

Abstract
To further explore the nature of the photo-Favorskii rearrangement and its commitment to substrate photorelease from p-hydroxyphenacyl (pHP), an array of ten new fluorinated pHP γ-aminobutyric acid (GABA) derivatives was synthesized and photolyzed. The effects of fluorine substitution on the chromophore and the photophysical and photochemical properties of these new chromophores were shown to be derived primarily from the changes in the ground state pKa of the phenolic groups. The quantum yields and rate constants for release are clustered around Φdis = 0.20 ± 0.05 and kr = 8 ± 7 × 107 s−1 (H2O), respectively. The triplet lifetimes of the pHP GABA derivatives were concentrated in the range of 0.4–6.0 ns (H2O). The corresponding deprotonated conjugate bases displayed reduced efficiencies by 50% or more (one exception) and exhibited a weak fluorescence in pH 8.2 buffer. Pump–probe spectroscopy studies have further defined the rates of intersystem crossing and the lifetimes of the reactive triplet state of the fluoro pHP chromophore.
Introduction
Substantial interest in the utility of photoremovable protecting groups (ppg’s) to control spatial and temporal release of substrates, chiefly those of biological interest, has arisen over the past two decades.1 Our group has focused, in particular, on the properties of ppg’s that control the rates of release, the quantum yields, and the changes in absorptivity during photorelease. The p-hydroxyphenacyl (pHP) moiety has consistently demonstrated efficient release of substrates such as neurotransmitters (GABA and Glu), oligopeptides (e.g., bradykinin), and nucleic acids (e.g., ATP and GTP).1,2 The photochemistry of pHP esters (see equation 1 in Fig. 1) is defined by facile substrate release and emergence of two major products, p-hydroxyphenylacetic acid and the leaving group, GABA. A “photo-Favorskii” mechanism has been postulated for substrate release from the pHP phototrigger, resulting in the formation of p-hydroxyphenylacetic acid.1 Requirements for clean, efficient photoconversions are the use of aqueous or mixed aqueous solvents and irradiation wavelengths λex < 350 nm. Prior reports1 have suggested that the quantum yields for release increased when electron-withdrawing groups were located meta to the pHP carbonyl. Fluorine, distinguished by its pronounced C–F bond polarity3 and enhanced lipophilicity in biological environments,4 affects the absorption and dispersion of its derivatives in vivo. Here, we report the synthesis of several fluorinated pHP derivatives (Fig. 1), address variations in quantum yields, Φ, and rate constants of GABA photorelease, kr, and compare these with the unsubstituted parent p-hydroxyphenacyl GABA’s (11) chemistry and photochemistry.5 The results confer mechanistic information regarding the photo-Favorskii rearrangement and the efficacy of fluoro substitution on the photochemistry.
Fig. 1.

Fluoro pHP GABA series.
Results
Ten fluorinated p-hydroxyphenacyl γ-aminobutyrates (pHP GABA esters 1–10) were synthesized from the commercially available fluorinated p-bromophenols (1a–10a). Reaction with benzyl bromide and potassium carbonate in CH3CN generated the benzyl-protected phenols (1b–10b), which were acety-lated under Stille conditions6 with Pd(PPh3)4 and tributyl(1-ethoxyvinyl)stannane to afford, after hydrolysis of the enol ether, 1c–10c. Subsequent treatment with dioxane-dibromide in CH2Cl2 at 0 °C over 2 h furnished 1d–10d, followed by SN2 substitution of the bromide with N-Boc-γ-aminobutyric acid using potassium carbonate in CH3CN over 24 h giving 1e–10e. Treatment with distilled TFA at room temperature for 24 h afforded 1–10 (Scheme 1). The pHP esters have good water solubility; for example, up to 11.8 mg (0.09 M) of 9 was readily soluble in 300 μl of H2O.
Scheme 1.
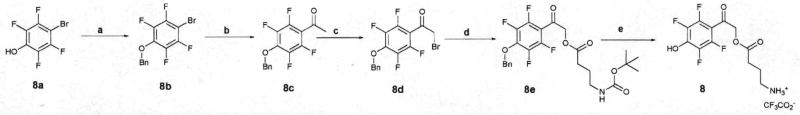
Synthesis of 8. (a) Benzyl bromide, K2CO2, CH3CN, 95%. (b) SnBu3 (1-ethoxyvinyl), Pd(Ph3)4, PhCH3, 100 °C, 85%. (c) Dioxane dibromide, CH2Cl2, 0 °C to rt, 93%. (d) N-Boc-γ-aminobutyric acid, K2CO3, CH3CN, 90%. (e) TFA, 0 °C to rt, 90%.
Physical and photophysical properties of fluoro pHP esters
The pka values of the fluoro pHP esters range from 7.2 for 2, to 3.9 for 8, and suggest that both the phenol and its conjugate base are present under the conditions for photolysis in aqueous, unbuffered media. A very weak fluorescence of neutral pHP in aqueous solution has been reported.7 For some of the pHP derivatives, a weak fluorescence was observed in unbuffered, aqueous media, which we assign to the conjugate base. In buffered media (pH 8.20) a bathochromic shift of the absorption maximum is accompanied by an increase in the fluorescence emission. A summary of pka, λmax (absorption wavelength maxima), and λem (emission wavelength maxima) is displayed in Table 1.
Table 1.
pka, λmax and λem for fluoro pHP esters
| pHP | R1 | R2 | R3 | R4 | λmax/nm (log ε/M−1 cm−1)a | λmax/nm (log ε/M−1 cm−1)b | λ/nmb | pKa |
|---|---|---|---|---|---|---|---|---|
| 1 | F | H | H | H | 274 (4.00), 335 (2.97) | 351 (3.67), 328 (3.96) | 395 | 6.5 |
| 2 | H | F | H | H | 271 (3.97) | 328 (3.90), 319 (3.91) | 382 | 7.2 |
| 3 | F | F | H | H | 272 (4.09), 328 (3.04) | 323 (4.07) | 390 | 5.9 |
| 4 | F | H | F | H | 278 (3.26), 326 (3.92) | 326 (4.12) | 385 | 5.7 |
| 5 | F | H | H | F | 331 (4.14) | 330 (4.14) | 399 | 5.3 |
| 6 | H | F | F | H | 274 (4.00) | 313 (4.06) | 374 | 6.8 |
| 7 | F | F | H | F | 324 (4.05) | 323 (4.01) | 392 | 4.5 |
| 8 | F | F | F | F | 316 (3.95) | 316 (4.10) | 384 | 3.9 |
| 9 | OCF3 | H | H | H | 274 (4.20), 331 (3.54) | 329 (4.18), 322 (4.18) | 395 | 6.7 |
| 10 | CF3 | H | H | H | 328 (4.11) | 328 (4.11) | 380 | 5.5 |
Determined in water.
Determined in 0.01 M HEPES, 0.1 M LiClO4, pH = 8.2. The quantum yields for fluorescence were not determined.
Photolysis experiments
Solutions of 1–10 (1–10 mM) in D2O were irradiated at 300 nm in an NMR tube and the reactions were monitored until complete (≤30 min). 1H NMR analysis confirmed the disappearance of the starting ester 1 and formation of two major photoproducts, p-hydroxyphenylacetic acid (1f) and GABA (12) along with a minor amount of p-hydroxybenzyl alcohol (1g) (Fig. 2). Identification of photoproduct 1f was achieved by spiking the photolysis mixture with an authentic sample of 2-(3-fluoro-4-hydroxyphenyl)acetic acid and with GABA (not shown). For the remaining pHP GABA esters 2–10, photolysis in D2O and analysis by 1H NMR of the corresponding product mixtures were compared with 1 and with the product mixture from the parent pHP GABA by 1H NMR chemical shifts or by spiking of authentic samples, when available. The minor product, p-hydroxybenzyl alcohol, 1g, was identified by comparison with the 1H NMR spectrum of unsubstituted p-hydroxybenzyl alcohol. All photoproducts were stable to photolysis conditions at prolonged irradiation. Even after 24 h of continuous irradiation, there was no significant change in the 1H NMR spectrum. It should be noted that the fluoro pHP derivatives proved to be resilient to hydrolysis under the same conditions in the dark over a 24 h period as indicated by the failure to observe disparate 1H NMR signals or changes in HPLC profiles.
Fig. 2.
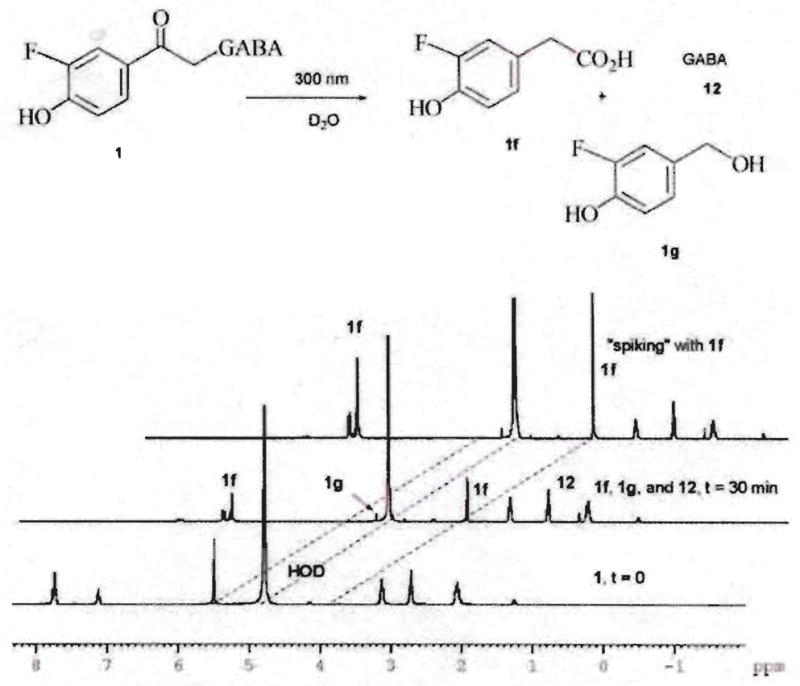
1H NMR analysis of the photolysis of 1 in D2O.
Quantum yield determinations
As shown for compound 1 in Fig. 2, the photoconversion of 1–10 is cleanly and efficiently achieved by irradiation at λ ~ 300 nm. The quantum yields for disappearance of fluoro pHP esters, Φdis, GABA appearance Φapp, and p-hydroxyphenylacetic acid appearance, Φacid, were determined in triplicate and light output measured by ferrioxalate actinometry.8 The p-hydroxybenzyl alcohol photoproduct, exiguously produced in aqueous media, was not quantified. The presence or absence of oxygen was found to be inconsequential, as an outgassed, oxygen-free solution caused little change in Φdis. Ester disappearance and product appearance from the photolyses of 1–10 in water and in buffered solutions (pH 7.3 and 8.2) are summarized in Table 2.
Table 2.
Quantum yield determination of fluoro pHP GABA’s
| Watera
|
pH 7.3b
|
pH 8.2c
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| pHP’s | Φdisd | Φappe | Φacidf | Φdis | Φapp | Φacid | Φdis | Φapp | Φacid |
| 1 | 0.16 ± 0.02 | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.12 ± 0.01 | 0.12 ± 0.01 | 0.11 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | < 0.01 |
| 2 | 0.28 ± 0.03 | 0.27 ± 0.03 | 0.26 ± 0.03 | 0.21 ± 0.02 | 0.20 ± 0.02 | 0.19 ± 0.02 | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.03 ± 0.02 |
| 3 | 0.24 ± 0.02 | 0.24 ± 0.02 | 0.22 ± 0.02 | 0.11 ± 0.01 | 0.11 ± 0.01 | 0.09 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 |
| 4 | 0.22 ± 0.02 | 0.21 ± 0.02 | 0.20 ± 0.02 | 0.10 ± 0.01 | 0.09 ± 0.01 | 0.09 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | < 0.01 |
| 5 | 0.11 ± 0.01 | 0.11 ± 0.01 | 0.10 ± 0.01 | 0.05 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | < 0.01 |
| 6 | 0.16 ± 0.02 | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.10 ± 0.01 | 0.09 ± 0.01 | 0.08 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.01 |
| 7 | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.04 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.01 | < 0.01 |
| 8 | 0.11 ± 0.01 | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.08 ± 0.01 | 0.09 ± 0.01 | 0.08 ± 0.01 | NDg |
| 9 | 0.09 ± 0.01 | 0.09 ± 0.01 | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | < 0.01 |
| 10 | 0.17 ± 0.02 | 0.16 ± 0.02 | 0.18 ± 0.02 | 0.12 ± 0.01 | 0.11 ± 0.01 | 0.10 ± 0.01 | 0.08 ± 0.01 | 0.08 ± 0.01 | NDg |
18 MΩ Ultrapure water.
0.01 M HEPES, 0.1 M LiClO4, pH = 7.3.
0.01 M HEPES, 0.01 M LiClO4, adjusted to pH = 8.2 with KOH.
Quantum yield for the disappearance of pHP ester.
Quantum yield for the appearance of GABA.
Quantum yield for the appearance of p-hydroxyphenylacetic acid.
ND = not determined.
The introduction of fluorine substituents did not significantly increase the quantum yields for the pHP esters in H2O over that of the parent pHP GABA (Φdis = 0.21) and in several cases resulted in lower quantum yields. One notable exception is the trifluoromethoxy pHP GABA. The quantum yield of GABA release from 9 is 0.09, twice that of the meta-methoxy analog (0.04).9 This increase may be due the reduced donating power of –OCF3 vs. –OCH3, an effect that we had reported earlier.1
Those pHP esters that have pKa greater than 5.5, i.e., 1–4, 6, and 10 are predominantly protonated in water and are characterized by λmax ~ 280 nm. This series had the higher release quantum yields of 0.16, 0.28, 0.24, 0.22, 0.16, and 0.20, respectively, whereas the esters that exist as their conjugate bases in H2O or in neutral pH, i.e., 5, 7, and 8 whose absorption maxima are shifted to >310 nm, have lower quantum yields of 0.11, 0.08, 0.11, respectively, i.e., a 50% reduction in Φdis. Interestingly, the pHP esters 2, 3, and 4 possessing only one fluorine in a meta position had higher quantum yields than the parent unsubstituted pHP GABA.
At pH 8.2, (HEPES buffer, Table 2), all but one of the fluorinated pHP esters gave lower efficiencies. The exception is 2 which is predominantly protonated and had a higher quantum yield, Φ = 0.21. The remaining ester series that were partially protonated in water, i.e., 1, 3, 4, 6 and 10, exhibited substantial ionization to their conjugate bases in this buffer and had lower quantum yields. The differences in the quantum yields cannot be ascribed solely to the degree of deprotonation, however, and may signal a small additional effect caused by this buffer. We observed larger variations in the quantum yields using other buffers.
Like 2, the unsubstituted parent pHP GABA is also fully protonated in both media and thus unchanged in efficiency in HEPES buffer vis-a-vis water at Φdis = 0.21 In exploring the factors that influence the efficacy of a phototrigger, the apparent trend between the pka of 1–10 and the release efficiencies strongly indicates that the quantum yields depend on the amount of ionization of the phenol to its conjugate base. The preponderance of conjugate base is seen for those derivatives that have pka’s of less than 7.
Mixed solvents also influence the quantum yields. For example, photolysis of 5, 7, and 8 in 75% CH3CN–25% H2O, where pHP is predominantly in its protonated form, the efficiencies are generally higher (0.16, 0.14, and 0.15, respectively), even higher than observed in pure aqueous solution runs.
Stern–Volmer quenching
Stern–Volmer quenching analyses performed with aqueous potassium sorbate solutions (0–0.1 M) of 1–10 (0.001 – 0.01 M) divulged a narrow range of the triplet lifetimes (Table 3). Estimated rate constants for release of GABA were obtained from the triplet lifetimes (3τ) which were derived from Stern–Volmer constants, Ksv, assuming diffusion-controlled quenching (kdiff = 7.4 × 109 M−1 s−1) and the quantum yields (i.e., Φrel/3τ) The rate constants spanned approximately an order of magnitude ranging from 1.6 × 108 s−1 for 2 to 2.0 × 107 s−1 for 9, signifying rapid substrate release for all fluoro pHP esters. In pH 7.30 buffer (Table 3), triplet lifetimes and release rates (107–108 s−1) for 1–10 were similar to those in water, confirming the rapidity of GABA release under buffered conditions.
Table 3.
Stern–Volmer quenching analyses of fluoro pHP GABA’s in H2O and 0.01 M HEPES, pH = 7.3
| Water
|
0.01 M HEPES pH = 7.3
|
|||||
|---|---|---|---|---|---|---|
| pHP’s | Ksv/M−1a | 3τ/nsb | kr/(107 s−1)c | KSV/M−1a | 3τ/nsb | kr/(107 s−1)c |
| 1 | 28 ± 2d | 3.8 ± 0.2 | 4.2 ± 0.2 | 23 ± 1 | 3.1 ± 0.3 | 3.6 ± 0.2 |
| 2 | 13 ± 0.2 | 1.9 ± 0.1 | 15.8 ± 0.1 | 19 ± 1 | 2.5 ± 0.1 | 8.4 ± 0.2 |
| 3 | 44 ± 2 | 6.0 ± 0.6 | 4.0 ± 0.4 | 39 ± 1 | 5.3 ± 0.4 | 2.1 ± 0.3 |
| 4 | 15 ± 0.8 | 2.1 ± 0.1 | 10.7 ± 0.1 | 51 ± 2 | 6.9 ± 0.2 | 1.6 ± 0.1 |
| 5 | 29 ± 0.8 | 4.0 ± 0.1 | 2.5 ± 0.1 | 32 ± 1 | 4.3 ± 0.1 | 1.1 ± 0.2 |
| 6 | 25 ± 0.5 | 3.3 ± 0.1 | 4.7 ± 0.1 | 8 ± 1 | 1.1 ± 0.1 | 8.9 ± 0.1 |
| 7 | 34 ± 2 | 4.6 ± 0.4 | 1.8 ± 0.1 | 36 ± 1 | 4.8 ± 0.2 | 1.3 ± 0.2 |
| 8 | 30 ± 0.5 | 4.1 ± 0.1 | 2.7 ± 0.1 | 14 ± 1 | 1.9 ± 0.3 | 5.5 ± 0.3 |
| 9 | 33 ± 0.7 | 4.4 ± 0.2 | 2.0 ± 0.1 | 29 ± 2 | 4.0 ± 0.3 | 1.4 ± 0.3 |
| 10 | 20 ± 2 | 2.7 ± 0.3 | 7.4 ± 0.7 | 30 ± 2 | 4.1 ± 0.5 | 3.0 ± 0.4 |
The Stern–Volmer constant, Ksv, was determined by the slope of the quantum yield ratios vs. concentration of potassium sorbate.
Triplet lifetime, 3τ, was estimated from KSV = kq3τ, where kq = kdiff = 7.4 × 109 M−1 s−1.
The release rate, kr, for GABA were estimated from kr = Φ/3τ.
Standard deviations.
Pump–probe spectroscopy
Triplet lifetimes of fluoro pHP esters were also derived from pump–probe optical spectroscopic data (Table 4). The pump wavelength was 266 nm.
Table 4.
LFP measured triplet lifetimes (3τ) and estimated kr’sa
| Water
|
10% H2O-CH3CN
|
||
|---|---|---|---|
| pHP’s | 3τ/ns | kr/(108 s−1) | 3τ/ns |
| 1 | ND | ND | 0.63 |
| 4 | 0.39 | 5.7 | 0.63 |
| 7 | 1.1 | 0.7 | 1.2 |
| 8 | ND | ND | 0.29 |
| 9 | 0.45 | 2.0 | 0.71 |
| 10 | 0.38 | 5.2 | 0.38 |
| 11 | 0.34 | 7.2 | ND |
kr values are estimated from kr = Φr/3τ (see Table 2).
Discussion
A photo-Favorskii mechanism has been proposed previously to explain the photochemical transformations that occur during the release of GABA from a pHP photoprotecting group1,2,10 (Scheme 2). Upon excitation, the pHP ester rapidly and efficiently intersystem crosses to the excited triplet state. Pump–probe studies have provided the rate of intersystem crossing, (kISC ~ 3 × 1011 s−1) with an efficiency near unity.
Scheme 2.

The photo-Favorskii mechanism.
Decay of the triplet and therefore the lifetimes obtained by LFP are shorter than those obtained by quenching with sorbate. The origin of this effect may be due to the nature of the quencher and the rate of diffusion assumed for the rate determinations. For example, it has been shown earlier by us10 and by Phillips7 that added organic solvents greatly increase the lifetime of the pHP acetate triplet. The diffusion rate constant may be an overestimate. The relative migrations of the two (the triplet of pHP GABA salt and the organic acid quencher) may not obey classical diffusion rates. A slower quenching diffusion rate will have a corresponding effect on the Stern–Volmer value which will consequently result in a lower estimated rate constant for release. As shown earlier by us11 and confirmed by Phillips,7,12 the triplet pHP ester is influenced by solvent H2O, primarily assisting in removal of the phenolic proton and also in the concomitant loss of the leaving group, GABA, through solvation with H2O.
Spin conservation requires that this process, which involves a heterolytic cleavage to liberate GABA as its zwitterion, lead to an oxyallyl-phenoxy triplet biradical. Recent studies revealed the signature absorption profile of this biradical intermediate through the appearance of three transitory bands at 320, 430, and 440 nm with a lifetime of 0.6 ns.10 The phenoxy-oxyallyl biradical triplet intersystem crosses to the singlet biradical followed by ring closure to form the putative spirodienedione 14 originally suggested by Anderson and Reese13 for the photo-Favorskii rearrangement.
The decrease in quantum yields observed when the photolyses are carried out in base suggests that the conjugate base of pHP esters may react by an alternative mechanism. The singlet slate of the conjugate base, although also rapidly intersystem crosses to the triplet state (kISC = 3 × 1011 s−1) exhibits a weak, but discernable blue fluorescence. This and the absence of a deprotonation step are clearly variations from the mechanism previously postulated for pHP esters. The lower efficiency is the consequence of the more efficient decay pathways available to the conjugate base. It is clear, however, that pHP esters do not ionize to their conjugate bases in their singlet excited states since they do not emit fluorescence on direct irradiation. Rapid proton loss might have been anticipated since it is known that triplet state of this chromophore has a significantly lower pka.11 Intersystem crossing, however, is too rapid (1011 s~−1) for deprotonation to be competitive.
A major feature of the photo-Favorskii pathway is a required out-of-plane rotation or twist of the substituted acetyl group possibly during the departure of the leaving group. This process in the triplet state would be tantamount to forming an orthogonal phenoxy-oxyallyl triplet biradical. The twisting motion was originally suggested by Philips7 in his initial report of the Time-Resolved Resonance Raman (TRR) spectra of pHP phosphate and acetate esters then abandoned in his later papers.7 To explore the effect of the out of plane motion of the oxyallyl group, we have computed the energies and the conformations of the series of fluorinated oxyallyl-phenoxy triplet biradicals using MP3 (6–31G* basis set). The phenoxy-oxyallyl triplet biradical lowest energy conformations are given in Table 5 and shown in Fig. 3 for 1, 8, 10, and 11. The planar phenoxy-oxyallyl biradical was also calculated and shown to be higher in energy (> 70 kJ mol−1) than the corresponding ground state spirodienedione intermediate 14. The singlet biradical had no barrier to closure and hence was not predicted to have a significant lifetime, decaying immediately to the spirodienone.10
Table 5.
Twist angles of 1. 8, 10 and the parent, 11, triplet biradicals and triplet biradical/singlet energy differences
S = singlet, T = triplet.
Out-of-plane twist.
Unsubstituted phenoxy-oxyallyl biradical.
Fig. 3.

Calculated lowest energy conformations of triplet biradicals and energy differences between triplet biradicals and the corresponding spirodienedione intermediates for (A) unsubstituted pHP (11), (B) meta-fluoro pHP (1), (C) meta-trifluoromethyl pHP (10), and (D) tetrafluoro pHP (8).
The biradicals are believed to be generated through out-of-plane rotation of the acyl group with rotations that range from 8.0 ° for 1 to 41.8 ° for 8. The large twist angle for 8 is intriguing in that it may suggest a potential link between extent of fluoro substitution, particularly at the ortho positions, and the stabilization of the oxyallyl-phenoxy biradical.
The predominant pathway for the decay of the spirodienedione 14 is hydrolysis with ring opening to the “Favorskii” product, p-hydroxyphenylacetic acid (1f–10f). A second pathway, decarbonylation, has recently been discovered10 through spectroscopic analyses and later by qualitative analysis of the reaction mixtures. This route produces a very modest yield, never more than 5%, of p-hydroxybenzyl alcohol from hydrolysis of p-quinone methide observed recently with our studies of pHP phosphates and tosylates.10
Conclusion
Several new fluoro pHP derivatives have been synthesized then employed in the caging and decaging of GABA. The mechanism of the photorelease reaction has been investigated for GABA release and the rearrangement of the chromophore. The fluoro pHP GABA’s displayed only a modest change in quantum yields of release (0.09–0.28) but maintained rapid release rates (107–108 s −1) when compared with unsubstituted pHP GABA. The conjugate bases of these phototriggers, however, had considerably lower quantum yields (0.03 to 0.12) while still maintaining fast substrate release rates. It is clear that the efficiency of GABA release and the degree of protonation (pKa) of pHP chromophore are interrelated. Fluorescence from neutral pHP GABA’s was not detected with a conventional spectrometer, whereas the conjugate bases do emit a weak, but measurable, fluorescence.
Experimental
Methods
Melting points were conducted with open ended capillary tubes using a non-calibrated Thomas-Hoover melting point apparatus. Lyophilization was performed using a Labconco FreezeZone device set at −52 °C and 0.04 mbar. Solution pH values were determined using a Fisher Science pH 510 meter calibrated with certified Fisher buffer solutions of pH 4, 7, and 10. Products of all reactions were assessed for purity using the following instrumental techniques: For 1H, 13C, and 19F NMR Bruker DRX400 and DRX 500 MHz spectrometers were utilized with trifluoroacetic acid as an internal standard (δ = −76.55 ppm).
GC-MS analysis was done on an Agilent 6890 N Network GC System equipped with a Quattro Micromass Triple Quadrupole Electron Impact mass spectrometer. Exact masses were performed on a Quattro Micromass Triple Quadrupole Electro Spray Ionization mass spectrometer. UV-Vis data were obtained on a Carey Bio 100 instrument using 1.5 ml quartz cuvettes. Fluorescence data were acquired on a Carey Eclipse Fluorescence Spectrophotometer using a 10 mm quartz cell. IR data were obtained using a Shimadzu FT-IR 8400 S instrument with pressed potassium bromide (KBr) pellets or a prefabricated sodium chloride (NaCl) chloroform cell for solid phase analysis, and NaCl prefabricated plates for oil analysis. Ground state pka’s were determined through sample titration with increasing amounts of 0.0461 M NaOH (standardized with potassium hydrogen phthalate), correlating pH vs. [OH−], and ascertaining the half equivalence point as the pKa. Product separation was achieved by flash chromatography using EM Science silica gel and gradient hexanes–ethyl acetate as eluting solvents. All reactions were run under ambient conditions unless otherwise stated. 1H NMR spectroscopy was utilized to monitor the progress of photolysis of all new pHP cages. In general, a 1–10 millimolar sample of pHP-caged GABA was prepared in 2 ml D2O, and approximately 1 ml was placed in an NMR tube. This was positioned in a photoreactor equipped with two 15 W, 3000 Å Rayonet lamps and a merry-go-round. Irradiation for 30 min generally led to 100% conversion as judged by the absence of the ester proton signals. Photoproduct identification was achieved through spiking the samples of the irradiated mixture with authentic samples of the resolved GABA.
Quantitative photolysis conditions for determination of quantum yields and Stern–Volmer quenching constants (kSV) were as follows: the lamp light output (in mEinstein min−1) was established using the potassium ferrioxalate method.7 Milligram quantities of caged compounds and caffeine or acetamidophenol were weighed out on a Fisher brand microbalance and dissolved in 4 ml of 18 MΩ ultrapure water, salt solutions of various concentrations, buffers with or without adjusted ionic strengths, or purified organic solvents were then added to a quartz tube and vortexed, resulting a homogenous solution of the caged compound and internal standard. Concentrations of the caged compounds ranged from 1–9 mM. These tubes were then placed in a carousel within a Rayonet Photochemical Reactor equipped with two 3000 Å, 15 W Rayonet Photochemical Reactor RPR3000 Mercury Lamps as light sources. 100 μl samples were removed at 30 s intervals up to 5 min. using a 250 μl Hamilton micro syringe and diluted to 1 ml with water using 1 ml volumetric flasks.
Picosecond pump–probe spectroscopy
Picosecond transient absorption was measured with the pump–supercontinuum probe technique using a Ti/Sa laser system (Clark MXR CPA-2001; 775 nm, pulse energy 0.9 mJ, full width at half maximum 150 fs, operating frequency 426 Hz). Part of the beam was fed into a Clark MXR NOPA. The output at 532 nm was frequency-doubled by a (β-barium borate (BBO) crystal to 266 nm and, after compression, provided pump pulses with an energy of 1 μJ and < 150 fs pulse width. A probe beam continuum was produced by focusing the 775 nm beam in front of a CaF2, of 4 mm path length. The pump and probe beams were focused to a 0.04 mm2 spot on the sample that was flowing in an optical cell of 0.3 mm thickness. The absorbance was in the range of 0.3–1, depending on the solubility of the compound. The probe beam and a reference signal (passing the solution besides the pump beam) were spectrally dispersed and registered with two photodiode arrays (512 pixels). Transient absorption spectra were calculated from ratio of the two beams. Spectra recorded with time delays ranging from 0 to 1.8 ns (to observe the triplet decay) were typically recorded with 20 ps steps, those ranging from 0–8 ps (to observe ISC) with 80 fs steps. The shorter measurements were corrected for chirp using a program (SPAN) kindly provided by Prof. N. Ernsting, Berlin. To improve the signal-to-noise ratio, the data were averaged over multiple pump–probe scans (3–6 scans with 400 shots per temporal point).
Quantum efficiency determinations
Quantitative analysis was achieved by HPLC-UV or HPLC-MS-MS. The LC-MS-MS instrument was a Waters 2695 Liquid Chromatographer equipped with a Quattro Micromass Triple Quadrupole Electro Spray Ionization Mass Spectrometer, outfitted with an autosampler. UV-Vis detection consisted of a Waters 2497 type with a dual wavelength detector set at 220 and 240 nm. The reservoirs used were as follows: (A) 99% water, 1% methanol, 10 mM ammonium formate and 0.06% formic acid. (B) 99% methanol, 1% water, 10 mM ammonium formate, and 0.06% formic acid. The column was a reverse-phase (C18), 4 μm mesh Altech Altima, 50 mm in length. Injections of 100 μl were made with an automated sampler for each run for a total of 3 injections per vial. A mobile phase gradient was utilized to optimize compound separation. The flow rate was set at 300 μl min−1. Data analysis was performed by Mass Lynx Ultima software and Microsoft Excel. Smoothing functions were used for peak analysis of the chromatographic peaks. Calibration curves to obtain R values from linear least-squares regression were determined at concentrations of the reactants and products in photolyses by systematic increases of pHP-caged GABA, free GABA, and p-hydroxyphenylacetic acid concentrations to determine correlations with internal standards caffeine or 4-acetamidophenol. The quantum efficiencies were then calculated from the ratio of the reactant or product concentrations to the photons absorbed using the actinometer values obtained as indicated above.
Lifetime and rate measurements
The Stern–Volmer quenching method14 was employed to determine the triplet lifetimes of pHP derivatives. Potassium sorbate served as the quenching agent. Solutions of pHP–GABA, 0.001–0.01 M, were diluted with sorbate solutions of increasing concentration (0–0.1 M), and photolyzed under the aforementioned conditions to ascertain the change in quantum efficiencies of GABA release. Φ0/Φ vs. [Q], and the Stern–Volmer constant, kSV, were determined. To determine the triplet lifetime, 3τ, the rate of quenching, kq, was assumed to be the rate of bimolecular diffusion, kdiff = 7.4 × 109 s−1 (water). The rates of photorelease, kr, were then computed as kr, = Φrel3τ.
Calculations
The atom coordinates for each molecule were introduced into SYBYL15 for molecular mechanics optimization using the Tripos Force Field.16 The ensuing structures withstood full geometric optimizations in Gaussian 0317 at the B3LYP18 level using the 6-31G* basis set19, soliciting the lowest energy triplet state in each case. The triplet stabilization energy was determined by comparing the final energy of optimized structure of each compound to that of the lowest energy singlet state at the triplet-optimized geometry. In order to examine differences in the carbonyl out-of-plane angles generated by the B3LYP calculation (vs. the MP2-level models reported elsewhere), a subset of these triplet-state species were geometrically optimized at the MP3 level20 in Gaussian using the 6-31G* basis set.
Materials
All starting materials were obtained from Aldrich or Matrix Scientific, unless otherwise indicated. Solvents were distilled prior to use, employing phosphorus pentoxide (P2O5) and stored in flame-dried, argon purged round-bottom flasks containing activated molecular sieves. Ultrapure (18 MΩ) water was used in all instances.
Syntheses
1-(Benzyloxy)-4-bromo-2-fluorobenzene, 1b
The general method of Frechét, et al.21 was followed. A solution of 4-bromo-2-fluorophenol (1a, 3.83 g, 20.0 mmol), benzyl bromide (2.38 ml, 20.0 mmol) and potassium carbonate (6.91 g, 50.0 mmol) in CH3CN (30 ml) was stirred at room temperature for 15 h. The solution was diluted with 50 ml CH2Cl2, washed with water (3 × 30 ml), dried (magnesium sulfate), and concentrated to give 5.24 g (93%) of 1-(benzyloxy)-4-bromo-2-fluorobenzene as a white precipitate: mp: 65–67 °C; IR (CHCl3): 3020, 2987, 2684, 2304, 1498, 1421, 1265, 1217, 1051, 896, 738, 695 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.41 (m, 5H), 7.27 (dd, J = 6.36, J = 2.28, 1H), 7.26 (dt, J = 4.37, J = 2.00, 1H), 6.89 (t, J = 8.76, 1H), 5.13 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 154.23, 151.73, 146.06, 136.02, 128.88, 127.73, 120.14, 117.23, 115.86, 113.02, 71.90; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −130.78. HRMS (EI): Calc’d for C13H10FBrO: 279.9899. Found: 279.9905.
1-(Benzyloxy)-3-fluorophenyl)ethanone, 1c22
The general method of Kosugi, et al.23 was used. Mp: 79–81 °C; IR (CHCl3): 3053, 2987, 1679, 1610, 1514, 1498, 1421, 1265, 1217, 1052, 896, 738, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ (ppm), 7.74 (dd, J = 11.33, J = 2.38, 1H), 7.69 (dd, J = 9.04, J = 1.59, 1H) 7.44, (m, 5H), 7.04 (t, J = 8.44, 1H), 5.23 (s, 2H), 2.55, (s, 3H). 13C NMR (125 MHz, CDCl3) δ 196.05, 153.66, 151.16, 135.82, 130.92, 128.95, 127.57, 125.76, 116.36, 114.22, 71.25, 26.52; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) −133.02. HRMS (M + H): Calc’d for C15H13FO2: 245.0978. Found: 245.0951.
1-(4-(Benzyloxy)-3-fluorophenyl)-2-bromoethanone, 1d
The general method of Paul, et al.24 was followed. A solution of 1c (700 mg, 2.86 mmol) in 20 ml CH2Cl2 was cooled to 0 °C in an ice bath. Dioxane dibromide (773 mg, 3.15 mmol) was added and the resulting mixture stirred for 30 min. The ice bath was removed with continued stirring for another 60 min. GC-MS indicated the reaction to be complete. The solution was washed with water and CH2Cl2, the aqueous layer removed, and the residual layer dried with anhydrous MgSO4, filtered, and evaporated under reduced pressure to yield an orange precipitate, 900 mg (2.58 mmol). Purification was cumbersome and unnecessary for completion of the supervening step, and hence was not pursued. IR (CHCl3): 3055, 2988, 1679, 1615, 1511, 1498, 1417, 1267, 1219, 1050, 891, 748, 696 cm−1; 1H NMR (400 MHz, CDCl3,) δ (ppm), 7.77 (dd, J= 10.00, J = 2.38, 1H), 7.72 (dd, J = 7.05, J = 2.78, 1H), 7.42 (m, 5H), 7.07 (t, J = 8.43, 1H), 5.24 (s, 2H), 4.37 (s, 2H);19F NMR (376 MHz, CDCl3+ a drop CF3CO2H) δ (ppm), −133.12.
2-(4-(Benzyloxy)-3-fluorophenyl)-2-oxoethyl-4-(tert-butoxycaronylamino)butanoate, 1e
The general method of Hiyama, et al.25 was utilized. A solution of 1-(4-(benzyloxy)-3-fluorophenyl)-2-bromoethanone, (1d, 900 mg, 2.78 mmol), potassium carbonate (1.15 mg, 8.35 mmol), 4-(tert-butoxycarbonylamino)butanoic acid (N-Boc-GABA, 678 mg, 3.33 mmol) in 50 ml of CH3CN was stirred for 24 h at room temperature. The solution was washed with EtOAc, water, the aqueous phase discarded, and the resulting phase evaporated under reduced pressure. Flash column chromatography (5:1 hexanes–EtOAc) afforded a white precipitate, 1.10 g (89%). Mp: 97–99 °C; IR (KBr): 3300, 3100–2800, 1742, 1701, 1600, 1517, 1437, 1400 1309, 1200, 1165, 953, 750, 700, 668 cm−1; 1H NMR (400 MHz, CD3CN) δ (ppm), 7.80 (dd, J = 7.05, J = 2.47, 1H), 7.78 (dd, J = 7.05, J = 1.99, 1H), 7.47 (dd, J = 4.45, J = 1.78, 1H), 7.42 (m, 5H), 5.31 (s, 2H), 5.25 (s, 2H), 2.97 (t, J = 6.79), 2.45 (t, J = 7.59 Hz, 2H), 1.76 (m, 2H), 1.40 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 190.67, 172.51, 155.60, 152.30, 150.80, 135.82, 128.39, 128.14, 127.68, 126.93, 125.60, 115.28, 114.82, 77.45, 70.36, 66.12, 39.64, 30.63, 28.22, 24.96; 19F NMR (376 MHz, CD3CN + drop CF3CO2H) δ (ppm), −133.44. HRMS (M + Na): Calc’d for C24H28FNO6Na: 468.1798. Found 468.1796.
4-(2-(3-Fluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 1f
The general method of Marsh, et al.26 was followed. In a flame-dried, 50 ml rb flask containing 2-(4-(benzyloxy)-3-fluorophenyl)-2-oxoethyl-4-(tert-butoxycarbonylamino)-butanoate, (1e, 850 mg, 1.91 mmol) was added 20 ml of freshly distilled TFA. Stirring continued for 24 h at room under ambient conditions. The solution was evaporated under reduced pressure and washed with EtOAc–water. The water layer was then extracted, frozen, then lyophilized, affording an adhesive precipitate, 550 mg (78%). IR (KBr): 3434, 3269, 3100–2800, 1741, 1693, 1610, 1553, 1420, 1201, 1050, 823, 759, 719 cm−1; 1H NMR (400 MHz, D2O) δ (ppm), 7.70 (d, J = 9.03, 2H), 7.12 (t, J = 9.94, 1H), 5.50 (s, 2H), 5.25 (s, 2H), 3.09 (t, J = 7.59, 2H), 2.66 (t, J = 7.08, 2H), 2.05 (m, 2H); 13C NMR (125 MHz, MeOD) δ (ppm), 190.05, 172.06, 161.60, 152.27, 151.07, 150.33, 126.03, 125.33, 117.30, 115.33, 65.95, 38.54, 30.02, 22.48. 19F NMR (376 MHz, D2O) δ (ppm), −133.58. HRMS (M+): Calc’d for C12H15FNO4: 256.0985. Found 256.0977.
The methodology for generating compounds 2–10 was analogous to that for 1 and gave comparable yields. Therefore, only spectroscopic data are included.
1-(4-Benzyloxy)-2-fluorophenyl)ethanone, 2c27
Same general method used as for 1d. 2b is commercially available. Mp: 97–99 °C; IR(CHCl3): 3056, 2992, 1701, 1616, 1510, 1487, 1415, 1275, 1207, 1055, 881, 728, 683 cm−1; 1H NMR (400 MHz, CDCl3) δ (ppm), δ 7.93 (t, J = 7.76, 1H), 7.40 (m, 5H), 6.85 (dd, J = 8.96, J = 2.49, 1H), 6.72 (dd, J = 13.25, J = 2.39, 1H), 5.15 (d, J = 4.98, 2H), 2.70 (s, 3H); 13C NMR (125 MHz, CDCl3) δ (ppm), 198.76, 165.25, 159.13, 135.54, 132.70, 128.78, 127.75, 118.13, 115.45, 114.03, 103.11, 70.96, 31.08; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −104.64. HRMS (M + H): Calc’d for C15H14FO2: 245.0978. Found: 245.0962.
1-(4-(Benzyloxy)-2-fluorophenyl)-2-bromoethanone, 2d
Oil. IR (NaCl): 3058, 2990, 1711, 1621, 1510, 1486, 1413, 1265, 1227, 1063, 890, 725, 680 cm−1; 1H NMR (400 MHz, CDCl3) δ (ppm), δ 7.95 (t, J = 8.84, 1H), 7.40 (m, 5H), 6.89 (dd, J = 8.84, J = 2.31, 1H), 6.73 (dd, J = 13.36, J = 2.50, 1H), 5.15 (s, 2H), 4.49 (d, J = 4.49, J = 2.50, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 189.31, 165.37, 159.17, 135.51, 132.68, 128.80, 127.75, 115.66, 115.45, 114.03, 102.99, 71.01, 35.73; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −104.68. HRMS (M + H): Calc’d for C15H13FO2Br: 323.0083. Found: 323.0101.
2-(4-(Benzyloxy)-2-fluorophenyl)-2-oxoethyl-4-(tert-butoxycarbonylamino)butanoate, 2e
Mp: 105–107 °C; IR (KBr): 3303, 3100–2800, 1742, 1701, 1614, 1437, 1408, 1309, 1203, 1167, 955, 747, 700, 668 cm−1; 1H NMR (400 MHz, d6-DMSO) δ (ppm), δ 7.85 (t, J = 8.94, 1H), 7.40 (m, 5H), 7.10 (dd, J = 13.59, J = 2.30, 1H), 7.01 (dd, J = 8.86, J = 2.42,1H), 6.88 (t, J = 5.70, 1H), 5.24 (s, 2H), 5.22 (d, J = 3.25, 2H), 2.98 (t, J = 6.81, 2H), 2.42 (t, J = 7.60, 2H), 1.70 (m, 2H), 1.38 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 188.97, 172.17, 164.26, 162.13, 155.60, 135.85, 131.75, 128.53, 127.96, 127.50, 115.05, 112.37, 102.82, 77.45, 70.04, 68.32, 39.30, 30.78, 28.22, 24.93; 19F NMR (376 MHz, CD3CN + drop CF3CO2H) δ (ppm), −106.01. HRMS (M + Na): Calc’d for C24H28FNO6Na: 468.1798. Found: 468.1787.
4-(2-(2-Fluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 2f
Adhesive solid. IR (KBr): 3434, 3244, 3100–2800, 1741, 1693, 1610, 1523, 1421, 1400, 1309, 1269, 1198, 1166, 1050, 824, 800, 760, 720 cm−1: 1H NMR (400 MHz, D2O) δ (ppm), δ 7.85 (t, J = 7.58, 2H), 6.73 (d, J = 9.09, 1H), 5.40 (d, J= 3.03, 2H), 3.10 (t, J = 7.64, 2H), 2.64 (t, J = 7.12, 2H), 2.09 (m, 2H); 13C NMR (125 MHz, D2O) δ (ppm), 189.35, 172.10, 165.34, 165.02, 163.13, 133.68, 113.65, 113.39, 102.58, 68.71, 38.60, 30.06, 22.48; 19F NMR (376 MHz,) δ (ppm), −106.45. HRMS (M+): Calc’d for C12H15FNO4: 256.0985. Found 256.0976.
1-(Benzyloxy)-4-bromo-2,3-difluorobenzene, 3b28
IR (CHCl3): 3058, 2982, 1600, 1510, 1465, 1250, 1202, 1050, 835, 740, 695 cm−1; 1H NMR (400 MHz, CDCl3) δ (ppm), 7.42 (m, 5H), 7.19 (dt, J = 8.05, J = 2.50, 1H), 6.90 (dt, J = 8.00, J = 1.82, 1H), 5.16 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 161.58, 149.74, 147.53, 135.60, 128.75, 127.50, 126.39, 117.38, 113.62, 101.35, 72.09. 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −129.51, −154.65. HRMS (EI): Calc’d for C13H9F2OBr: 297.9805. Found: 297.9822.
1-(4-(Benzyloxy)-2,3-difluorophenyl)ethanone, 3c
Mp: 96–98 °C; IR (KBr): 3055, 2990, 1695, 1614, 1464, 1448, 1380, 1275, 1202, 1051, 880, 725, 680 cm−1;1H NMR (400 MHz, CDCl3) δ (ppm), 7.69 (dt, J = 9.39, J = 2.24, 1H), 7.39 (m, 6H), 6.90 (dt, J = 8.00, J = 1.82, 1H), 5.26 (s, 2H), 2.71 (d, J = 5.38, 3H); 13C NMR (125 MHz, CDCl3) δ (ppm), 198.21, 160.44, 141.88, 140.59, 134.96, 128.75, 127.46, 118.93, 116.46, 113.62, 109.67, 71.67, 30.86; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −132.74, −158.39. HRMS(M + H): Calc’d for C15H13F2O2: 263.0884. Found: 263.0887.
1-(4-(Benzyloxy)-2,3-difluorophenyl)-2-bromoethanone, 3d
Oil. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.71 (dt, J = 8.44, J = 2.38, 1H), 7.41 (s, 5H), 6.92 (dt, J = 7.80, J = 1.90, 1H), 5.27 (s, 2H), 4.46 (d, J = 2.48, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 198.61, 160.37, 153.65, 140.38, 135.11, 128.93, 125.60, 117.97, 113.44, 109.93, 71.96, 31.02; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −132.93, −158.06.
2-(4-(Benzyloxy)-2,3-difluorophenyl)-2-oxoethyl-4-(tert-butoxycarbonylamino)butanoate, 3e
Mp: 115–117 °C; IR (KBr): 3300, 3100–2800, 1742, 1701, 1664, 1619, 1436,1406, 1200, 1169, 1136, 955, 749, 700, 670, cm−1; 1H NMR (400 MHz, d6DMSO) δ (ppm), 7.71 (dt, J = 8.38, J = 2.22, 1H), 7.41 (m, 5H), 7.29 (dt, J = 8.22, J = 1.57, 1H), 6.88, (t, J = 5.76, 1H), 5.34 (s, 2H), 5.25 (d, J= 2.82, 2H), 2.96 (t, J = 6.85, 2H), 2.44 (t, J = 7.55, 2H), 1.67 (m, 2H), 1.37 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 188.76, 172.13, 155.60, 152.08, 149.66, 142.12, 141.00, 139.04, 128.60, 128.02, 124.86, 116.21, 110.59, 72.91, 69.81, 69.72, 40.79, 32.09, 29.01, 26.49; 19F NMR (376 MHz, CD3CN + drop CF3CO2H) δ (ppm), −158.62, −133.05. HRMS (M + Na): Calc’d for C24H27F2NO6Na: 486.1704. Found: 486.1689.
4-(2-(2,3-Difluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 3f
Adhesive solid; IR (KBr): 3435, 3246, 3100–2800, 1739, 1694, 1610, 1525, 1420, 1400, 1310, 1269, 1200, 1165, 1049, 825, 800, 759, 720. cm−1; 1H NMR (400 MHz, D2O) δ (ppm), 7.43 (d, J = 6.57, 1H), 6.75 (d, J = 9.34, 1H), 5.18 (d, J = 2.64, 2H), 3.04 (t, J = 6.81, 2H), 2.60 (t, J = 7.53, 2H), 1.97 (m, 2H); 13C NMR (125 MHz, D2O) δ (ppm), 191.36, 173.96, 163.08, 151.57, 140.64, 138.70, 128.71, 119.79, 117.47, 113.20, 68.83, 38.34, 30.09, 21.61; 19F NMR (376 MHz, D2O) δ (ppm), −133.71, −162.58. HRMS (M+): Calc’d for C12H14FNO4: 274.0891. Found: 274.0884.
1-(Benzyloxy)-4-bromo-2,5-difluorobenzene, 4b
Mp: 58–60 °C. IR (CDCl3): 3002, 2943, 1618, 1508, 1419, 1406, 1375, 1334, 1188, 1170, 1039, 840,748, 702.1 H NMR (400 MHz, CDCl3) δ (ppm), 7.37 (m, 5H), 7.29 (dd, J = 6.64, J = 3.82, 1H), 6.82 (dd, J = 7.19, J = 2.68, 1H), 5.12 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 154.82, 150.45, 148.49, 147.28, 135.96, 129.35, 128.03, 120.47, 104.54, 98.93, 72.22; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −111.53, −137.66. HRMS (EI): Calc’d for C13H9F2BrO: 297.9805. Found: 297.9833.
1-(4-(Benzyloxy)-2,5-difluorophenyl)ethanone, 4c
Mp: 89–91 °C; IR (CDCl3): 3060, 3002, 2943, 1681, 1625, 1508, 1411, 1398, 1375, 1334, 1186, 1159, 1039, 744, 702.1H NMR (400 MHz, CD3CN) δ (ppm), 7.60 (dd, J = 6.88, J = 4.78, 1H), 7.43 (m, 5H), 7.00 (dd, J = 6.73, J = 5.93, 1H), 5.20 (s, 2H), 2.54 (d, J = 5.18, 3H); 13C NMR (125 MHz, CD3CN) δ (ppm), 194.65, 159.45, 158.50, 148.92, 136.38, 130.69, 129.43, 118.88, 117.28, 115.01, 104.46, 72.74, 31.59; 19F NMR (376 MHz, CD3CN + drop CF3CO2H) δ (ppm), −110.97, −140.38. HRMS (EI): Calc’d for C15H12F2O2: 262.0805. Found: 262.0791.
1-(4-(Benzyloxy)-2,5-difluorophenyl)-2-bromoethanone, 4d
Oil, IR (NaCl): 3055, 3008, 2942, 1691, 1624, 1511, 1406, 1395, 1357, 1332, 1181, 1150, 1039, 790,744,702. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.70 (dd, J = 6.69, J = 4.59, 1H), 7.43 (m, 5H), 6.78 (dd, J = 6.50, J = 5.73, 1H), 5.22 (s, 2H), 4.46 (d, J = 2.89, 3H).
2-(4-(Benzyloxy)-2,5-difluorophenyl)-2-oxoethyl-4-(tert-butoxycarbonylamino)butanoate, 4e
Mp 118–120 °C; IR (KBr): 3303, 3100–2800, 1741, 1701, 1657, 1623, 1512, 1437, 1407, 1203, 1165, 1144, 953, 897, 750, 699, 667, cm−1; 1H NMR (400 MHz, d6-DMSO) δ (ppm), 7.69 (dd, J = 6.66, J = 4.91, 1H), 7.43 (m, 6H), 6.89 (t, J = 5.62, 1H), 5.32 (d, 2H), 5.23 (d, J = 3.33, 2H), 2.97 (t, J = 6.79, 2H), 2.44 (t, 2H), 1.68 (m, 2H), 1.38 (s, 9H); 13CNMR(125MHz, d6-DMSO) δ (ppm), 188.50, 172.14, 159.73, 155.59, 151.91, 149.05, 147.12, 135.26, 128.48, 128.10, 115.36, 114.11, 103.96, 77.46, 70.96, 68.25, 38.97, 30.61, 28.22, 24.92; HRMS (M + Na): Calc’d for C24H27F2NO6Na: 486.1704. Found: 486.1702.
4-(2-(2,5-Difluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 4f
Adhesive solid. IR (KBr): 3440, 3246, 3100–2800, 1736, 1695, 1610, 1525, 1422, 1398, 1310, 1269, 1200, 1167, 1049, 825, 800, 760, 718. cm−1; 1H NMR (400 MHz, MeOD) δ (ppm), 7.62 (dd, J = 6.40, J = 4.73, 1H), 6.81 (d, J = 6.75, 1H), 5.26 (d, J = 3.54, 2H), 3.10 (t, J = 7.59, 2H), 2.67 (t, J = 7.08, 2H), 2.05 (m, 2H); 13C NMR (125 MHz, MeOD) δ (ppm), 188.62, 172.02, 160.69, 158.70, 152.48, 149.40, 147.48, 115.53, 113.04, 104.71, 68.77, 38.55, 29.99, 22.48; 19F NMR (376 MHz, MeOD) δ (ppm), −110.81, −141.90. HRMS (M+): Calc’d for C12H14NF2O4: 274.0891. Found: 274.0878.
2-(Benzyloxy)-5-bromo-1,3-difluorobenzene, 5b
Oil. IR (NaCl): 3091, 2948, 1595, 1581, 1494, 1454, 1382, 1220, 1190, 1037, 840, 750, 696. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.41, (m, 5H), 7.08 (d, J = 7.70, 2H), 5.17 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 155.74, 136.37, 134.91, 129.60, 118.28, 116.52, 113.03, 109.48, 66.85; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −125.93. HRMS (EI): Calc’d for C13H9F2BrO: 297.9805. Found: 297.9815.
1-(4-(Benzyloxy)-3,5-difluorophenyl)ethanone 5c
Oil. IR (NaCl): 3033, 2927, 1689, 1616, 1577, 1508, 1454, 1433, 1360, 1197, 1041, 975, 750, 696. 1H NMR (400 MHz, CD3CN) δ (ppm), 7.49 (dd, J = 9.24, 2H), 7.41 (m, 5H), 7.36 (d, 1H), 5.30 (s, 2H) 2.57 (s, 3H); 13C NMR (125 MHz, CD3CN) δ (ppm), 194.88, 156.50, 154.11, 135.99, 131.77, 128.39, 128.18, 113.32, 111.00, 75.48, 25.49; 19F NMR (376 MHz, CD3CN + drop CF3CO2H) δ (ppm), −127.25. HRMS (EI): Calc’d for C15H12F2O2: 262.0805. Found: 262.0816.
1-(4-(Benzyloxy)-3,5-difluoropheny1)-2-bromoethanone, 5d
Oil IR(NaCl): 3035, 2922, 1692, 1621, 1577, 1516, 1446, 1425, 1369, 1205, 1041, 965, 749, 696. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.55 (d, J = 6.54, 2H), 7.43 (m, 5H), 5.34 (s, 2H) 4.41 (s, 3H). 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −127.88.
2-(4-(Benzyloxy)-3,5-difluorophenyl)-2-oxoethyl-4-(tertbutoxycarbonylamino)butanoate, 5e
Adhesive solid. IR (KBr) 3302, 3230, 3100–2800, 1742, 1700, 1660, 1621, 1518, 1426, 1407, 1202, 1152, 951, 748, 698, 672 cm−1; 1H NMR (400 MHz, CD3CN) δ (ppm), 7.56 (d, J = 6.32, 2H), 7.43 (m, 5H), 5.32 (s, 2H), 5.08 (s, 2H), 3.09 (t, J = 6.83, 2H), 2.45 (t, J = 7.58, 2H), 1.76 (m, 2H), 1.40 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 193.45, 172.21, 160.02, 156.10, 151.72, 149.22, 146.32, 135.30, 128.76, 128.18, 115.54, 113.99, 105.01, 76.91, 69.95, 68.01, 39.30, 30.61, 28.20, 24.85;.19F NMR (376 MHz, CD3CN) δ (ppm), −127.82. HRMS (M + Na): Calc’d for C24H27F2NO6Na: 486.1704. Found: 486.1700.
4-(2-(3,5-Difluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium2,2,2-trifluoroacetate, 5f
Mp: 85–87 °C; IR (KBr): 3435, 3238, 3100–2800, 1740, 1695, 1608, 1524, 1420, 1399, 1310, 1270, 1200, 1165, 1050, 823, 800, 759, 720. cm−1; 1H NMR (400 MHz, MeOD) δ (ppm), 7.63 (d, J = 7.41, 2H), 5.39 (d, 2H), 3.07 (t, J = 7.33, 2H), 2.64 (t, J = 7.09, 2H), 2.05 (m, 2H); 13C NMR (125 MHz, MeOD) δ (ppm), 194.16, 175.94, 157.12, 155.17, 155.12, 144.00, 132.59, 128.13, 115.39, 69.88,42.47, 33.93, 26.42; 19F NMR (376 MHz, MeOD) δ (ppm), −133.62. HRMS (M+): Calc’d for C12H14F2NO4: 274.0891. Found: 274.0884.
1-(4-Benzyloxy)-2,6-difluorophenyl)ethanone, 6c
5-(Benzyloxy)-2-bromo-1,3-difluorobenzene, 6b, was generated by the same method as for Ib, determined to be pure by 1H NMR and GC-MS, and utilized in the synthesis of 6c. Oil. IR (NaCl): 3035, 2930, 1696, 1610, 1571, 1500, 1451, 1433, 1360, 1199, 1043, 978, 748, 695. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.39 (m, 5H), 6.62 (dt, J = 8.07, J = 5.08, 2H), 5.08 (s, 2H) 2.56 (dd, J = 1.85, 3H); 13C NMR (125MHz, CDCl3) δ (ppm), 193.78, 163.12, 162.20, 161.02, 134.26, 128.74, 127.74, 111.05, 99.54; 19FNMR (376 MHz, CD3CN +drop CF3CO2H) δ (ppm), −108.33. HRMS (EI): Calc’d for C15H12F2O2: 262.0805. Found: 262.0812.
1-(4-Benzyloxy)-2,6-difluorophenyl)-2-bromoethanone, 6d
Oil. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.45 (m, 5H), 6.61 (dt, J = 10.47, J = 5.32, 2H), 5.10 (s, 2H), 4.35 (s, 2H).
2-(4-(Benzyloxy)-2,6-difluorophenyl)-2-oxoethy1-4-(tert-butoxycarbonylamino)butanoate, 6e
Mp 69–71 °C; IR (KBr): 3304, 3100–2800, 1742, 1703, 1652, 1630, 1437, 1407, 1201, 1153, 1133, 1049, 953, 750, 699, 667, cm −1; 1H NMR (400 MHz, d6-DMSO) δ (ppm), 7.43 (m, 5H), 6.97 (dd, J = 11.14, J = 4.46, 1H), 6.89, (t, J = 5.95, 1H), 5.22 (s, 2H), 5.08 (s, 2H), 2.95 (t, J = 6.80, 2H), 2.41 (t, J = 7.59, 2H), 1.65 (m, 2H), 1.37 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 188.75, 172.12, 163.10, 162.68, 160.75, 155.59, 135.51, 128.88, 128.33, 128.06, 106.77, 106.49, 100.06, 76.33, 69.63, 67.69, 38.01, 29.40, 24.70; 19F NMR (376 MHz, d6-DMSO) δ (ppm), −108.41. HRMS (M + Na): Calc’d for C24H27F2NO6Na: 486.1704. Found: 486.1682.
4–2-(2,6-Difluoro-4-hydroxyphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 6f
Mp: 85–87 °C; IR (KBr): 3438, 3245, 3100–2800, 1741, 1694, 1610, 1525, 1420, 1400, 1310, 1268, 1200, 1165, 1050, 825, 800, 758, 720. cm−1; 1H NMR (400 MHz, MeOD) δ (ppm), 7.41 (d, 1H), 6.54 (d, J = 13.06, 2H), 5.10 (s, 2H), 3.09 (t, J = 7.59, 2H), 2.59 (t, J = 7.05, 2H), 2.00 (m, 2H); 13C NMR (125 MHz, D2O) δ (ppm), 188.59, 171.98, 163.70, 162.48, 161.56, 128.75, 117.38, 99.68, 68.98, 38.52, 30.07, 22.45; 19F NMR (376 MHz, D2O) δ (ppm), −109.55. HRMS (M+): Calc’d for C12H14F2NO4: 274.0891. Found: 274.08879.
2-(Benzyloxy)-5-bromo-1,3,4-trifluorobenzene, 7b
Oil. IR (NaCl): 3033, 2952, 1629, 1598, 1487, 1475, 1456, 1190, 1089, 1018, 821, 744, 696 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.38 (m, 5H), 7.11–7.09 (qd, J = 2.54, 1H), 5.22 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 152.73, 150.75, 135.53, 129.06, 128.65, 128.45, 115.38, 114.56, 113.11, 102.41, 98.93, 66.31; 19F NMR (376 MHz, CDCl3 + drop CF2CO2H) δ (ppm), −131.82, −133.66, −147.45. HRMS (EI): Calc’d for C13H8F3BrO: 315.9711. Found: 315.9712.
1-(4-(Benzyloxy)-2,3,5-trifluorophenyl)ethanone, 7c
Mp: 45–47 °C; IR (CDCl3): 3002, 2943, 1691, 1627, 1500, 1375, 1346, 1081, 1039, 918, 750, 702 cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.45 (m, 6H), 5.37 (s, 2H), 2.68 (s, 3H);13C NMR (125 MHz, CDCl3) δ (ppm), 194.74, 152.20, 150.23, 147.94, 143.62, 135.58, 128.77, 119.68, 118.10, 115.83, 113.56, 66.45, 31.39; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −131.10, −136.69, −149.56. HRMS (EI): Calc’d for C15H11F3O2: 280.0711. Found: 280.0717.
1-(4-(Benzyloxy)-2,3,5-trifluoropheny1)-2-bromoethanone, 7d
IR(CDCl3): 3000, 2945, 1698, 1624, 1502, 1369, 1352, 1081, 1045, 921, 750, 699. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.53–7.51 (qd, J = 2.30, 1H), 7.40 (m, 5H), 5.25 (d, 2H), 4.45 (s, 2H).
2-(4-(Benzyloxy)-2,3,5-trifluoropheny1)-2-oxoethyl-4-(tert-butoxycarbonylamino)butanoate, 7e
Mp: 110–112 °C; IR (KBr) 3302, 3100–2800, 1743, 1703, 1651, 1629, 1437, 1407, 1202, 1170, 1147, 950, 748, 698, 667 cm−1; 1H NMR (400 MHz, d6-DMSO) δ (ppm), 7.62–7.60 (qd, J = 2.30, 1H), 7.41 (m, 5H), 6.88, (t, J = 6.00, 1H), 5.40 (s, 2H), 5.26 (s, 2H), 2.96 (t, J = 6.82, 2H), 2.44 (t, J = 7.60, 2H), 1.67 (m, 2H), 1.38 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 188.033, 172.09, 155.60, 151.48, 149.53, 148.47, 146.44, 144.63, 142.83, 140.05, 128.55, 116.87, 110.58, 77.46, 75.71, 68.22, 30.53, 28.21, 24.89; HRMS (M + Na): Calc’d for C24H26F3NO6Na: 504.1610. Found: 504.1593.
4-Oxo-4-(2-oxo-2-(2,3,5-trifluoro-4-hydroxyphenyl) ethoxy) butan-1-aminium 2,2,2-trifluoroacetate, 7f
Mp: 131–133 °C; IR (KBr): 3434, 3244, 3100–2800, 1738, 1695, 1610, 1523, 1420, 1400, 1309, 1267, 1199, 1166, 1049, 824, 800, 759, 720 cm−1; 1H NMR (400 MHz, D2O) δ (ppm), 7.52–7.50 (qd, J = 2.35, 1H), 5.37 (d, 2H), 3.14 (t, J = 7.06, 2H), 2.72 (t, J = 6.71, 2H), 2.07 (m, 2H); 13C NMR (125 MHz, D2O) δ (ppm), 190.89, 174.05, 163.12, 149.21, 147.28, 141.03, 139.81, 117.19, 115.16, 109.89, 69.88, 42.47, 33.93, 26.42; 19F NMR (376 MHz, D2O) δ (ppm), −136.48, −137.81, −156.79. HRMS (M+): Calc’d for C12H13F3NO4: 292.0797. Found: 292.0794.
1-(Benzyloxy)-4-bromo-2,3,5,6-tetrafluorobenzene, 8b
Mp: 43–45 °C; IR (CDCl3): 3002, 2943, 1560, 1458, 1442, 1407, 1380, 1039, 918, 750. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.39 (m, 5H), 5.28 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm), 146.18, 144.22, 144.11, 140.70, 135.25, 129.04, 128.72, 93.26, 76.51; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −131.37, −155.12. HRMS (EI): Calc’d for C13H7F4BrO: 333.9616. Found: 333.9606.
1-(4-(Benzyloxy)-2,3,5,6-tetrafluorophenyl)ethanone, 8c
A solution of 8b (1.00 g 2.98 mmol), tributyl(1-ethoxyvinyl)stannane (1.44 ml, 4.27 mmol) and 25 ml toluene was degassed for 30 min while stirring. Tetrakistriphenylphosphine palladium(0) (205 mg, 0.178 mmol) was then added and the solution heated to 100 °C overnight with stirring. Fifty ml of 50% HCl (aq) were added and the solution stirred for 6 h, followed by filtration through Celite that effectively removed the palladium black. The resulting toluene filtrate was charged with 50 ml of saturated aqueous potassium fluoride and continuously stirred for 12 h. The insoluble tributylflourostannane was then filtered, the aqueous layer removed, and the organic layer concentrated. Flash column chromatography (6: 1 Hexanes-Et2O) afforded l-(4-(benzyloxy)-2,3,5,6-tetrafluorophenyl)ethanone, 8d in 85% yield (755 mg) as a faint yellow solid. Mp: 47–49 °C; IR (CDCl3): 2921, 2852, 1712, 1643, 1461, 1377, 1190, 1100, 742, 723, 696 cm−1. 1HNMR (400 MHz, CDCl3) δ (ppm), 7.38 (m, 5H), 5.27 (s, 2H), 2.59 (t, J = 1.90, 3H);13C NMR (125 MHz, CDCl3) δ (ppm), 192.07, 146.39, 143.95, 142.52, 139.89, 135.28, 129.27, 128.46, 66.07, 32.70; 19F NMR (376 MHz, CDCl3 + drop CF3CO2H) δ (ppm), −144.22, −161.88. HRMS (M + H): Calc’d for C15H11F4O2: 299.0695. Found: 299.0702.
1-4-(Benzyloxy)-2,3,5,6-tetrafluorophenyl)-2-bromoethanone, 8d
IR (CDCl3): 2919, 2856, 1721, 1645, 1453, 1379, 1190, 1107, 745, 715, 696. 1H NMR (400 MHz, CDCl3) δ (ppm), 7.40 (m, 5H), 5.35 (s, 2H), 4.32 (t, J = 1.20, 2H).
2-(4-(Benzyloxy)-2,3,5,6-tetrafluorophenyl)-2-oxoethy1-4-(tert-butoxycarbonylamino)butanoate, 8e
Mp: 106–108 °C; IR (KBr) 3301, 3100–2800, 1744, 1703, 1655, 1615, 1487, 1437, 1407, 1210, 1153, 1050, 749, 700, 667 cm−1; 1H NMR (400 MHz, d6-DMSO) δ (ppm), 7.42 (m, 5H), 6.88, (t, J = 5.92, 1H), 5.43 (s, 2H), 5.02 (t, J = 1.43, 2H), 2.93 (t, J = 6.74, 2H), 2.41 (t, J = 7.57, 2H), 1.65 (m, 2H), 1.37 (s, 9H); 13C NMR (125 MHz, d6-DMSO) δ (ppm), 188.04, 172.14, 155.59, 145.97, 143.96, 141.32, 139.47, 135.39, 128.64, 128.39, 109.10, 77.46, 76.14, 68.81, 30.39, 28.82, 24.82; HRMS (M + Na): Calc’d for C24H25F4NO6Na: 522.1516. Found: 522.1493.
4-Oxo-4-(2-oxo-2-(2,3,5,6-tetrafluoro-4-hydroxyphenyl) ethoxy)butan-1-aminium2,2,2-trifluoroacetate, 8f
Mp: 107–109 °C; IR (KBr): 3440, 3241, 3100–2800, 1735, 1690, 1610, 1525, 1420, 1400, 1310, 1265, 1200, 1166, 1050, 824, 800, 759, 720. cm−1; 1H NMR (400 MHz, D2O) δ (ppm), 5.25 (t, J = 1.67, 2H), 3.07 (t, J = 7.61, 2H), 2.66 (t, J = 7.06, 2H), 2.01 (m, 2H); 13C NMR (125 MHz, D2O) δ (ppm), 189.10, 173.25, 162.36, 147.43, 139.18, 129.31, 117.78, 113.12, 104.46, 69.40, 38.59, 30.13, 22.22; 19F NMR (376 MHz, D2O) δ (ppm), −142.76, −163.05. HRMS (M+): Calc’d for C12H12F4NO4: 310.0702. Found: 310.0701.
4-(2-(4-Hydroxy-3-trifluoromethoxyphenyl-2-oxoethoxy)-4-oxobutan-1-aminium-2,2,2-trifluoroacetate, 9f
The synthesis of this will be reported in a future publication
4-(2-(4-Hydroxy-3-trifluoromethylphenyl)-2-oxoethoxy)-4-oxobutan-1-aminium 2,2,2-trifluoroacetate, 10f
The synthesis of this will be reported in a future publication.
Acknowledgments
We thank the NIH (RO1GM72910, P50-GMO69663) for financial support. We thank Dr Gerry Lushington for molecular modelling of the pHP derivatives and Dr Todd Williams for assistance with HPLC and MS determinations.
Contributor Information
Jakob Wirz, Email: J.Wirz@unibas.ch.
Richard S. Givens, Email: givensr@ku.edu.
Notes and references
- 1.Goeldner MG, Givens R, editors. Dynamic studies in Biology, Phototriggers, Photoswitches, and Caged Compounds. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.Park CH, Givens RS. New Photoactivated Protecting Groups. 6. p-Hydroxyphenacyl: A New Phototrigger for Chemical and Biological Probes. J Am Chem Soc. 1997;119:2453–2463. [Google Scholar]
- 3.Lemal DM. Perspective on Fluorocarbon Chemistry. J Org Chem. 2004;69:1–11. doi: 10.1021/jo0302556. [DOI] [PubMed] [Google Scholar]
- 4.Jeschke P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. Chem Bio Chem. 2004;5:570–589. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- 5.Conrad PG, II, Givens RS, Weber JFW, Kandler K. New Phototriggers 10: Extending the π, π*. Absorption to Release Peptides in Biological Media. Org Lett. 2000;2:1545–1547. doi: 10.1021/ol005856n. [DOI] [PubMed] [Google Scholar]
- 6.Stille JK. The Palladium Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Angew Chem, Int Ed Engl. 1986;25:508–524. [Google Scholar]
- 7.Ma C, Chan WS, Kwok WM, Zuo P, Phillips DL. Time-Resolved Resonance Raman Study of the Triplet State of the p-Hydroxyphenacyl Acetate Model Phototrigger Compound. J Phys Chem B. 2004;108:9264–9276. doi: 10.1021/jo049331a. and subsequent papers. [DOI] [PubMed] [Google Scholar]
- 8.Hatchard CG, Parker CA. A New Sensitive Chemical Actinometer. II. Potassium, Ferrioxalate as a Standard Chemical Actinometer. Proc R Soc London, Ser A. 1956;235:518–536. [Google Scholar]
- 9.Conrad PG, II, Givens RS, Weber JFW, Kandler K. New Phototriggers: Extending the p-Hydroxyphenacyl π–π*Absorption Range. Org Lett. 2000;2(11):1545–1547. doi: 10.1021/ol005856n. [DOI] [PubMed] [Google Scholar]
- 10.Givens RS, Heger D, Kamdzhilov Y, Mac M, Cope E, Lee JI, Wirz J. The Photo-Favorskii Reaction of p-Hydroxyphenacyl Compounds is Initiated by Water-Assisted, Adiabatic Extrusion of a Triplet Biradical. J Am Chem Soc. 2008;130:3307–3309. doi: 10.1021/ja7109579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conrad PG, Givens RS, Hellrung B, Rajesh CS, Ramseier M, Wirz J. P-Hydroxyphenacyl Phototriggers: The Reactive Excited State of Phosphate Photorelease. J Am Chem Soc. 2000;122:9346–9348. [Google Scholar]
- 12.Zuo P, Ma C, Kwok WM, Chan WS, Phillips DL. Time Resolved Resonance Raman and Density Functional Theory Study of the Deprotonation Reaction of the Triplet State of p-Hydroxyacetophenone in Water Solution. J Org Chem. 2005;70:8661–8675. doi: 10.1021/jo050761q. [DOI] [PubMed] [Google Scholar]
- 13.Anderson JC, Reese CB. Tetrahedron Lett. 1962;1:1–4. [Google Scholar]
- 14.Desilets DJ, Kissinger PT, Lytle FE. Improved Method for Determination of Stern-Volmer Quenching Constants. Anal Chem. 1987;59:1244–1246. [Google Scholar]
- 15.SYBYL 7. 2. The Tripos Associates; St. Louis, MO: 2007. [Google Scholar]
- 16.Clark M, Cramer RD, Van Opdenbosch N. Validation of the General Purpose Tripos 5.2 Force Field. J Comput Chem. 1989;10:982–1012. [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski J, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03 (Revision C.02) Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 18.Becke AD. Density-Functional Thermochemistry III. The, Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 19.Ditchfield R, Hehre WJ, Pople JA. Self-Consistent Molecular-Orbital Molecules IX. Extended, Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J Chem Phys. 1971;54:724–728. [Google Scholar]
- 20.Pople JA, Seeger R, Krishan R. Variational Configuration Interaction Methods and Comparisons with Perturbation Theory. Int J Quant Chem Symp. 1977;11:149–163. [Google Scholar]
- 21.Hawker CJ, Lee R, Frechet JM. One Step Synthesis of Hyperbranched Dendridic Polyesters. J Am Chem Soc. 1991;113:4583–4588. [Google Scholar]
- 22.Machin PJ, Hurst DN, Bradshaw RM, Blaber LC, Burden DT, Fryer AD, Melarange RA, Shivdasani C. (β1-Selective Adrenoceptor Antagonists 2. 4-Ether-Linked Phenoxypro-panolamines. J Med Chem. 1983;26:1570–1576. doi: 10.1021/jm00365a005. [DOI] [PubMed] [Google Scholar]
- 23.Kosugi M, Sumiya T, Obara Y, Suzuki M, Sano H, Migita T. (α-Ethoxyvinyl) tributyltin; an efficient reagent for the nucleophilic acetylation of organic halides via palladium catalysis. Bull Chem Soc Jpn. 1987;60:767–768. [Google Scholar]
- 24.Paul S, Gupta V, Gupta R, Loupy A. Microwave-Induced Selective Synthesis of α-Bromo and α, α-Dibromoalkanones Using Dioxane-Dibromide and Silica Gel under Solvent Free Conditions. Tetrahedron Lett. 2003;44:439–442. [Google Scholar]
- 25.Hiyama T, Fujita M. Fluoride Ion Catalyzed of Aldehydes and Ketones with Hydrosilanes. Synthetic and Mechanistic Aspects and an Application to the Threo-Directed Reduction of α-Substituted Alkanoncs. J Org Chem. 1988;53:5405–5415. [Google Scholar]
- 26.Marsh JP, Goodman L. Removal of O-Benzyl Blocking Groups with Trifluoroacetic Acid. J Org Chem. 1965;30:2491–2492. [Google Scholar]
- 27.Dyck B, Zhao L, Tamiya J, Pontillo J, Hudson S, Ching B, Heise CE, Wen J, Norton C, Madan A, Schwartz D, Wade W, Goodfellow VS. Substituted Chromones and Quinolines as Potent Melanin-Concentrating Hormone Receptor 1 Antagonists. Bioorg Med Chem Lett. 2006;16:4237–4242. doi: 10.1016/j.bmcl.2006.05.075. [DOI] [PubMed] [Google Scholar]
- 28.Combi-Blocks, LLC. 7949 Silverton Avenue, Suite 915, San Diego, CA, 92126 USA.