends and peritrichous flagella [Figure 2]. Spores of D. kuznetsovii are spherical (1.3 μm in diameter) and centrally located causing swelling of the cells. D. kuznetsovii grows between 50 and 85°C, but the optimal growth temperature is 60-65°C. The substrates D. kuznetsovii can grow with are completely oxidized to CO2. Suitable electron acceptors are sulfate, thiosulfate and sulfite. D. kuznetsovii is also able to grow by fermentation of pyruvate and fumarate. A summary of the classification and general features of D. kuznetsovii is presented in Table 1 [1].
Figure 2.

Scanning electron microscopic photograph of D. kuznetsovii.
Table 1. Classification and general features of D. kuznetsovii DSM 6115 according to the MIGS recommendations [4].
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [5] | |
| Phylum Firmicutes | TAS [6-8] | ||
| Class Clostridia | TAS [9,10] | ||
| Order Clostridiales | TAS [11,12] | ||
| Family Peptococcaceae | TAS [11,13] | ||
| Genus Desulfotomaculum | TAS [11,14,15] | ||
| Species Desulfotomaculum kuznetsovii | TAS [1,16] | ||
| Type strain 17 | |||
| Gram stain | Positive | TAS [1] | |
| Cell shape | Rods | TAS [1] | |
| Motility | peritrichous flagella | TAS [1] | |
| Sporulation | oval, terminal or subterminal, slightly swelling the cell. | TAS [1] | |
| Temperature range | 50-85°C | TAS [1] | |
| Optimum temperature | 60-65°C | TAS [1] | |
| Carbon source | CO2 (autotrophic) and organic substrates (heterotrophic) | TAS [1] | |
| Energy source | Sulfate-dependent growth and fermentative growth with pyruvate and fumarate. | TAS [1] | |
| Electron acceptor | Sulfate, thiosulfate and sulfite. | TAS [1] | |
| MIGS-6 | Habitat | Geothermal groundwater, sediment and hot solfataric fields. | TAS [1,17,18] |
| MIGS-6.3 | Salinity | 2-3% NaCl | TAS [1] |
| MIGS-22 | Oxygen | Obligate anaerobes | TAS [1] |
| MIGS-15 | Biotic relationship | Free living | TAS [1] |
| MIGS-14 | Pathogenicity | None | |
| MIGS-4 | Geographic location | Sukhumi, Georgia | TAS [1] |
| MIGS-5 | Sample collection time | 1987 or before | TAS [1] |
| MIGS-4.1 | Latitude | 43.009 | TAS [1] |
| MIGS-4.2 | Longitude | 40.989 | TAS [1] |
| MIGS-4.3 | Depth | 2800-3250 m | TAS [1] |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from the Gene Ontology project [19].
Genome sequencing and annotation
Genome project history
D. kuznetsovii was selected for sequencing in the DOE Joint Genome Institute Community Sequencing Program 2009, proposal 300132_795700 'Exploring the genetic and physiological diversity of Desulfotomaculum species', because of its phylogenetic position in one of the Desulfotomaculum sub-groups, its important role in bioremediation, and its ability to use propionate, acetate and methanol for growth. The genome project is listed in the Genome OnLine Database (GOLD) [20] as project Gc01781, and the complete genome sequence was deposited in Genbank. Sequencing, finishing and annotation of the D. kuznetsovii genome were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Four genomic libraries: one Illumina shotgun library, one 454 standard library, two paired end 454 libraries |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 Titanium |
| MIGS-31.2 | Fold coverage | 158.2 × illumina; 30.6 × pyrosequencing |
| MIGS-30 | Assemblers | VELVET version 0.7.63; Newbler version 2.3; phrap version SPS - 4.24 |
| MIGS-32 | Gene calling method | Prodigal 1.4, GenePRIMP |
| INSDC ID | CP002770.1 | |
| Genome Database release | July 20, 2012 | |
| Genbank Date of Release | May 24, 2011 | |
| MIGS-13 | GOLD ID NCBI project ID Source material identifier |
Gc01781 48313 DSM 6115T |
| Project relevance | Obtain insight into the phylogenetic and physiological diversity of Desulfotomacum species, and bioremediation. |
Growth conditions and DNA isolation
D. kuznetsovii was grown anaerobically at 60oC in bicarbonate buffered medium with propionate and sulfate as substrates [1]. DNA of cell pellets was isolated using the standard DOE-JGI CTAB method recommended by the DOE Joint Genome Institute (JGI, Walnut Creek, CA, USA). In short, cells were resuspended in TE (10 mM tris; 1 mM EDTA, pH 8.0). Subsequently, cells were lysed using lysozyme and proteinase K, and DNA was extracted and purified using CTAB and phenol:chloroform:isoamylalcohol extractions. After precipitation in 2-propanol and washing in 70% ethanol, the DNA was resuspended in TE containing RNase. Following a quality and quantity check using agarose gel electrophoresis in the presence of ethidium bromide, and spectrophotometric measurement using a NanoDrop ND-1000 spectrophotometer (NanoDrop® Technologies, Wilmington, DE, USA).
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [21]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 81 contigs in five scaffolds was converted into a phrap [22] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (570.2 Mb) was assembled with Velvet [23] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 134.6 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [22] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [21], Dupfinisher [24], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 400 additional reactions and one shatter library were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [25]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 188.8 × coverage of the genome. The final assembly contained 323,815 pyrosequence and 15,594,144 Illumina reads.
Genome annotation
Genes were identified using Prodigal [26] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [27]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [28].
Genome properties and genome comparison with other strains
The genome of D. kuznetsovii consists of a circular chromosome of 3,601,386 bp with 54.88% GC content (Table 3 and Figure 3). Pseudogenes comprise 4.66% of the genes identified. Of the 3,625 genes predicted, 3,567 are protein-coding genes of which 2,560 are assigned to COG functional categories. The distribution of these genes into COG functional categories is presented in Table 4.
Table 3. Genome statistics.
| Attribute | Value | % of totala |
|---|---|---|
| Genome size (bp) | 3,601,386 | 100.00 |
| Genome coding region (bp) | 3,057,959 | 84.91 |
| Genome G+C content (bp) | 1,976,601 | 54.88 |
| Total genes | 3,625 | 100.00 |
| RNA genes | 58 | 1.60 |
| Protein-coding genes | 3,567 | 98.40 |
| Genes in paralog clusters | 1,373 | 37.88 |
| Genes assigned to COGs | 2,560 | 70.62 |
| Pseudo genes | 169 | 4.66 |
| Genes with signal peptides | 582 | 16.06 |
| Genes with transmembrane helices | 748 | 20.63 |
Figure 3.

Graphical map of the chromosome of D. kuznetsovii. From outside to the center: Genes on the forward strand (colored by COG categories), Genes on the reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | %agea | Description |
|---|---|---|---|
| J | 148 | 5.32 | Translation |
| A | 0 | 0.00 | RNA processing and modification |
| K | 184 | 6.61 | Transcription |
| L | 207 | 7.44 | Replication, recombination and repair |
| B | 2 | 0.07 | Chromatin structure and dynamics |
| D | 60 | 2.16 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.00 | Nuclear structure |
| V | 35 | 1.26 | Defense mechanisms |
| T | 177 | 6.36 | Signal transduction mechanisms |
| M | 122 | 4.38 | Cell wall/membrane biogenesis |
| N | 79 | 2.84 | Cell motility |
| Z | 2 | 0.07 | Cytoskeleton |
| W | 0 | 0.00 | Extracellular structures |
| U | 75 | 2.69 | Intracellular trafficking and secretion |
| O | 81 | 2.91 | Posttranslational modification, protein turnover, chaperones |
| C | 261 | 9.38 | Energy production and conversion |
| G | 106 | 3.81 | Carbohydrate transport and metabolism |
| E | 197 | 7.08 | Amino acid transport and metabolism |
| F | 55 | 1.98 | Nucleotide transport and metabolism |
| H | 158 | 5.68 | Coenzyme transport and metabolism |
| I | 89 | 3.20 | Lipid transport and metabolism |
| P | 127 | 4.56 | Inorganic ion transport and metabolism |
| Q | 122 | 3.58 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 331 | 11.89 | General function prediction only |
| S | 257 | 9.23 | Function unknown |
| - | 1,065 | 29.38 | Not in COGs |
The genome of D. kuznetsovii has 58 RNA genes of which, three are 16S rRNA genes. This is one more than the previously described rrnA and rrnB [29]. These two rRNA genes contained two large inserts. One at the variable 5’terminal region and one at the variable 3’terminal region. The main differences between the two rRNA genes were found in these inserts. These inserts were hypothesized to be involved in the operation of ribosomes at high temperatures. However, more research is needed to assess the function of these inserts. All three rRNA genes of D. kuznetsovii have a size of approximately 1,700 nucleotides. This suggests that the third rRNA gene might also contain inserts. Alignment of the 16S rRNA genes confirmed the presence of inserts in all three 16S rRNA genes (data not shown).
BLAST analysis [30,31] of the genes of D. kuznetsovii against genes in the KEGG Sequence Similarity DataBase revealed similarity with other Desulfotomaculum strains (Table 5), D. acetoxidans, D. carboxydivorans, “D. reducens” and D. ruminis, but interestingly also with non-Desulfotomaculum strains. D. kuznetsovii contains 873 genes with high similarity to genes of Pelotomaculum thermopropionicum, which is more than to any of the sequenced Desulfotomaculum species. Moreover, we identified the conserved proteins of D. kuznetsovii across three related fully sequenced species (Table 6). The bidirectional best blast hits showed that despite the smaller genome of P. thermopropionicum it contained more homologous predicted proteins with D. kuznetsovii (1,406) compared to D. acetoxidans (1,309) and “D. reducens” (1330). This suggests a strong physiological similarity between D. kuznetsovii and P. thermopropionicum.
Table 5. Taxonomic distribution of the top KEGG hits of D. kuznetsovii genes based on BLAST against KEGG database.†.
| Kingdom | Category | Species | Hits |
|---|---|---|---|
| Archaea | 91 | ||
| Crenarchaeota | 9 | ||
| Euryarchaeota | 81 | ||
| Thaumarchaeota | 1 | ||
| Bacteria | 2,963 | ||
| Acidobacteria | 2 | ||
| Actinobacteria | 16 | ||
| Alphaproteobacteria | 13 | ||
| Bacteroidetes | 5 | ||
| Betaproteobacteria | 14 | ||
| Cyanobacteria | 19 | ||
| Deinococcus-Thermus | 16 | ||
| Deltaproteobacteria | 62 | ||
| Epsilonproteobacteria | 1 | ||
| Firmicutes | 2,728 | ||
| Ammonifex degensii | 170 | ||
| Carboxydothermus hydrogenoformans | 58 | ||
| Desulfotomaculum acetoxidans | 310 | ||
| Candidatus Desulforudis audaxviator | 154 | ||
| Desulfotomaculum carboxydivorans | 268 | ||
| Desulfotomaculum reducens | 111 | ||
| Desulfotomaculum ruminis | 132 | ||
| Moorella thermoacetica | 183 | ||
| Pelotomaculum thermopropionicum | 873 | ||
| Thermincola potens JR | 104 | ||
| Fusobacteria | 2 | ||
| Gammaproteobacteria | 12 | ||
| Green nonsulfur bacteria | 20 | ||
| Green sulfur bacteria | 4 | ||
| Hyperthermophilic bacteria | 31 | ||
| OtherProteobacteria | 1 | ||
| Spirochaetes | 9 | ||
| Synergistetes | 6 | ||
| Verrucomicrobia | 2 | ||
| Eukaryotes | 3 | ||
| Plants | 1 | ||
| Protists | 2 | ||
| Null | 342 | ||
| Total | 3,399 |
†Species that had more than 50 genes similar to D. kuznetsovii were included in this table, others were only summarized in categories.
Table 6. Proteins of D. kuznetsovii conserved across three related species with fully sequenced genomes†.
| Subject DB Input Query | D. acetoxidans | D. kuznetsovii | D. reducens | P. thermopropionicum |
|---|---|---|---|---|
| Desulfotomaculum acetoxidans | 4,068 | 1,539 | 1,525 | 1,486 |
| 1,309 | 1,316 | 1,255 | ||
| Desulfotomaculum kuznetsovii | 1,509 | 3,398 | 1,518 | 1,645 |
| 1,309 | 1,330 | 1,406 | ||
| Desulfotomaculum reducens | 1,537 | 1,571 | 3,276 | 1,438 |
| 1,316 | 1,330 | 1,211 | ||
| Pelotomaculum thermopropionicum | 1,430 | 1,600 | 1,395 | 2,919 |
| 1,255 | 1,406 | 1,211 |
† BLAST analyses were performed using standard settings and best hits were filtered for 40% identity over an alignment length of 75 amino acids as a minimum requirement. The values show the number of predicted proteins that are homologous to the query species in each row. The number of similar proteins obtained with a unidirectional BLAST is indicated in light blue. Bidirectional best blast hits are indicated in dark blue. Proteomes were obtained from ftp.ncbi.nih.gov/Bacteria/. Accession numbers are in parenthesis: D. acetoxidans (NC_013216); D. kuznetsovii (NC_015573); “D. reducens” (NC_009253); Pelotomaculum thermopropionicum (NC_009454).
Insights into the genome
Involvement of the acetyl-coA pathway in growth with acetate and methanol
D. kuznetsovii oxidizes acetate completely to CO2. The pathway of acetate degradation has not been studied yet, but sulfate reducers may employ the tricarboxylic acid (TCA) cycle or the acetyl-CoA pathway for acetate degradation, as exemplified by Desulfobacter postgatei and Desulfobacca acetoxidans, respectively [32]. Most genes predicted to code for enzymes of the TCA cycle are present in the genome of D. kuznetsovii, but genes with similarity to those coding for an ATP-dependent citrate synthase and isocitrate dehydrogenase are missing. This suggests that the TCA cycle is not complete and that the TCA cycle enzymes have mainly an anabolic function or a function in other catabolic pathways, such as the propionate degradation pathway. Genes with similarity to those coding for enzymes involved in the acetyl-CoA pathway are all present in the genome of D. kuznetsovii (Figure 4), which suggests its involvement in acetate oxidation. However, there are no genes similar to those that code for acetate kinase and phosphate acetyltransferase present in the genome. The reaction from acetate to acetyl-CoA is likely performed by acetyl-CoA synthetase (Desku_1241).
Figure 4.

Pathway of acetate oxidation to CO2 by D. kuznetsovii. Enzymes in this figure are in bold italic and their locus tags are included. Genes with the locus tags Desku_1488 and Desku_1490 putatively code for the small subunit and the large subunit of the iron-sulfur protein, respectively. This protein is involved in transferring the methyl from acetyl-CoA to tetrahydrofolate. Abbreviations: A-CoA S, acetyl-CoA synthetase; AcsA, carbon-monoxide dehydrogenase; AcsB, acetyl-CoA synthase; CFeSP, iron-sulfur protein; CH3, methyl; THF, tetrahydrofolate; MeTr, methyltransferase.
D. acetoxidans is an acetate-oxidizing Desulfotomaculum species, positioned in sub-group 1e (Figure 1), that also uses the acetyl-CoA pathway for acetate oxidation to CO2 [33]. The genes involved in acetate oxidation in D. acetoxidans are similar to those in D. kuznetsovii, but there are some exceptions. The genome of D. acetoxidans does not contain a gene that putatively codes for acetyl-CoA synthetase, similar to D. kuznetsovii, but contains genes that putatively code for an acetate kinase and a phosphate acetyltransferase [34]. Additionally, putative carbon-monoxide dehydrogenase complex coding genes involved in the acetyl-CoA pathway show differences between the two Desulfotomaculum species. D. kuznetsovii lacks a ferredoxin coding gene that is located between cooC (Desku_1493) and acsE (Desku_1487), which in contrast is present in the genome of D. acetoxidans (Dtox_1273). Moreover, three genes similar to heterodisulfide reductase encoding genes (Desku_1486-1484) are located upstream of acsE in D. kuznetsovii, which is not the case in the genome of D. acetoxidans.
Methanol metabolism
Growth of D. kuznetsovii with methanol and sulfate was studied [35]. In that study the activity of methyltransferase, an enzyme that is involved in methanol metabolism in methanogens and acetogens [36,37], could not be assessed, while low activities of an alcohol dehydrogenase could be measured. An alcohol dehydrogenase with a molecular mass of 42 kDa was partially purified and showed activity with methanol [35]. The genome of D. kuznetsovii contains several alcohol dehydrogenase genes (Desku_0165, 0619, 0624, 0628, 2955, 3082) that each code for an enzyme with a size of approximately 42 kDa. In the genome, genes with similarity to those coding for a methanol methyltransferase mtaA (Desku_0050, 0055, 0060), mtaB (Desku_0051) and mtaC (Desku_0048, 0049, 0052, 0056) were also found, suggesting a methanol metabolism as described in Moorella thermoacetica [36]. Further studies are needed to obtain information about the diversity of the methanol-degradation pathways in D. kuznetsovii.
Comparison of D. kuznetsovii and P. thermopropionicum genomes
Genomic comparison revealed that a large number of D. kuznetsovii genes show similarity to genes of Pelotomaculum thermopropionicum, a syntrophic propionate-oxidizing thermophile (Table 5 and 6). Interestingly, among them are genes that putatively code for enzymes involved in propionate metabolism (Table 7). Moreover, the genetic organization of the methylmalonyl-CoA (mmc) cluster in the genome of both bacteria is similar (Figure 5). However, D. kuznetsovii lacks tps, mmcA and mmcM in the mmc cluster. mmcA codes for a response regulator and mmcM for pyruvate ferredoxin oxidoreductase.
Table 7. Genes in D. kuznetsovii that are annotated as enzymes involved in propionate metabolism†.
| Homologous protein in P. thermopropionicum |
||||
|---|---|---|---|---|
| Gene symbol | Locus tag | Function | Identity (%) | Locus tag |
| sdhB | Desku_0434 | Succinate dehydrogenase, FeS protein | 76 | PTH_1018 |
| sdhA | Desku_0435 | Succinate dehydrogenase, flavoprotein | 76 | PTH_1017 |
| sdhC | Desku_0436 | Succinate dehydrogenase, cytochrome b | 51 | PTH_1016 |
| citE | Desku_1348 | Citrate lyase | 57 | PTH_1335 |
| sdhA | Desku_1353 | Succinate dehydrogenase, flavoprotein | 83 | PTH_1491 |
| sdhB | Desku_1354 | Succinate dehydrogenase, FeS protein | 75 | PTH_1490 |
| mmcB | Desku_1358 | Fumarase, N-terminal domain | 73 | PTH_1356 |
| mmcC | Desku_1359 | Fumarase, C-terminal domain | 77 | PTH_1357 |
| mmcD2 | Desku_1361 | Succinyl-CoA synthetase, alpha subunit | 78 | PTH_1359 |
| mmcE | Desku_1362 | Methylmalonyl-CoA mutase, N-terminal domain |
77 | PTH_1361 |
| mmcF | Desku_1363 | Methylmalonyl-CoA mutase, C-terminal domain |
82 | PTH_1362 |
| mmcG | Desku_1364 | Methylmalonyl-CoA epimerase | 86 | PTH_1363 |
| mmcH | Desku_1365 | Methylmalonyl-CoA decarboxylase, alpha subunit |
75 | PTH_1364 |
| mmcI | Desku_1366 | Methylmalonyl-CoA decarboxylase, epsilon subunit |
82 | PTH_1365 |
| mmcJ | Desku_1367 | Methylmalonyl-CoA decarboxylase, gamma subunit |
56 | PTH_1366 |
| mmcK | Desku_1368 | Malate dehydrogenase | 75 | PTH_1367 |
| mmcL | Desku_1369 | Transcarboxylase 5S subunit | 66 | PTH_1368 |
| pykF | Desku_1651 | Pyruvate kinase | 73 | PTH_2214 |
| ppsA | Desku_2615 | Pyruvate phosphate dikinase | 78 | PTH_0903 |
| citE | Desku_2747 | Citrate lyase | 56 | PTH_1335 |
†Corresponding homologs in P. thermopropionicum are included.
Figure 5.
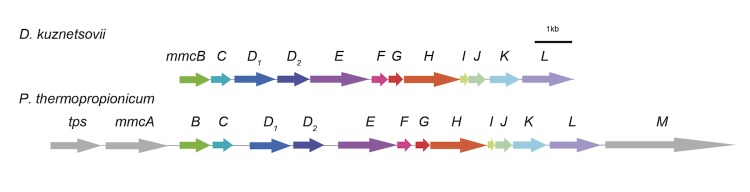
Gene organization of the mmc cluster in D. kuznetsovii and P. thermopropionicum. Names of the genes can be found in table 6, except for tps, which is a transposase gene.
Based on 16S rRNA gene sequences, D. kuznetsovii and P. thermopropionicum group in cluster group c and h of the Desulfotomaculum cluster 1, respectively (see Figure 1). P. thermopropionicum is known for its ability to grow with propionate and ethanol in syntrophic association with methanogens. It is not able to grow by sulfate respiration, despite the presence of sulfate reduction genes in the genome [38]. In contrast, D. kuznetsovii is able to grow with propionate (Figure 6) and ethanol with sulfate. However, in the absence of sulfate, it cannot grow in syntrophic association with methanogens. Therefore, differences are expected in genes coding for hydrogenases, formate dehydrogenases, and those involved in sulfate reduction.
Figure 6.

Propionate degradation pathway in D. kuznetsovii based on genomic data. Enzymes are depicted in bold italic. Next to these enzymes are the possible encoding genes, and their locus tags. GCT, Glutaconate CoA-transferase; MCD, Methylmalonyl-CoA decarboxylase; PCC, Propionyl-CoA carboxylase; MCE, Methylmalonyl-CoA epimerase; MCM, Methylmalonyl-CoA mutase; SCS, Succinyl-CoA synthetase; SDH, Succinate dehydrogenase; FHT, Fumarase; MDH, Malate dehydrogenase; OAD, Oxaloacetate decarboxylase; PFL, Pyruvate formate lyase.
Sulfate reduction genes: Figure 7 depicts the sulfate reduction pathway of the two strains. In the genome of D. kuznetsovii two genes (Desku_2103; Desku_3527) are annotated as phosphoadenosine phosphosulfate reductase encoding genes whose corresponding proteins might be involved in assimilatory sulfate metabolism. The P. thermopropionicum genome lacks these genes [39]. Instead, the P. thermopropionicum genome contains an adenylylsulfate kinase gene (PTH_0238). In the dissimilatory sulfate reduction pathway, the two strains both have genes that code for enzymes to reduce sulfate to H2S. However, P. thermopropionicum is missing the gene that codes for an adenylylsulfate reductase beta subunit, which is present in the D. kuznetsovii genome (Desku_1073). Moreover, the gene labeled as a dissimilatory sulfite reductase (dsr) alpha and beta subunit in the P. thermopropionicum genome (PTH_0242) is not similar to dsrA or dsrB from D. kuznetsovii or any other Desulfotomaculum strain. However, it has high similarity to the dsrC gene from D. kuznetsovii, indicating that it is not a dsrA or dsrB gene but a dsrC gene (data not shown). Therefore, the inability of P. thermopropionicum to grow by sulfate respiration is most likely caused by the absence of an adenylylsulfate reductase beta subunit encoding gene and the dsrAB genes.
Figure 7.

Sulfate reduction pathway of D. kuznetsovii and P. thermopropionicum. Depicted in green are genes that code for sulfate reduction enzymes that are present in the genome. Dashed arrows indicate the presence of a subunit encoding gene, but not the presence of all genes required for the enzyme. Dashed dotted arrows are used when no genes were found for the reaction. Abbreviations: APS, adenylylsulfate; DP, diphosphate; PAPS, 3’-Phosphoadenylyl-sulfate (PAPS); redA, reduced acceptor; oxA, oxidized acceptor.
Hydrogenase and formate dehydrogenase genes: Schut and Adams (2009) [40] showed that the trimeric [FeFe]-hydrogenase from Thermotoga maritima oxidizes NADH and ferredoxin simultaneously to produce H2. Similar bifurcating / confurcating [FeFe]-hydrogenases and formate dehydrogenases are present in Syntrophobacter fumaroxidans and P. thermopropionicum [41]. Both generate NADH and ferredoxin during propionate degradation via the methylmalonyl-CoA pathway and might use confurcating hydrogenases and formate dehydrogenases to drive the unfavorable re-oxidation of NADH (E0’=-320mV) by the exergonic re-oxidation of ferredoxin (E0’=-398mV) to produce hydrogen (E0’= -414mV) or formate (E0’= -432mV) that are subsequently transferred to hydrogen and formate scavenging methanogens. Additionally, up-regulation of genes encoding hydrogenases and formate dehydrogenases in P. thermopropionicum was shown during syntrophic growth [42]. The P. thermopropionicum genome contains three [FeFe]-hydrogenases, one [NiFe]-hydrogenase and two formate dehydrogenases. One [FeFe]-hydrogenase (PTH_0668-0670) was shown to be down-regulated during syntrophic growth, while the other two [FeFe]-hydrogenases (PTH_1377-1379 and PTH_2010-2012) were up-regulated. The two formate dehydrogenases of P. thermopropionicum (I, PTH_1711-1714 and II, PTH_2645-2649) were both up-regulated during syntrophic growth [42]. According to TMHMM server v. 2.0 [43] formate dehydrogenase I of P. thermopropionicum has transmembrane helices. Therefore, it might play an essential role in the interspecies transfer of reducing equivalents in syntrophic growth.
The genome of D. kuznetsovii was screened for hydrogenase and formate dehydrogenase encoding gene clusters with BLAST analysis. Pfam search [44] was used to identify motifs in the amino acid sequences and the TMHMM Server v. 2.0 [43] was used to screen for transmembrane helices. The TatP 1.0 Server was used to screen for twin-arginine translocation (Tat) motifs in the N-terminus to predict protein localization in the cell [45]. The incorporation of selenocysteine (SeCys) was examined by RNA loop predictions with Mfold version 3.2 [46, 47]. The predicted RNA loop in the 50-100 bp region downstream of the UGA-codon was compared with the consensus loop described earlier [48].
Compared to P. thermopropionicum, D. kuznetsovii lacks membrane associated formate dehydrogenases and hydrogenases and also lacks [NiFe]-hydrogenase. This might explain why D. kuznetsovii cannot grow in syntrophic relation with methanogens. The genome of D. kuznetsovii indicates the presence of a confurcating selenocysteine-incorporated formate dehydrogenase (Desku_2987-2991), two trimeric confurcating [FeFe]-hydrogenases (Desku_2307-2309, Desku_2995-2997) and two [FeFe]-hydrogenases (Desku_0995, Desku_2934-2935) without NADH-binding sites (Figure 8). Several subunits of these enzymes are related to subunits of NADH dehydrogenase (complex I), including the NADH-binding proteins related to NuoF (Desku_2990, 2308 and 2996) and the electron transfer subunits related to NuoE (Desku_2991, 2935, and 2997) and to NuoG (Desku_2989). In three of the [FeFe]-hydrogenases this NuoG-like domain is fused with the catalytic subunit (Desku_2995, 2307 and 2934). Two of the multimeric hydrogenases are found next to [FeFe]-hydrogenases containing PAS-sensor domains (Desku_2932 and Desku_2994), suggesting they are involved in the regulation of the synthesis of those hydrogenases. All complexes are predicted to be cytoplasmic and not membrane bound.
Figure 8.

Schematic representation of a putative confurcating formate dehydrogenase, two putative confurcating [FeFe]-hydrogenases and two ferredoxin re-oxidizing [FeFe]-hydrogenases in Desulfotomaculum kuznetsovii. Gene locus tag numbers and α-, β-, and γ-subunits are depicted. Moreover, predicted iron-sulfur clusters and metal-binding sites are indicated.
Apart from a possible involvement in the acetate oxidation pathway (Figure 4), it remains unclear for which purpose D. kuznetsovii uses its confurcating formate dehydrogenase and hydrogenases because our genome analysis indicates that pyruvate oxidation during propionate degradation generates formate instead of ferredoxin (Figure 6).
Vitamin synthesis
D. kuznetsovii is able to grow in medium without vitamins [1]. This indicates that D. kuznetsovii is able to synthesize all the vitamins that are required for its metabolism and that vitamin synthesis genes should be present in the genome. Vitamin B12 is essential for the methylmalonyl-CoA pathway and the acetyl-coA pathway. The biosynthesis of cobalamin (vitamin B12) is known to occur from uroporphyrinogen-III to adenosylcobalamin via two possible pathways, the aerobic and anaerobic pathway of the corrinoid ring [49,50]. The D. kuznetsovii genome contains all genes needed for the anaerobic pathway: cysGA (Desku_1520), cysGB (Desku_1460, Desku_1523), cbiA (Desku_1765, Desku_2368), cbiBCDEFGHJLPT (Desku_2369, 1459, 1468, 1467, 1464, 1463, 1462, 1461, 1465, 2370 and 1466, respectively), cobalt reductase (Desku_2757), btuR (Desku_0004, 1209), cobS (Desku_2367) and cobU (Desku_2371). Moreover, D. kuznetsovii has genes to convert glutamyl tRNA to uroporphyrinogen-III, hemABCDL (Desku_1522, 1518, 1521, 1520 and 1522, respectively). The genome also contains some unassigned cobalamin synthesis genes (P47K, Desku_0046, 0053; cbiM, Desku_2905), corrinoid transport proteins (Desku_0693, 702, 2237-2239, 2902-2904, 3025-3027) and, interestingly, two cobN genes (Desku_2189, 2227), genes involved in the aerobic pathway. It is unclear why D. kuznetsovii has these cobN genes, since all anaerobic pathway genes are present in the genome, and it is unclear if the products of these two genes are used for cobalamin synthesis by D. kuznetsovii.
Other vitamin synthesis genes present in the genome of D. kuznetsovii are genes involved in biotin synthesis (vitamin H) (Desku_1295-1297, 2246-2247, 2317), nicotinamide (vitamin B3) synthesis (Desku_0433, 0614, 0662, 0815, 1248, 1417, 1472, 1499, 1925, 1951, 3103, 3121, 3227, 3228, 3231, 3246, 3337), thiamin (vitamin B1) synthesis (0372, 0543, 0545, 2253, 2363, 2639), riboflavin (vitamin B2) synthesis (Desku_1244-1247), and pantothenate (vitamin B5) synthesis (Desku_3262). The genes involved in coenzyme A production from pantothenate are also present in the D. kuznetsovii genome (Desku_1254, 1307, 3145, 3200). Moreover, genes involved in the biosynthesis of pyridoxine (vitamin B6) via the deoxyxylulose 5-phosphate (DXP) independent route were found to be in the genome (Desku_0007, 0008). These genes code for two enzymes that facilitate the conversion of glutamine to the active form of vitamin B6, pyridoxal 5’-phosphate [51].
Menaquinone (vitamin K) and ubiquinone (coenzyme Q10) biosynthesis is important because of the electron transport function in the membranes. The genes that code for the biosynthesis enzymes from polyprenyldiphosphate to menaquinone and ubiquinone are present in the D. kuznetsovii genome (Desku_0124, 0126, 0629, 1551-1554, 1829, 2629 and 3525), except for the genes that code for a 2-polyprenyl-6-methoxyphenol 4-monooxygenase (UbiH) and 2-polyprenyl-3-methyl-6-methoxy-1,4-benzoquinone hydroxylase (UbiF). Additionally, three genes (Desku_1548-1550) could be identified as putative menaquinone biosynthesis genes and are part of a menaquinone biosynthesis gene cluster (Desku_1548-1554). The products of those three genes could be involved in the reactions of the missing UbiH and UbiF encoding genes.
Folate (vitamin B9) biosynthesis is also of great importance for D. kuznetsovii, because it is an essential part of the acetyl-CoA pathway. It is involved in the transfer of one-carbon compounds and can be biosynthesized from chorismate and guanosine triphosphate (GTP) [52-55]. Both pathways use a dihydropteroate synthase to produce dihydropteroate. The genome of D. kuznetsovii contains the genes encoding the enzymes involved in the pathway from chorismate to dihydropteroate (Desku_0219, 2268-2269) and from GTP to dihydropteroate (Desku_0210, 0219-0221 and 1419). The gene encoding a phosphatase (Desku_0210) in the D. kuznetsovii genome is probably involved in the removal of phosphate groups from dihydropterine triphosphate as a substitute for an alkaline phosphatase encoding gene, which is not present in the genome. Additionally, the genome contains a bifunctional protein encoding gene (Desku_404) that is expected to be responsible for the production of dihydrofolate (DHF) and the addition of multiple glutamate moieties to DHF or tetrahydrofolate (THF). However, the D. kuznetsovii genome lacks the DHF reductase encoding gene, which is required to reduce DHF to THF. The DHF reductase encoding gene appears to be absent in many microorganisms [56]. Levin et al. (2004) propose that in Halobacterium salinarum a dihydrofolate synthase and a dihydropteroate synthase domain is able to replace the function of the DHF reductase. Additionally, the authors show that when using a BLAST search, homologs of polypeptides can be found in organisms that lack a DHF reductase [56]. However, BLAST results showed no homologous protein encoding gene in the genome of D. kuznetsovii (data not shown). How in D. kuznetsovii DHF is reduced to THF can currently not be deduced from the genome sequence.
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and was also supported by grants CW-TOP 700.55.343 and ALW 819.02.014 of the Netherlands Science Foundation (NWO) and grant 323009 of the European Research Council.
References
- 1.Nazina TN, Ivanova AE, Kanchaveli LP, Rozanova EP. A new sporeforming thermophilic methylotrophic sulfate-reducing bacterium, Desulfotomaculum kuznetsovii sp. nov. Mikrobiologiya 1988; 57:823-827 [Google Scholar]
- 2.Stackebrandt E, Sproer C, Rainey FA, Burghardt J, Pauker O, Hippe H. Phylogenetic analysis of the genus Desulfotomaculum: evidence for the misclassification of Desulfotomaculum guttoideum and description of Desulfotomaculum orientis as Desulfosporosinus orientis gen. nov., comb. nov. Int J Syst Bacteriol 1997; 47:1134-1139 10.1099/00207713-47-4-1134 [DOI] [PubMed] [Google Scholar]
- 3.Plugge CM, Zhang W, Scholten JC, Stams AJM. Metabolic flexibility of sulfate-reducing bacteria. Front Microbiol 2011; 2:81-87 10.3389/fmicb.2011.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1-6 10.1099/00207713-28-1-1 [DOI] [Google Scholar]
- 7.Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119-169. [Google Scholar]
- 8.Murray RGE. The Higher Taxa, or, a Place for Everything...? In: Holt JG (ed), Bergey's Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams and Wilkins Co., Baltimore, 1984, p. 31-34. [Google Scholar]
- 9.List Editor List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol 2010; 60:469-472 10.1099/ijs.0.022855-0 [DOI] [PubMed] [Google Scholar]
- 10.Rainey FA. Class II. Clostridia class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 736. [Google Scholar]
- 11.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 12.Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D, Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et Cie, Paris, 1953, p. 1-692. [Google Scholar]
- 13.Rogosa M. Peptococcaceae, a new family to include the Gram-positive, anaerobic cocci of the genera Peptococcus, Peptostreptococcus and Ruminococcus. Int J Syst Bacteriol 1971; 21:234-237 10.1099/00207713-21-3-234 [DOI] [Google Scholar]
- 14.Campbell LL, Postgate JR. Classification of the spore-forming sulfate-reducing bacteria. Bacteriol Rev 1965; 29:359-363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell LL. Genus IV. Desulfotomaculum Campbell and Postgate 1965, 361. In: Buchanan RE, Gibbons NE (eds), Bergey's Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 572-573. [Google Scholar]
- 16.Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 35. Int J Syst Bacteriol 1990; 40:470-471 10.1099/00207713-40-4-470 [DOI] [PubMed] [Google Scholar]
- 17.Isaksen MF, Bak F, Jorgensen BB. Thermophilic sulfate-reducing bacteria in cold marine sediment. Microb Ecol 1994; 14:1-8 10.1111/j.1574-6941.1994.tb00084.x [DOI] [Google Scholar]
- 18.Goorissen HP, Boschker HT, Stams AJM, Hansen TA. Isolation of thermophilic Desulfotomaculum strains with methanol and sulfite from solfataric mud pools, and characterization of Desulfotomaculum solfataricum sp. nov. Int J Syst Evol Microbiol 2003; 53:1223-1229 10.1099/ijs.0.02476-0 [DOI] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40(Database issue):D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.JGI website. http://www.jgi.doe.gov
- 22.The Phred/Phrap/Consed software package. http://www.phrap.com
- 23.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: H.R. A, H. V, editors2006 June 26-29, 2006. CSREA Press. p 141-6. [Google Scholar]
- 25.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. Marco Island, FL: AGBT; 2008. [Google Scholar]
- 26.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 28.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 29.Tourova TP, Kuznetsov BB, Novikova EV, Poltaraus AB, Nazina TN. Heterogeneity of the nucleotide sequences of the 16 S rRNA genes of the type strain of Desulfotomaculum kuznetsovii. Mikrobiologiya 2001; 70:788-795 [PubMed] [Google Scholar]
- 30.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389-3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. Protein database searches using compositionally adjusted substitution matrices. FEBS J 2005; 272:5101-5109 10.1111/j.1742-4658.2005.04945.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goevert D, Conrad R. Carbon isotope fractionation by sulfate-reducing bacteria using different pathways for the oxidation of acetate. Environ Sci Technol 2008; 42:7813-7817 10.1021/es800308z [DOI] [PubMed] [Google Scholar]
- 33.Spormann AM, Thauer RK. Anaerobic acetate oxidation to CO2 in Desulfotomaculum acetoxidans. Arch Microbiol 1988; 150:374-380 10.1007/BF00408310 [DOI] [Google Scholar]
- 34.Spring S, Lapidus A, Schroder M, Gleim D, Sims D, Meincke L, Glavina Del Rio T, Tice H, Copeland A, Cheng JF, et al. Complete genome sequence of Desulfotomaculum acetoxidans type strain (5575). Stand Genomic Sci 2009; 1:242-253 10.4056/sigs.39508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goorissen HP, Stams AJM, Hansen TA. Methanol dissimilation in Desulfotomaculum kuznetsovii PhD dissertation: Thermophilic methanol utilization by sulfate reducing bacteria 2002;Chapter 3:55-61. [Google Scholar]
- 36.Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, Richardson P, Brettin TS, Das A, Ljungdahl LG, et al. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ Microbiol 2008; 10:2550-2573 10.1111/j.1462-2920.2008.01679.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Meijden P, Heythuysen HJ, Pouwels A, Houwen F, van der Drift C, Vogels GD. Methyltransferases involved in methanol conversion by Methanosarcina barkeri. Arch Microbiol 1983; 134:238-242 10.1007/BF00407765 [DOI] [PubMed] [Google Scholar]
- 38.Imachi H, Sekiguchi Y, Kamagata Y, Hanada S, Ohashi A, Harada H. Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate-oxidizing bacterium. Int J Syst Evol Microbiol 2002; 52:1729-1735 10.1099/ijs.0.02212-0 [DOI] [PubMed] [Google Scholar]
- 39.Kosaka T, Kato S, Shimoyama T, Ishii S, Abe T, Watanabe K. The genome of Pelotomaculum thermopropionicum reveals niche-associated evolution in anaerobic microbiota. Genome Res 2008; 18:442-448 10.1101/gr.7136508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schut GJ, Adams MW. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol 2009; 191:4451-4457 10.1128/JB.01582-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller N, Worm P, Schink B, Stams AJM, Plugge CM. Syntrophic butyrate and propionate oxidation processes: from genomes to reaction mechanisms. Environmental Microbiology Reports 2010; 2:489-499 10.1111/j.1758-2229.2010.00147.x [DOI] [PubMed] [Google Scholar]
- 42.Kato S, Kosaka T, Watanabe K. Substrate-dependent transcriptomic shifts in Pelotomaculum thermopropionicum grown in syntrophic co-culture with Methanothermobacter thermautotrophicus. Microb Biotechnol 2009; 2:575-584 10.1111/j.1751-7915.2009.00102.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DTU. 2009 Center for biological sequence analysis: Technical University of Denmark. http://www.cbs.dtu.dk/services/TMHMM
- 44.Sanger Institute CU 2009. http://pfam.sanger.ac.uk/search
- 45.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinformatics 2005; 6:167 10.1186/1471-2105-6-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 1999; 288:911-940 10.1006/jmbi.1999.2700 [DOI] [PubMed] [Google Scholar]
- 47.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Gladyshev VN. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics 2005; 21:2580-2589 10.1093/bioinformatics/bti400 [DOI] [PubMed] [Google Scholar]
- 49.Roth JR, Lawrence JG, Bobik TA. Cobalamin (coenzyme B12): synthesis and biological significance. Annu Rev Microbiol 1996; 50:137-181 10.1146/annurev.micro.50.1.137 [DOI] [PubMed] [Google Scholar]
- 50.Roessner CA, Santander PJ, Scott AI. Multiple biosynthetic pathways for vitamin B12: variations on a central theme. Vitam Horm 2001; 61:267-297 10.1016/S0083-6729(01)61009-4 [DOI] [PubMed] [Google Scholar]
- 51.Fitzpatrick TB, Amrhein N, Kappes B, Macheroux P, Tews I, Raschle T. Two independent routes of de novo vitamin B6 biosynthesis: not that different after all. Biochem J 2007; 407:1-13 10.1042/BJ20070765 [DOI] [PubMed] [Google Scholar]
- 52.Bermingham A, Derrick JP. The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays 2002; 24:637-648 10.1002/bies.10114 [DOI] [PubMed] [Google Scholar]
- 53.Dosselaere F, Vanderleyden J. A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Crit Rev Microbiol 2001; 27:75-131 10.1080/20014091096710 [DOI] [PubMed] [Google Scholar]
- 54.Hu SI, Drake HL, Wood HG. Synthesis of acetyl coenzyme A from carbon monoxide, methyltetrahydrofolate, and coenzyme A by enzymes from Clostridium thermoaceticum. J Bacteriol 1982; 149:440-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stupperich E, Konle R. Corrinoid-Dependent Methyl Transfer Reactions Are Involved in Methanol and 3,4-Dimethoxybenzoate Metabolism by Sporomusa ovata. Appl Environ Microbiol 1993; 59:3110-3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levin I, Giladi M, Altman-Price N, Ortenberg R, Mevarech M. An alternative pathway for reduced folate biosynthesis in bacteria and halophilic archaea. Mol Microbiol 2004; 54:1307-1318 10.1111/j.1365-2958.2004.04339.x [DOI] [PubMed] [Google Scholar]