

Abstract
Population studies have suggested an association between diabetes and the symptoms of testosterone deficiency. Recently, the expression of the androgen receptor (AR) has been shown to be decreased in diabetic patients. Furthermore, diabetes has been shown to induce global methylation. In this study, we used an animal model to investigate whether diabetes results in increased methylation of the AR promoter and whether these changes are associated with the decreased expression of AR in penile cavernosal smooth muscle tissue. Twenty C57BL/6J mice were divided into two groups, receiving either high- (mature diabetic) or low- (mature control) caloric meals for 14 weeks. Another 10 mice were killed at 1 week (young control). Animals in the mature diabetic group showed decreased testosterone levels, although this was not statistically significant. In both control groups, no significant methylation was observed in the AR promoter region CpG island (−85 to +339). In the mature diabetic group, significant methylation was observed at +185 and +200 of the AR promoter. These changes were associated with increased homeostatic model assessment for insulin resistance (HOMA-IR) and decreased corpus cavernosal tissue mass and expression of AR mRNA and protein. We conclude that in these animals, insulin resistance increased the methylation of the GC-rich regions of the AR promoter, leading to decreased AR expression.
Keywords: androgen receptor (AR), diabetes, DNA methylation
Introduction
Patients with insulin resistance (IR) constitute a significant portion of patients with sexual dysfunction. Observational studies have shown a significant inverse relationship between the total serum testosterone levels and IR in men.1 Erectile dysfunction is more common in diabetic men, with a prevalence of as high as 85%.2,3 Phosphodiesterase type 5 inhibitors are effective in approximately 55%–60% of erectile dysfunction patients with diabetes.4 Testosterone replacement combined with phosphodiesterase type 5 inhibitors have shown variable but improved responses for the duration of treatment.5,6 Recent studies have suggested that this may be associated with the decreased expression of androgen receptors (AR) in diabetic men.7 The decreased expression may limit the effectiveness of testosterone replacement therapy in these patients.
Several mechanisms may interfere with the normal expression of AR, ranging from transient physiologic changes to overt genetic mutations.8,9 While no single factor currently seems to account for this process, recent studies have shown that epigenetic modifications during the lifetime of an organism may also have significant effects on the pathogenesis of decreased androgenic responsiveness. Monozygotic twin studies have shown that epigenetic profiles deviate following birth, leading to different phenotypes from the same genotype.10 Prolonged hyperglycaemic states have been shown to induce epigenetic changes in pro-inflammatory genes, leading to persistent risks of macrovascular complications in diabetics.11,12 Studies have shown that global DNA methylation is also correlated with increases in the indices of IR, such as the homeostatic model assessment for insulin resistance (HOMA-IR). This indicates some degree of causal association between hyperglycaemia and epigenetic modifications.13
The AR gene contains a CpG island that covers the proximal promoter region and the first exon.14 The methylation of the AR promoter at the CpG island has been correlated with the loss of AR mRNA expression in human AR-negative prostate cancer cell lines and tissues.14,15,16 The goal of this study is to examine AR promoter methylation in the cavernosal smooth muscle of C57BL/6J mice that had been induced to develop diabetes and to show whether these transformations translate to a subsequent decrease in AR expression.
Materials and methods
Animals, diets and experimental design
Thirty 4-week-old C57BL/6J mice were maintained at 24±2 °C with free access to food and drinking water and a 12-h light cycle (7 a.m. to 7 p.m.). They were divided into three groups of 10 mice each. After 1 week on a low-caloric diet (Rodent Diet with 10% kcal% fat, No. D12450B; Research Diets Inc., New Brunswick, NJ, USA), one group was killed immediately (young control group). The second group was started on a low-caloric diet (mature control group), while the third group was given a high-caloric diet (Rodent Diet with 60% kcal% fat, No. D12492; Research Diets Inc.) (mature diabetic group) for 14 weeks. The animals had ad libitum access to water and food during the experimental trial, and their food intake and weight gain were recorded daily. After 14 weeks, the mice were anaesthetized in the fasted state with ketamine (50 mg kg−1) and xylazine (6.7 mg kg−1). Blood was collected by cardiac puncture.
Tissue collection
The penis was excised by incising the skin overlying the penis and dissection at the bilateral penile crura. The urethra, the ischiocavernous muscle and fascia were dissected and removed. The glans, urethra and os penis were removed. The tunica albuginea was opened, and the corpus cavernosal tissue was harvested and weighed. The tissues were then washed in phosphate-buffered saline and frozen at −20 °C. Frozen sections were cut into 10-µm sections using a standard cryostat. Laser capture microdissection of smooth muscle cell bundles was performed using the Leica AS LMD (Leica Microsystems, Wetzlar, Germany) according to the manufacturer's instructions. Samples from a single animal were collected and lysed with 350 µl RP1 lysis buffer (Macherey-Nagel, Duren, Germany) and 3.5 µl β-mercaptoethanol. The samples were then centrifuged at 8900g for 1 min and homogenized with 350 µl ethanol. The samples were then used for reverse transcriptase polymerase chain reaction (RT-PCR), Western blot and sodium bisulphite modification for pyrosequencing. One sample from each animal was used for each process.
Serum insulin, testosterone and glucose
Serum insulin was measured with the mouse insulin ELISA kit (Linco Research Inc., St Charles, MO, USA), and testosterone was measured by mouse testosterone ELISA (Calbiotech, Spring Valley, CA, USA). Serum glucose was measured with an enzymatic kinetic assay for hexokinase (Roche, Basel, Switzerland). HOMA-IR was estimated by glucose (mmol l−1)×insulin (µIU ml−1)/22.5.
RNA isolation and RT-PCR
Total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). The RNA was extracted with chloroform (Sigma Chemical, St Louis, MO, USA) and precipitated with ethanol. The RNA was quantified using a NanoPhotometerTM (Implen GmbH, Munchen, Germany). For an RT reaction, 3 µg total RNA was used as a template to synthesize cDNA. The total RNA was mixed with 4 µg random hexamers (GeneChem, Shanghai, China), incubated at 65 °C for 10 min and cooled on ice for 2 min. The RT reaction was carried out in a final volume of 50 µl with 2 units of M-MLV RT (Invitrogen) at 42 °C for 2 h, followed by heating at 95 °C for 5 min to terminate the reaction. PCR was carried out in 20 µl reaction mixture containing 2 µl cDNA template, 25 pmol each of gene-specific primer and 10 µl Atlas HotTaq 2× PCR Mix (BioAtlas, Tartu, Estonia). A varying number of PCR cycles were performed at 95 °C for 60 s, 55 °C for 60 s and 72 °C for 60 s, followed by 72 °C for 5 min. The resultant PCR products were resolved on a 2% agarose gel containing ethidium bromide. The gel was scanned with Gel Logic 212 pro (Kodak, Rochester, NY, USA) on a UV illuminator, and the DNA bands were quantitated using Kodak MI software (Kodak, Rochester, NY, USA).
Western blot
The tissue harvested from microdissection was lysed and washed with 500 µl 50% ethanol, and 20–100 µl protein solving buffer (Macherey-Nagel, Duren, Germany) was added. The samples were then incubated for 3 min at 95–98 °C. The proteins were separated on an SDS–polyacrylamide gel. The proteins on the gel were transferred onto a nitrocellulose membrane. The membranes were incubated with blocking solution (5% skim milk), and then with rabbit anti-mouse AR antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) in 0.1% Tween 20-Tris-buffered saline. The hybridized primary antibodies were detected using a horseradish peroxidase-conjugated IgG antibody (Thermo Scientific, Waltham, MA, USA). The bands were visualized by enhanced chemiluminescence (Thermo Scientific). Quantitative analysis graphs were made using the TINA quantitative program.
Sodium bisulphite modification
Bisulphite modified gDNA was prepared using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. The bisulphite reaction was carried out on 500 ng DNA. The reaction volume was adjusted to 20 µl with sterile water, and 130 µl CT conversion reagent was added. The sample tubes were placed in a thermal cycler (MJ Research, St Bruno, Quebec, Canada) and incubated for 10 min at 98 °C and 2 h 30 min at 64 °C. The samples were then stored at 4 °C.
The DNA was purified using the reagent provided in the EZ DNA Methylation-Gold Kit (Zymo Research). The converted samples were added into the Zymo-Spin ICTM Columns containing 600 µl of the M-binding buffer and then mixed by inverting the column several times. The column was centrifuged at full speed (13 000g) for 30 s, and the flow-through was discarded. The column was washed by adding 200 µl M-wash buffer, centrifuged at full speed, and 200 µl M-desulphonation buffer was added to the column. The column was allowed to stand at room temperature (20–30 °C) for 15–20 min. After incubation, the column was centrifuged at full speed for 30 s. The column was washed by adding 200 µl M-wash buffer and centrifuged at full speed. The converted gDNA was eluted by adding 20 µl M-elution buffer into the column and centrifuging it. The DNA samples were stored at −20 °C.
Pyrosequencing analysis
We used the bisulphite pyrosequencing method for methylation analyses of the AR gene. Each primer was designed using the PSQ assay design program (Biotage, Charlotte, NC, USA). The primer sequences are listed in Table 1. Each PCR reaction was carried out in a volume of 20 µl with 50 ng converted gDNA, PCR premixture (Enzynomics, Daejon, Korea), 2 µl of 10 pmole µl−1 Primer-S and 2 µl of 10 pmole µl−1 biotinylated-Primer-As. The amplification was carried out according to the general guidelines suggested by pyrosequencing as follows: denaturation at 95 °C for 10 min, followed by 45 cycles at 95 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s and a final extension at 72 °C for 5 min. The PCR products (4 µl) were separated by electrophoresis on a 2% agarose gel and visualized by ethidium bromide staining.
Table 1. Primers used for RT-PCR analysis of AR mRNA and pyrosequencing of CpG islands.
| Gene | Primer (5′–3′) | Size (bp) | |
|---|---|---|---|
| AR mRNA | Forward | GGAGAACTCTTCAGAGCAAG | 495 |
| Reverse | AGCTGAGTCATCCTGATCTG | ||
| 28S rRNA | Forward | TTGAAAATCCGGGGGAGAG | 100 |
| Reverse | ACATTGTTCCAACATGCCAG | ||
| AR (pyrosequencing) | Forward | GGGTATTAGAGGGGAAAAGATTGAGTT | 212 |
| Biotinylated-reverse | ATCCTACCCAACACTTTCCTTACTTCC | ||
| Sequencing primer | GATAGTTAAAGTTTGTTGTAG | ||
Abbreviations: AR, androgen receptor; RT-PCR, reverse transcriptase polymerase chain reaction.
An ssDNA template was prepared from 15−20 µl biotinylated PCR product using streptavidin Sepharose HP beads (Amersham Biosciences, Piscataway, NJ, USA) according to the PSQ 96 sample preparation guide using multichannel pipets. Fifteen picomoles of the respective sequencing primers were added for analysis. Sequencing was performed on a PyroMark ID system with the Pyro Gold reagents kit (Biotage, Charlotte, NC, USA) according to the manufacturer's instructions without further optimisation.
Statistical analysis
The data are expressed as the mean±s.e.m. The differences between the groups were compared by t-test and one-way ANOVA, with P<0.05 considered to be statistically significant. Statistical analyses were performed with R (v 2.12.1. 2010-12-16, R Development Core Team 2010, Vienna, Austria).
Results
Confirmation of the diabetic animal model
Mice from the mature diabetic group showed the characteristics of insulin resistance, higher body weight and higher HOMA-IR compared to the mature control group. While there was a significant difference in overall body weight between the groups, the highest weight gain was observed in the mature diabetic group, and the corpus cavernosal tissue weight was significantly decreased in these mice compared to the mature control animals. There was no significant difference in testosterone levels between the groups (Table 2).
Table 2. Basic outcome measures.
| Young Control (n=10) | Mature Control (n=10) | Mature Diabetic (n=10) | P | |
|---|---|---|---|---|
| Weight (g) | 21.58±0.41 | 27.44±0.42 | 45.73±2.64 | <0.001 |
| Testis weight (g) | 1.3±0.1 | 1.4±0.1 | 1.2±0.1 | 0.377 |
| CC tissue weight (g) | 0.9±0.1 | 1.2±0.1 | 0.8±0.1 | 0.001 |
| Insulin (µU ml-1) | 34.87±16.80 | 8.30±1.95 | 196.71±53.32 | 0.001 |
| Glucose (mg dl-1) | 284.70±6.04 | 190.70±6.50 | 291.20±20.11 | <0.001 |
| HOMA-IR | 20.13±9.23 | 3.65±0.96 | 135.88±38.12 | 0.001 |
| Testosterone (ng ml-1) | 4.29±2.00 | 1.19±0.49 | 2.92±1.19 | 0.292 |
CpG methylation
Using the PSQ assay design program, we identified one island in the AR promoter, located at −85 to 339 bp, that was susceptible to epigenetic regulation. This region was surrounded by the consensus transcription start site, which contained 22 CpGs susceptible to methylation. In the analysis, a CpG island was considered only if it had a minimum length of 100 bp, a percentage of CpGs over 50% and a calculated-versus-expected CpG distribution higher than 0.6 of the abundance of CpG islands. The mean methylation of all susceptible positions was 1.53%±0.54%, 2.68%±1.25% and 5.70%±2.53%, for the young control, mature control and mature diabetic groups (P<0.01), respectively. Three sites, located at +185, +194 and +200 bp, showed significant differences in methylation between the groups (P=0.001, 0.043 and 0.030, respectively), and two sites, located at +185 and +200, showed more than 5% methylation. In the mature diabetic group, the AR promoter was more methylated at site +185 compared to the mature control and the young control groups (P=0.003 and 0.001, respectively). Another site, +200, showed higher methylation in the mature diabetic group, although this difference was not significant when compared to the young control group (P=0.065). The other sites did not show differences between the groups and were generally unmethylated (<5% methylated) (Figure 1). In addition, there was a statistically significant correlation between HOMA-IR and methylation at position +185 (Figure 2).
Figure 1.
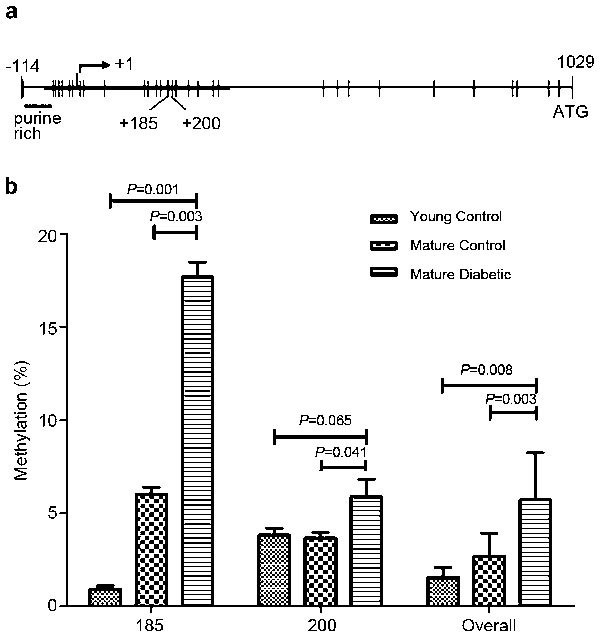
(a) A schematic representation of the 59 region of the mouse AR gene (X 36.0). The AR CpG island is depicted by the horizontal thick line extending across the transcription start site (right angle arrow at +1). Vertical tic represents CpG site. (b) The comparison of methylation between young, mature control and diabetic mice at sites +185, +200 and the overall methylation of 22 CpG sites. AR, androgen receptor.
Figure 2.
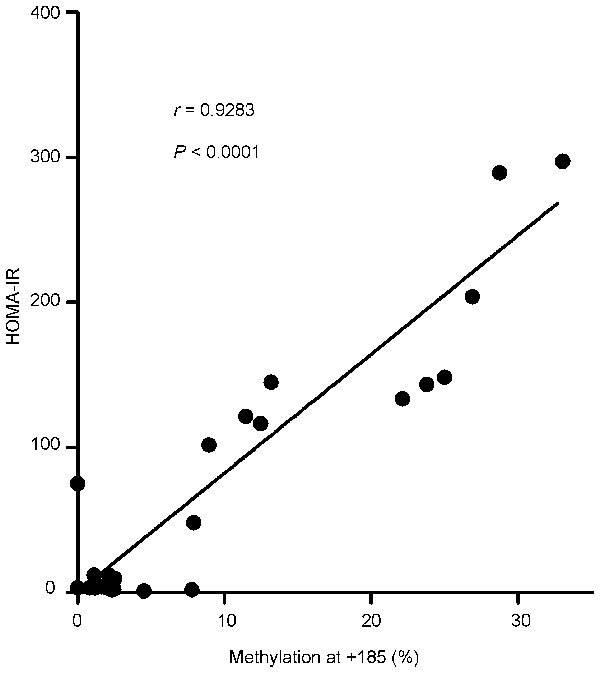
The correlation analysis of HOMA-IR and methylation at CpG site +185. The AR DNA methylation status at site +185 is highly correlated with HOMA-IR. AR, androgen receptor; HOMA-IR, homeostatic model assessment for insulin resistance.
AR mRNA and protein expression
AR mRNA expression, as normalized to the expression of 28S rRNA, was significantly decreased in mature diabetic mice. This was 0.68±0.04, 0.52±0.05 and 0.63±0.04 for the young control, mature control and mature diabetic groups, respectively (P=0.018 vs young control, P=0.045 vs mature control) (Figure 3). AR protein expression, as normalized to the expression of GAPDH, was also decreased. This was 0.43±0.32, 0.29±0.22 and 0.11±0.07 for the young control and matured control and mature diabetic groups, respectively (P=0.012 vs young control, P=0.041 vs mature control) (Figure 4).
Figure 3.
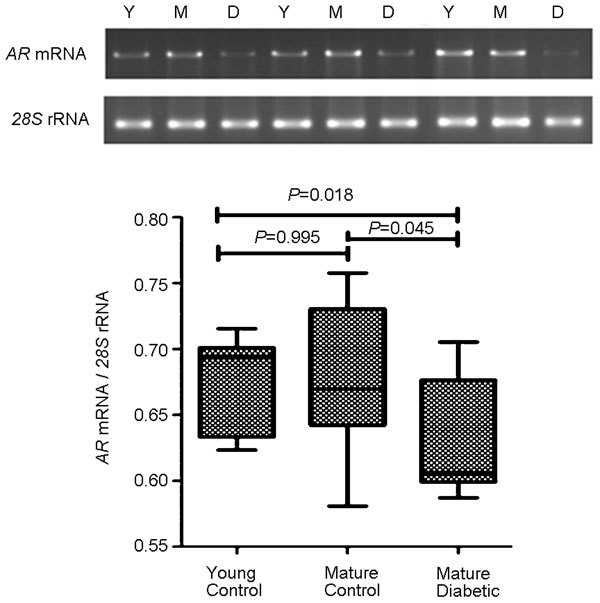
(a) The RT-PCR analysis of AR mRNA, as normalized to 28S rRNA expression. (b) The mature diabetic group showed significantly decreased mRNA transcription compared to the young control and mature control groups. AR, androgen receptor; RT-PCR, reverse transcriptase polymerase chain reaction.
Figure 4.
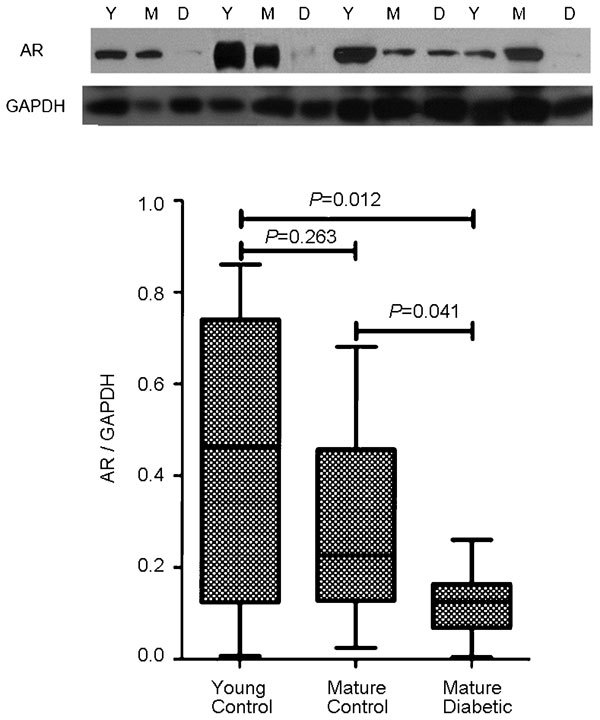
(a) Western blot detection of AR protein compared to GAPDH. (b) The mature diabetic group showed significantly decreased AR expression compared to the young control and mature control groups. AR, androgen receptor; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase.
Discussion
The current study reports AR promoter methylation pattern changes in mice with diabetes. The resulting methylation of CpG sites within the promoter DNA led to decreased mRNA and protein expression in these mice. Following a classical approach to study DNA methylation, we conclude that the AR promoter is slightly more methylated in mice with diabetes. This was especially the case at one position, +185 bp, where large differences in methylation proportions were observed. We also observed a high degree of correlation between HOMA-IR and AR promoter methylation at site +185.
A few previous studies have elucidated the functional characteristics of the promoter region of AR. The AR has been previously shown to contain a promoter located approximately 1000 bp upstream of the translation start site.17 A second promoter is located 13 bp away from this site.18 While both promoters lack TATA and CAAT boxes,17 these regions contain several potential transcription factor binding sites, including an activating protein 2 binding site, a purine-rich consensus sequence (PU box) and two guanine cytosine rich consensus sequences (GC box). The GC boxes are immediately upstream of the specificity protein 1 (Sp1) transcription factor binding site19 and Sp1- and GC-rich sequences are known to form a pre-initiation complex in promoters lacking TATA or CAAT.20
Methylation of the AR promoter at these GC-rich sequences has been correlated with the loss of AR mRNA expression in human AR-negative prostate cancer cell lines and tissues.14,15,16 Using AR-negative and AR-positive human prostate cancer cell lines, Kinoshita et al.21 showed that the methylation of such minimal promoter sites was associated with decreased AR expression in advanced prostate cancer. They reported significantly higher levels of methylation of the first promoter. However, the GC-rich sequence adjacent to the second promoter was not investigated. Our analysis covered both GC boxes, and we found significant methylation of the second GC box, which was associated with decreased AR mRNA and protein expression in diabetic mice. While it is premature to assign a functional significance to this particular site because the overall methylation was also increased in diabetic mice compared to controls, our observations of increased methylation and decreased expression of AR may be one of several processes accounting for the decreased erectile response and penile structural atrophy in diabetics.22
Because our study was not based on clonal cell colonies, the decreased penetrance of epigenetic changes observed is not unexpected. The level of methylation observed in our study is similar to the increased levels of global methylation that have been demonstrated in the insulin-resistant twin of a monozygotic pair.13 This may suggest that the methylation of the AR gene in diabetic mice may have occurred through the nonspecific methylation of susceptible sites, although other studies have suggested that Sp1 binding sites are generally kept methylation-free.23 While the processes of how these housekeeping activities occur remain to be elucidated, in this study, sufficient physiologic stress may have allowed significant methylation to occur.
Clinically, diabetes is associated with decreased sexual function. This has been associated with both decreased testosterone levels and the decreased responsiveness of the erectile tissue.24 While the association between testosterone deficiency and insulin resistance has been widely demonstrated in population studies, the strength of association is moderate, with only a 2–3 nmol l−1 (0.58–0.87 ng ml−1) decrease in total testosterone being associated with diabetes.25 The lack of a stronger association suggests that other mechanisms may alter the direct responsiveness of the target tissues. Cao et al.7 previously demonstrated decreased AR expression in the peripheral leukocytes of diabetic men. Our study suggests that the decreased expression of AR through the methylation of the AR promoter may decrease the erectile response, as suggested by the decreased corpus cavernosal tissue mass. While other measures of erectile function would have been more representative of functionality, practical considerations of preserving the tissue for methylation analysis impeded further investigation.
Finally, while conventional models would dictate an age-matched diabetic control using streptozotocin to exclude the effects of obesity, previous studies have shown that streptozotocin can induce DNA methylation. Thus, we excluded streptozotocin when designing this study, as shown by the lack of a young diabetic group.26
Recent progress in the understanding of testosterone and its receptor has shown that they also function in the overall balance of energy metabolism.27,28 Furthermore, the relationship between testosterone and AR has been shown to be complex and dynamic.9 Epigenetic mechanisms interfering with AR expression further complicate this relationship, and further investigation regarding their effects, reversibility and possible therapeutic potential is warranted. Studies elucidating the effects of testosterone replacement or identifying changes that may have confounded our results, including aging and obesity, may help to elucidate the effect of insulin resistance on AR promoter methylation.
Conclusion
The results of the present study indicate that insulin resistance caused by a high-calorie diet induces DNA methylation at the CpG islands of the AR promoter. Furthermore, methylation of these sites was shown to decrease the expression of AR mRNA and protein. Therefore, diabetes not only affects the hormonal levels of androgens, but may also disrupt receptor mechanics, resulting in the overall decrease of the androgenic phenotype.
Author contributions
JWK carried out the studies and drafted the manuscript. MMO performed the statistical analysis. CYY participated in the molecular genetic studies. JHB participated in the tissue harvesting and molecular studies. JJK and DGM designed the study and reviewed the manuscript. DGM is also the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Acknowledgments
This study was supported by a grant from Korea University.
The authors declare no competing financial interests.
References
- Stellato R, Feldman H, Hamdy O, Horton E, McKinlay JB. Testosterone, sex hormone-binding globulin, and the development of type 2 diabetes in middle-aged men: prospective results from the Massachusetts male aging study. Diabetes Care. 2000;23:490–4. doi: 10.2337/diacare.23.4.490. [DOI] [PubMed] [Google Scholar]
- Isidro ML. Sexual dysfunction in men with type 2 diabetes. Postgrad Med J. 2012;88:152–9. doi: 10.1136/postgradmedj-2011-130069. [DOI] [PubMed] [Google Scholar]
- Lu CC, Jiann BP, Sun CC, Lam HC, Chu CH, et al. Association of glycemic control with risk of erectile dysfunction in men with type 2 diabetes. J Sex Med. 2009;6:1719–28. doi: 10.1111/j.1743-6109.2009.01219.x. [DOI] [PubMed] [Google Scholar]
- Rendell MS, Rajfer J, Wicker PA, Smith MD. Sildenafil for treatment of erectile dysfunction in men with diabetes. JAMA. 1999;281:421–6. doi: 10.1001/jama.281.5.421. [DOI] [PubMed] [Google Scholar]
- Kalinchenko S, Kozlov G, Gontcharov N, Katsiya G. Oral testosterone undecanoate reverses erectile dysfunction associated with diabetes mellitus in patients failing on sildenafil citrate therapy alone. Aging Male. 2003;6:94–9. [PubMed] [Google Scholar]
- Jones TH, Arver S, Behre HM, Buvat J, Meuleman E, et al. Testosterone replacement in hypogonadal men with type 2 diabetes and/or metabolic syndrome (the TIMES2 study) Diabetes Care. 2011;34:828–37. doi: 10.2337/dc10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Li J, Hao W, Li X, Wang H, et al. Correlation of sex hormone and androgen receptor with diabetes mellitus in elderly men. Aging Male. 2011;14:162–7. doi: 10.3109/13685538.2011.575479. [DOI] [PubMed] [Google Scholar]
- Härkönen K, Huhtaniemi I, Mäkinen J, Hübler D, Irjala K, et al. The polymorphic androgen receptor gene CAG repeat, pituitary–testicular function and andropausal symptoms in ageing men. Int J Androl. 2003;26:187–94. doi: 10.1046/j.1365-2605.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010;42:813–27. doi: 10.1016/j.biocel.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Nat Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers J, MacMahon S, Patel A, Cooper M, Ferrannini E, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- Gerstein H, Miller M, Byington R, Goff D, Bigger J, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Goldberg J, Bremner JD, Vaccarino V. Global DNA Methylation Is Associated With Insulin Resistance. Diabetes. 2012;61:542–6. doi: 10.2337/db11-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, et al. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res. 1998;58:5310–4. [PubMed] [Google Scholar]
- Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. Prostate. 2001;47:66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Inaguma S, Sakakibara M, Cho YM, Suzuki S, et al. DNA methylation in the androgen receptor gene promoter region in rat prostate cancers. Prostate. 2002;52:82–8. doi: 10.1002/pros.10099. [DOI] [PubMed] [Google Scholar]
- Faber P, King A, van Rooij H, Brinkmann A, de Both N, et al. The mouse androgen receptor. Functional analysis of the protein and characterization of the gene. Biochem J. 1991;278:269–78. doi: 10.1042/bj2780269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann ME, Lindzey J, Blok L, Perry JE, Kumar MV, et al. The mouse androgen receptor gene contains a second functional promoter which is regulated by dihydrotestosterone. Biochemistry. 1994;33:14594–600. doi: 10.1021/bi00252a027. [DOI] [PubMed] [Google Scholar]
- Kumar MV, Jones EA, Grossmann ME, Blexrud MD, Tindall DJ. Identification and characterization of a suppressor element in the 5′-flanking region of the mouse androgen receptor gene. Nucleic Acids Res. 1994;22:3693–8. doi: 10.1093/nar/22.18.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courey AJ, Holtzman DA, Jackson SP, Tjian R. Synergistic activation by the glutamine-rich domains of human transcription factor Sp1. Cell. 1989;59:827–36. doi: 10.1016/0092-8674(89)90606-5. [DOI] [PubMed] [Google Scholar]
- Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, et al. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60:3623–30. [PubMed] [Google Scholar]
- Abidu-Figueiredo M, Ribeiro IC, Chagas MA, Cardoso LEM, Costa WS, et al. The penis in diabetes: structural analysis of connective tissue and smooth muscle alterations in a rabbit model. BJU Int. 2011;108:400–4. doi: 10.1111/j.1464-410X.2010.09944.x. [DOI] [PubMed] [Google Scholar]
- Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Gene Dev. 1994;8:2282–92. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- Traish AM, Saad F, Guay A. The dark side of testosterone deficiency: II. Type 2 diabetes and insulin resistance. J Androl. 2009;30:23–32. doi: 10.2164/jandrol.108.005751. [DOI] [PubMed] [Google Scholar]
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metabol. 2011;96:2341–53. doi: 10.1210/jc.2011-0118. [DOI] [PubMed] [Google Scholar]
- Murata M, Takahashi A, Saito I, Kawanishi S. Site-specific DNA methylation and apoptosis: induction by diabetogenic streptozotocin. Biochem Pharmacol. 1999;57:881–7. doi: 10.1016/s0006-2952(98)00370-0. [DOI] [PubMed] [Google Scholar]
- Haren M, Siddiqui A, Armbrecht H, Kevorkian R, Kim M, et al. Testosterone modulates gene expression pathways regulating nutrient accumulation, glucose metabolism and protein turnover in mouse skeletal muscle. Int J Androl. 2011;34:55–68. doi: 10.1111/j.1365-2605.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- Mutoh E, Senba K, Akieda-Asai S, Miyashita A, Poleni P, et al. Sex differences in energy metabolism in pre-pubescent, early pubescent and adult rats. Obes Res Clin Pract. 2011;5:e119–e28. doi: 10.1016/j.orcp.2010.12.006. [DOI] [PubMed] [Google Scholar]