

Abstract
Genetic analysis of TP63indicates that ΔNp63 isoforms are required for preservation of regenerative stasis within diverse epithelial tissues. In squamous carcinomas, TP63 is commonly amplified, and ΔNp63α confers a potent survival advantage. Genome-wide occupancy studies demonstrate that ΔNp63 promotes bidirectional target gene regulation by binding >5000 sites throughout the genome; however, the subset of targets mediating discreet activities of TP63 remains unclear. We report that ΔNp63α activates BMP signaling by inducing the expression of BMP7. Immunohistochemical analysis indicates that hyper-activation of BMP signaling is common in human breast cancers, most notably in the basal molecular subtype, as well as in several mouse models of breast cancer. Suppression of BMP signaling in vitro with LDN193189, a small molecule inhibitor of BMP Type I Receptor kinases, represses clonogenicity and diminishes the cancer stem cell enriched ALDH1+ population. Importantly, LDN193189 blocks reconstitution of mixed ALDH1+/ALDH1- cultures indicating that BMP signaling may govern aspects of cellular plasticity within tumor hierarchies. These results show that BMP signaling enables reversion of committed populations to a stem-like state, potentially supporting progression and maintenance of tumorigenesis. Treatment of a mouse model of breast cancer with LDN193189 caused reduced expression of markers associated with epithelial to mesenchymal transition (EMT). Furthermore, in vivo limiting dilution analysis assays revealed that LDN193189 treatment suppressed tumor-initiating capacity and increased tumor latency. These studies support a model in which ΔNp63α-mediated activation of BMP signaling governs epithelial cell plasticity, EMT, and tumorigenicity during breast cancer initiation and progression.
Keywords: BMP signaling, Breast Cancer, ΔNp63α, Epithelial to Mesenchymal Transition, Mammary Stem Cells
Introduction
TP63 plays important roles in tumorigenesis, senescence, and epithelial stem cell regulation. It is rarely mutated in human cancers; however, genomic amplification of TP63 is observed in 10% of head and neck squamous cell carcinomas, 13% of serous ovarian carcinomas, 23% of squamous cervical carcinomas and 28% of lung squamous cell carcinomas (1). TP63-/- mice exhibit developmental defects, including limb truncations, craniofacial abnormalities, and loss of stratified epithelia(2, 3). Additional analysis indicates that ΔNp63 isoforms preserve long-term regenerative stasis in epithelial tissues(4). ΔNp63α is the predominant isoform in normal and malignant breast tissue, and is selectively expressed in the basal/myoepithelial cell layer that houses mammary stem cells(2, 5, 6). In basal breast cancers and head and neck squamous cell carcinomas (HNSCCs), ΔNp63α confers a survival advantage by actively repressing pro-apoptotic transcriptional programs (7, 8). Additionally, ΔNp63α suppresses oncogene-induced senescence, suggesting it plays a role in cancer initiation (9, 10). These findings implicate ΔNp63α in cancer initiation and progression and suggest that this gene may be targeted for therapeutic benefit.
Bone morphogenic proteins (BMPs) are members of the TGFβ superfamily of cytokines that regulate diverse cellular processes; including cell-fate specification, embryonic patterning, and maintenance of developmental potency in embryonic and adult stem cells (11). BMPs bind to heterotetrameric complexes consisting of two type II receptors and two type I receptors. BMP-specific type II receptors (BMPRII, ActRII, and ActRIIB) possess constitutively active kinase domains that, upon tetramerization, phosphorylate, and activate, BMP-specific type I receptor kinases (ALK2, ALK3, and ALK6). Activated type I receptors phosphorylate BMP specific receptor SMADs (SMADS 1,5 and 8). This promotes interaction with SMAD4, and subsequently causes nuclear translocation and target gene transcription (12). The pathway is negatively regulated by secreted antagonists such as Noggin and Gremlin, and by inhibitory SMADs (SMAD6 and SMAD7) (12). Beyond its role in embryonic patterning, BMP signaling preserves prolonged replicative potential of adult stem cells by promoting cellular quiescence (13-15). In these studies, BMP signaling ablation caused increased proliferation of stem cell populations (13-15).
The role of BMP signaling in breast cancer initiation and progression remains unclear. Conflicting studies report that BMP signaling exerts either tumor suppressive or tumor promoting effects (16-22). The human genome encodes >20 BMP ligands and 10 BMP antagonists (23) suggesting that context-specific receptor-ligand combinations account for distinct cellular responses. Recent studies have focused on the effect of individual ligands or receptors on cellular processes; however, the comprehensive effect of BMP signaling on breast cancer progression remains unknown. LDN193189, a potent inhibitor of BMP type I receptor kinases, provides the opportunity to evaluate the effects of BMP signaling in diverse developmental, physiological and pathophysiological processes. Here we describe a regulatory relationship in which ΔNp63α activatesBMP signaling via increased expression of BMP7 in the mammary epithelium. We show that BMP signaling is elevated in human breast tumors relative to matched normal tissue controls, and in diverse mouse models of breast cancer. Suppression of BMP signaling with LDN193189 reduced clonogenicity and ALDH1 activity of mammary epithelial cells, indicating that LDN193189 inhibits the cancer stem cell phenotype (24, 25). Administration of LDN193189 to a murine breast cancer model decreased tumor incidence, tumor size, and expression of the EMT marker vimentin. These studies support a model in which ΔNp63α-mediated induction of BMP7 contributes to the tumor stem cell phenotype. They also demonstrate that pharmacologic inhibition of BMP signaling may represent an opportunity to target cancer stem cells.
Materials and Methods
Cell Culture and Reagents
Breast cancer cell lines, MCF7, MDA-MB-231, SUM149, SUM102, SKBR3, BT20, HCC1937, and ZR75 were maintained according to ATCC guidelines. The immortalized mammary epithelial cell (IMEC) line has been previously described (26). Mouse HC11 cells were grown in RPMI-1640 supplemented with 10% fetal bovine serum, 10 ng/ml EGF, 5 μg/ml insulin, 100 units/ml penicillin, and 100 μg/ml streptomycin. MMTV-Myc primary tumor cells were cultured in Mammary Epithelial Cell Growth Medium (Lonza) without Bovine Pituitary Extract (BPE).
LDN193189 was purchased from Stemgent Technologies and solubilized in DMSO. For in vivo studies, LDN193189 was synthesized by Aberjona Laboratories, Inc and solubilized in sterile H20 for injection. Recombinant BMP7 (used at 50 ng/ml) and recombinant Noggin (used at 125 ng/ml) were purchased from R&D systems. For neutralizing experiments, a rabbit polyclonal antibody to BMP7 was used at 500 ng/ml (Abcam).
Western Blot Analysis
Cell lysates were prepared in NETN buffer (100 nM Tris-Cl [pH 7.8], 1 mM EDTA, 100 mM NaCl, and 0.1% Triton X-100) supplemented with protease and phosphatase inhibitors (Roche). Proteins were resolved by SDS-PAGE and transferred onto PVDF membranes. Membranes were immunoblotted for P-SMAD1/5/8 (Cell Signaling), total SMAD1/5/8 (Cell Signaling), p63 (4A4 clone; Sigma), Vimentin (Lab Vision), E-Cadherin (BD Biosciences), and β-Actin (Cell Signaling), and visualized by enhanced chemiluminescence (Amersham Pharmacia).
Colony Formation Assay
Immortalized and transformed mammary epithelial cell lines were plated at 500 cells per well in six-well plates. Cells were treated with the indicated concentrations of LDN193189 or vehicle control every 48 hours for 10-14 days. Cells were then fixed in methanol and stained with 0.1% crystal violet.
Quantitative RT-PCR Analysis
RNA was isolated with the RNeasy Kit (Qiagen). cDNA was prepared using the iScript cDNA synthesis kit (BioRad). Quantitative PCR was performed with iQ SYBR Green Super mix (BioRad) and oligonucleotide primers specific to each target gene. Relative changes in gene expression were obtained using the 2-Δ ΔCT method normalizing to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Animals
FVB wild-type (wt) and MMTV-Myc transgenic mice were maintained according to institutional guidelines. Animal protocols were approved by the Institutional Animal Care and Use Committee at Dartmouth Medical School.
Human Tissue Samples
Formalin-fixed paraffin-embedded (FFPE) human breast tumors and normal mammary tissue were obtained from the Tissue and Tumor Bank at Dartmouth Hitchcock Medical Center. The molecular subtype of human breast tumor samples was identified by the Department of Pathology at Dartmouth Hitchcock Medical Center.
Mouse Tissue Samples
Normal mammary glands from FVB wild-type and MMTV-Myc mice were fixed in Bouin's solution for 24 hours. Tumor tissue from MMTV-Myc, MMTV-Wnt-1, MMTV-HER2/Neu, MMTV-PyMT and BRCA1Co/Co; MMTV-Cre; p53+/- mice were fixed in 10% formalin for 24 hours. Fixed tissues were dehydrated through a series of graded alcohols and embedded in paraffin. Five μm sections were applied to charged glass microscope slides.
Immunohistochemisty
Tissue sections were deparaffinized in xylene and rehydrated in a series of graded alcohol steps. Antigen retrieval was performed by incubating sections in 10 μmol/L sodium citrate buffer for 20 min in a microwave oven. Peroxidase activity was quenched by incubating in 0.3% H2O2 for 30 min at RT. Sections were blocked for two hours in 5% goat serum/0.5% Tween-20/PBS at RT followed by incubation with P-SMAD1/5/8 primary antibody (1:100) diluted in blocking buffer at 4°C overnight in a humidified chamber. Sections were incubated with biotinylated secondary antibodies (Vector Laboratories) for 1 hour. Targets were detected with streptavidinlinked peroxidase (1:400, Vector Laboratories) for 30 min at RT and developed using the DAB staining kit and counterstained with hematoxylin.
Immunofluorescence
Following deparaffinization, rehydration, antigen retrieval, and blocking, sections were incubated in primary antibodies overnight at 4°C in a humidified chamber. Primary antibodies used were; p63 (1:100, 4A4 clone, Sigma), P-SMAD1/5/8 (1:100, Cell Signaling), and Vimentin (1:100, Lab Vision). Sections were washed in PBS/0.1% Tween-20 and incubated in antimouse-Alexa Fluor 488 and anti-rabbit-Alexa Fluor 555, (1:500, Invitrogen). Sections were mounted in Vectashield with DAPI (Vector Laboratories) and imaged by fluorescence microscopy.
Breast Cancer Allografts
MMTV-Myc tumors were resected and single cell suspensions were prepared by incubation in collagenase/hyaluronidase for 6 hours at 37°C with periodic resuspension of the tissue. Cells were collected and incubated with Trypsin-EDTA followed by Dispase and DNase 1. Cells were then filtered through 40 μM cell strainers and resuspended in NH4Cl to lyse all red blood cells. For transplantation, cells were embedded in 100μl of Matrigel and transplanted into the dorsal right flanks of female FVB recipients. Mice were injected intraperitoneally with 2.5 mg/kg LDN193189 or vehicle control daily for 16 days and sacrificed two hours after the last injection.
Aldefluor Assay
Aldehyde dehydrogenase1 (ALDH1) activity was assessed in IMECs, HC11s, and primary MMTV-Myc tumor cells using the ALDEFLUOR kit (STEMCELL Technologies) per manufacturer's protocol. Flow cytometric gates were based DEAB-treated negative controls. Samples were analyzed on a BD FACScan instrument using CellQuest software (BD Biosciences).
Statistical Methods
Quantitative data is presented as mean values of triplicate points and error bars represent the standard error of the mean (SEM).
Results
ΔNp63α activates the BMP signaling pathway via enhanced expression of BMP7
Several lines of evidence support a role for BMP signaling in stem cell regulation where it maintains the proliferative potential of adult stem cells (27). Recent reports demonstrate an interaction between TP63 and BMP signaling. BMP7 expression is lost in TP63-/- embryos(28), which is consistent with the finding that BMP7 is a transcriptional target of p53 family members, including ΔNp63α (21). Additionally, ΔNp63α suppresses the inhibitory SMAD, SMAD7, in primary keratinocytes (29). To address the relevance of this relationship to breast cancer, expression levels of ΔNp63 and BMP7 were measured by Q-PCR in breast cancer cell lines. Results indicated a positive correlation between ΔNp63α and BMP7 expression (Fig. 1A). Ectopic ΔNp63α in human (IMEC) and mouse (HC11) immortalized mammary epithelial cell lines caused an 8-fold and 5.5-fold induction of BMP7 mRNA respectively (Fig. 1B), indicating that ΔNp63α was able to enhance BMP7 expression. Ectopic ΔNp63α activates SMAD1/5/8 phosphorylation in a manner that is sensitive to either Noggin or BMP7 neutralizing antibodies (Fig. 1C). Ectopic TAp63γ also induced BMP signaling in IMECs (Supplemental Fig. S1A); however, ΔNp63 expression is several thousand-fold greater than TAp63 expression in the mammary epithelium (Supplemental Fig. S1B). Treatment of IMECs with recombinant BMP7 rapidly and potently increased P-SMAD1/5/8. This effect could be inhibited by LDN193189, a selective inhibitor of BMPR1 kinase activity (Fig. 1D). LDN193189 also suppressed endogenous levels of BMP signaling in IMECs (compare lane1 with lanes 4-6 in Fig. 1D). LDN193189 did not alter phosphorylation of TGFβ-specific SMADs (Fig. 1D), indicating specificity for BMP signaling over other TGFβ superfamily members. Additionally, LDN193189 prevented ΔNp63α-mediated phosphorylation of SMAD1/5/8 (Fig. 1E). Treatment of IMECs with recombinant BMP7 caused increased expression of the canonical target genes, Id1, Id2 and Id3 (Supplemental Fig. S2A). Ectopic ΔNp63α enhanced expression of Id2, (Supplemental Fig. S2B) and this induction was suppressed by LDN193189, indicating that ΔNp63α-mediated induction of Id2 partially depends on BMP type I receptor activity. These data support the conclusion that ΔNp63α activates canonical BMP signaling via expression of BMP7 in the mammary epithelium.
Figure 1. ΔNp63α induces canonical BMP signaling in mammary epithelial cells via induction of the BMP7 ligand.
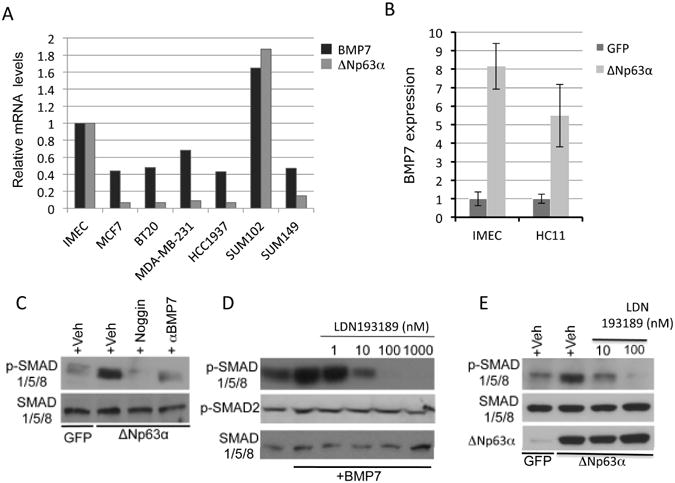
A. Quantitative PCR analysis of ΔNp63 and BMP7 expression in human breast cancer cell lines. Expression levels are normalized to normal hTERT-immortalized human mammary epithelial cells (IMECs) and to GAPDH. B. Ectopic ΔNp63α induces the expression of BMP7 mRNA in IMECs and HC11 cells. C. Western blots were prepared with extracts from IMECs infected with adenoviral ΔNp63α or GFP as a control, and treated with either vehicle, rhNoggin (250 ng/ml), or BMP7-neutralizing antibodies. Blots were probed with antibodies againstP-SMAD1/5/8 (identifying active BMP signaling), and total SMAD1/5/8 as a loading control. D. Western blot analysis of IMECs pre-treated with vehicle or LDN193189 for 10 minutes, followed by stimulation with vehicle orrhBMP7 (50ng/ml) for 45 minutes. Blots were probed with antibodies directed against P-SMAD1/5/8 and P-SMAD2. Total SMAD1/5/8 is used as a loading control. E. IMECs were infected with adenoviruses programmed to express GFP (control) or ΔNp63α. Following infection, cells were treated with vehicle (control) or the indicated doses of LDN193189. Blots were probed with antibodies directed against P-SMAD1/5/8. β-Actin is used as a loading control. Data is presented as mean values of triplicate points ± S.E.
BMP signaling is elevated in human and mouse mammary tumors
The relationship between ΔNp63α and BMP signaling coupled to the role of ΔNp63α in breast cancer initiation and progression, suggested that BMP signaling might be hyper-activated in breast cancer. To address this, a tissue microarray containing breast cancer samples representing five molecular subtypes of breast cancer (30) was subjected to immunofluorescent analysis of P-SMAD1/5/8 expression. Results demonstrated that BMP signaling was elevated in the majority of breast cancers relative to matched normal controls and was preferentially activated in basal breast tumors (Supplemental Fig. S3). Notably, 12 of 15 cases of the basal breast cancer subtype had levels of immuno-detectable P-SMAD1/5/8 greater than the median expression level (Supplemental Fig. S3). To confirm this observation, two-color immunofluorescence was used to determine BMP signaling status in normal human mammary epithelia and basal-subtype breast tumors. This analysis indicated that BMPR1A (Fig. 2A) and P-SMAD1/5/8 (Fig. 2B) levels are elevated in basal breast tumors relative to normal mammary gland tissue. (Negative controls for immunohistochemical and immunofluorescent analysis can be found in Supplementary Fig. S4). Robust levels of P-SMAD1/5/8 were also noted in tumors from MMTV-Myc, MMTV-PyMT, MMTV-Wnt-1, and MMTV-ErbB2/Neu mice relative to normal mammary gland controls (Fig. 2C). Interestingly, we noted that P-SMAD1/5/8 expression was widespread and not confined to specific cell types within the tumor tissue. This data indicates that BMP signaling is elevated in breast cancer and may be targeted for therapeutic benefit.
Figure 2. BMP Signaling is elevated in basal subtype breast tumors and in mouse models of human breast cancer.
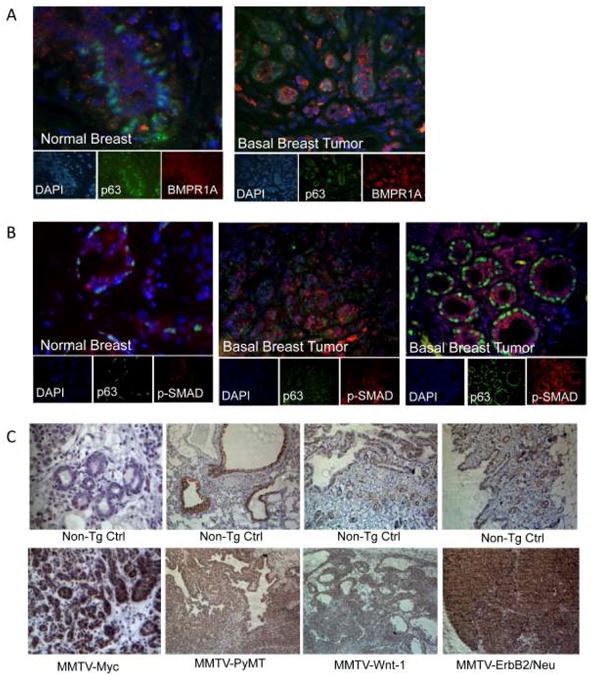
A. BMP type 1 Receptor A (BMPR1A) expression (red), and p63 expression (green) detected by immunofluorescence in normal breast and triple-negative type tumor tissue. B. Phosphorylated-SMAD1/5/8 (red) and p63 (green) expression detected by immunofluorescence in normal breast and triple negative tumor tumors; N=5 per group. Nuclei are stained with DAPI. C. Immunohistochemical staining of phospho-SMAD1/5/8 on MMTV-Myc, MMTV-PyMT, MMTV-Wnt-1, and MMTV-ErbB2/Neu mouse tumor tissue and non-transgenic normal tissue controls; sections were counterstained with hematoxylin; N=3 per group.
Activation of BMP signaling is an early event in breast cancer
There is an urgent need to identify pharmacologically accessible pathways that contribute to early stages of breast cancer. To address this, clinically defined samples of ductal carcinoma in situ (DCIS) were evaluated for levels of phospho-SMAD1/5/8. Co-staining with p63 revealed increased BMP signaling in luminal epithelial cells within DCIS lesions (Fig.3A) suggesting that BMP activation may be an early event in breast cancer. This result also suggests a paracrine model in which ΔNp63α promotes BMP7 expression in basal epithelia resulting in activation of BMP signaling in neighboring luminal epithelia. To determine if BMP signaling is hyper-activated in a premalignant state, phospho-SMAD1/5/8 levels were evaluated in two mouse models of breast cancer with distinct stages of pre-malignancy. In the MMTV-Myc model, parity shortens tumor latency and increases tumor penetrance to nearly 100%, suggesting that the parous MMTV-Myc mammary gland represents a model of pre-malignancy(31). Immunofluorescent analysis of P-SMAD1/5/8 in the mammary glands of nulliparous and parous MMTV-Myc mice revealed that BMP signaling is hyper-activated following parity and remains elevated through tumor progression (Fig. 3B). A second model of pre-malignancy in the mammary gland is the BRCA1Co/Co; MMTV-Cre;p53+/- model of basal breast cancer (32). Analysis of BMP signaling in this model showed robust levels of phospho-SMAD1/5/8 in unaffected mammary glands of tumor bearing mice as well as in the tumor tissue, suggesting that BMP signaling is elevated prior to cancer initiation (Fig. 3C). Collectively, these studies indicate that hyper-activation of BMP signaling is an early event in tumorigenesis.
Figure 3. BMP Signaling is elevated in mouse models of breast premalignancy and is upregulated at early stages of breast cancer progression.
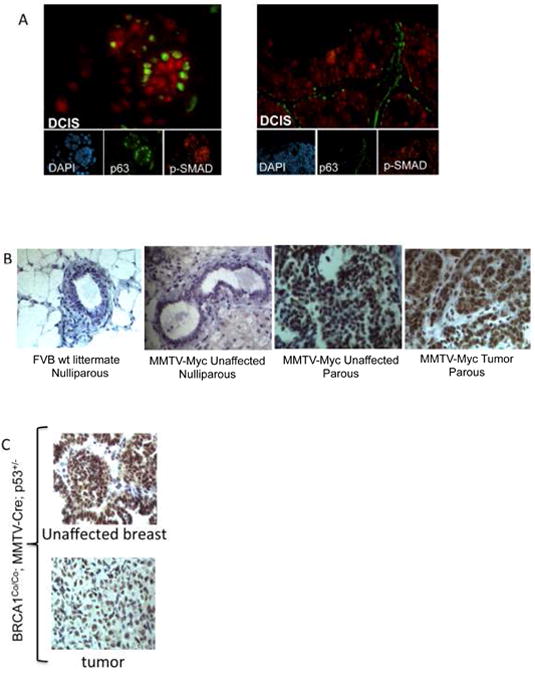
A. Phosphorylated-SMAD1/5/8 (red) and p63 (green) expression detected by immunofluorescence in ductal carcinoma in situ (DCIS); N=10. B. Phospho-SMAD1/5/8 expression detected by immunohistochemical analysis in mammary tissue sections derived from mice at various stages during MMTV-Myc tumor progression. Sections were counterstained with hematoxylin. C. Phospho-SMAD1/5/8 expression, detected by immunohistochemical analysis, in tumors and unaffected mammary glands from tumor bearing mice with the BRCA1Co/Co; MMTV-Cre; p53+/-genotype.
LDN193189 inhibits cancer stem cell activity in vitro
The established role for BMP signaling in preservation of replicative capacity and developmental potency of embryonic stem cells suggested a similar role in cancer stem cell regulation. To address this, we sought to determine if disruption of BMP signaling affected aldehyde dehydrogenase1 positive (ALDH1+) populations, which have been shown to be enriched for stem cells (25). Flow-cytometric analysis of ALDH1 activity in IMEC andHC11 cells revealed that LDN193189 reduced the ALDH1+ populations in both cell lines (Fig. 4A and S5A). Similar effects were observed in MMTV-Myc primary tumors cells where LDN193189 reduced the ALDH1+ population from 5.49% to 0.225% (Fig. 4B). These effects are dose dependent and evident at doses at which LDN193189 is highly selective for BMP type I receptors (33). Additionally, LDN193189 potently inhibits colony formation in IMECs (Fig. 4D) as well several immortalized and malignant mammary epithelial cells (Supplemental Fig. S5C,D). Conversely, treatment of IMECs with rHBMP7 increased clonogenic capacity (Supplement Fig. S5B) suggesting that the anticlonogenic effects of LDN193189 are due to disruption of BMP signaling. These data indicate that LDN193189 inhibits stem cell populations and clonogenic capacity in established mammary epithelial cell lines and primary murine tumor cells.
Figure 4. Pharmacologic inhibition of BMP signaling reduces normal and cancer stem cell populations in vitro.
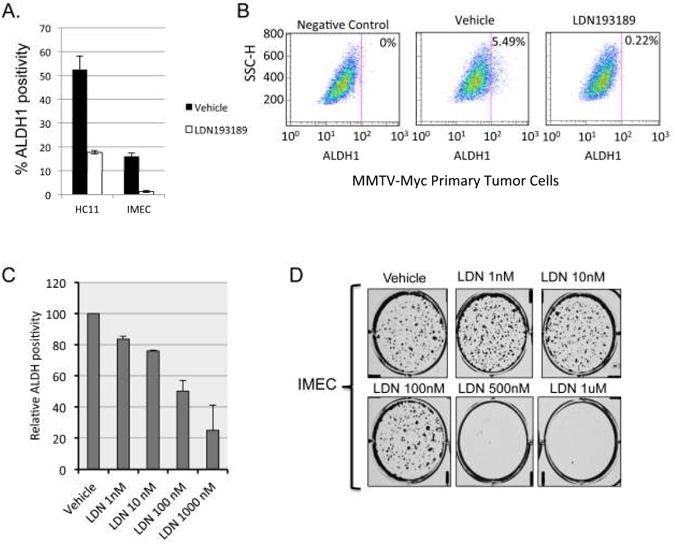
A. Graphic display of FACS-based analysis of ALDH1 activity in HC11 cells and IMECs treated with vehicle or 1uM LDN193189 for 48 hours. Cells were incubated with DEAB, an ALDH1 inhibitor, and used as a negative control. Gates were set according to the negative control. B. FACS analysis of ALDH1 activity in primary MMTV-Myc tumor cells treated with vehicle or 1μM LDN193189 for 48 hours. Cells were incubated with DEAB, an ALDH1 inhibitor, and used as a negative control. Gates were set according to the negative control. C. HC11 cells were treated with the indicated doses of LDN193189 for 48 hours and assayed for ALDH1 activity by flow cytometry. Data are expressed as the percentage of ADLH1+ cells relative to vehicle-treated controls. D. Colony formation assay in IMECs treated with vehicle control and the indicated concentrations of LDN193189. Cells were stained with crystal violet 14 days after plating at 500 cells/well in 6-well plates.
LDN193189 promotes Mesenchymal to Epithelial Transition and restricts plasticity of mammary epithelial cells
EMT is an important cellular process that contributes to metastasis in response to ionizing radiation and chemotherapeutics (34) suggesting that EMT may represent an adaptive mechanism that enables cells to evade therapeutic assault. The ability of cells to undergo both EMT and the reverse process, Mesenchymal to Epithelial Transition (MET), implies a state of cellular plasticity that may contribute to features of cancer stem cells. EMT is associated with decreased expression of epithelial cell-adhesion proteins, notably E-cadherin and zonula occludens protein 1 (ZO-1), and a corresponding increase in mesenchymal markers, such as vimentin and N-cadherin. It also renders cells more migratory, invasive, and resistant to apoptosis(34-36). The previous data in this study demonstrate that LDN193189 prevents colony formation. During these studies, we noted thatLDN193189 causes IMECs to undergo morphological changes that result in colonies that stain more intensely with crystal violet, indicating greater cell density and reduced migratory capacity (Fig. 5A). Staining of these colonies withZO-1revealed that LDN193189 treatment increases ZO-1 expression and promotes tight junction formation (Fig. 5A). Consistent with this observation, treatment of IMECs with LDN193189 for 72 hours lead to increased expression of the epithelial marker E-cadherin, and a corresponding decrease in expression of the mesenchymal marker, vimentin (Fig. 5B). These results implicate BMP signaling in the promotion of EMT in mammary epithelial cells. Recent reports have demonstrated a direct link between induction of EMT and the acquisition of tumor initiating stem cell-like properties (37). Thus, this observation suggests that inhibition of BMP signaling opposes EMT and is consistent with the ability of LDN193189 to reduce the ALDH1+ stem-cell enriched population. To test if LDN193189 is sufficient to restrict plasticity of epithelial cell populations, ALDH1+ and ALDH-HC11 fractions were cultured for 72 hours in the absence or presence of LDN193189. ALDH- cells efficiently reconstituted the ALDH1+/ALDH1- parental culture and this effect was blocked by LDN193189 (Fig. 5C). The ability of theALDH1+ fraction to reconstitute the ALDH1+/ALDH1- parental culture was accelerated in the presence of LDN193189, which is consistent with previous data demonstrating that LDN193189 reduces the ALDH1+ population (Fig. 5C). These results indicate that BMP signaling confers plasticity and supports the cancer stem cell state (Fig. 5D).
Figure 5. LDN193189 promotes Mesenchymal to Epithelial Transition (MET) and restricts cellular plasticity.
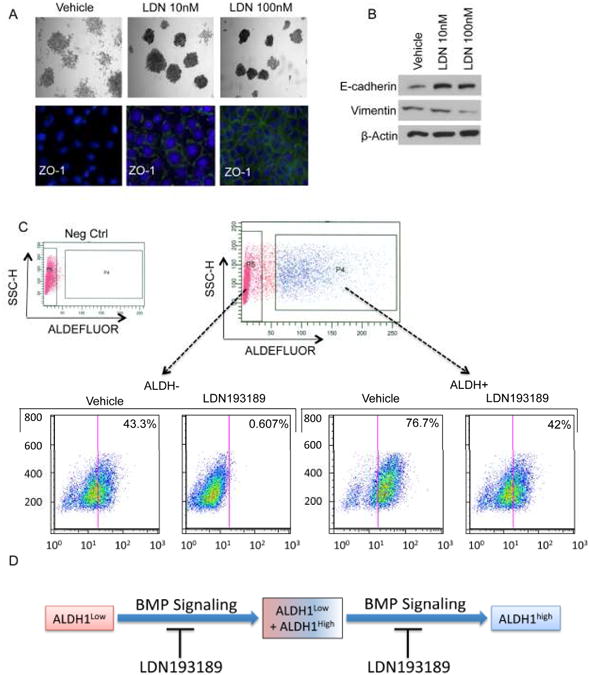
A, top. IMECs were cultured at clonogenic density and treated with the indicated concentrations of LDN193189. Colonies were stained with crystal violet 10-14 days after plating. A, bottom. Immunofluorescent analysis of the tight junction protein ZO-1 in colonies formed in the presence or absence of LDN193189. B. Western blot analysis of E-cadherin and vimentin levels indicate that treatment of IMECs with LDN193189 causes changes that are consistent with MET. C. HC11 cells were FACS-segregated based on ALDH1 activity and re-cultured in the presence of vehicle or LDN193189 for 72 hours. Cells were then reassayed for ALDH1 activity to determine the ability of each fraction to reconstitute the mixed parental ALDH1+/ALDH1- culture. D. Schematic summarizing the results of data shown in C.
LDN193189 exhibits in vivo activity in a syngeneic allograft model of MMTV-Myc driven mammary tumorigenesis
To investigate the in vivo biological activity of LDN193189, we established syngeneic allografts of MMTV-Myc driven breast cancer in 40 female FVB wildtype recipients. Two days post-transplantation, mice were randomized and injected IP with 2.5 mg/kg LDN193189 or vehicle once daily for 16 days. Analysis of tumor lysates revealed that LDN193189 reduces phospho-SMAD1/5/8 levels and expression of ΔNp63α (Fig. 6A). The reduction in ΔNp63α is consistent with results showing that LDN193189 can inhibit the ΔNp63α-enriched ALDH1+ population. This effect was also observed in IMEC and HC11 cells treated with LDN193189 in vitro (Supplemental Fig. S6A,B). Additionally, immunofluorescent analysis of these tumors showed that LDN193189 treatment decreased phospho-SMAD1/5/8 (shown in green) and vimentin (shown in red) expression indicating that suppression of BMP signaling disrupts the EMT phenotype (Fig. 6B). Hematoxylin and Eosin (H&E) staining of these tumors showed similar tumor morphology amongst both treatment groups (Supplemental Fig. S6).
Figure 6. LDN193189 exhibits pharmacologic activity in vivo and causes reversion of EMT.
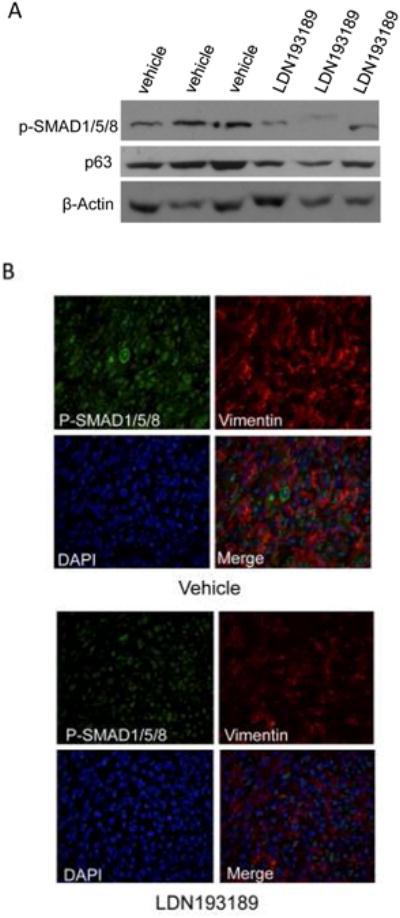
One thousand primary MMTV-Myc tumor cells were embedded in matrigel and transplanted into the dorsal right flanks of female syngeneic (FVB/N) wt recipients; N=40. At two days post transplant, mice were injected intraperitoneally with 2.5 mg/kg LDN193189 or vehicle control daily for 16 days and then sacrificed two hours after the last injection. A. Western blot analysis of 3representative tumor lysates from vehicle and LDN193189 treatment groups probing for phospho-SMAD1/5/8 and p63. β-Actin is used as a loading control. B. Representative images of 2-color Immunofluorescent analysis of mouse tumor tissue probing for P-SMAD1/5/8 (green) and Vimentin (red), a marker of EMT. Nuclei are stained with DAPI.
LDN193189 reduces tumor initiating ability and increases tumor latency
To assess the effect of LDN193189 on tumor-initiating capacity, limiting dilution transplants of cells pre-treated withLDN193189 was performed.; Primary MMTV-Myc tumor cells were treated ex vivo with LDN193189 for 48 hours, sharply diminishing the ALDH1+ population compared to vehicle control (Fig. 7A). Treated and untreated cells were embedded in matrigel and transplanted at limiting dilutions into female FVB recipients. Recipients of LDN193189-treated cells were treated with 2.5 mg/kg LDN193189 daily for 24 days. These mice displayed a marked decrease in tumor incidence and increased tumor latency (Fig. 7B). Tumor incidence results (Supplemental Fig.7B) highlight the increased latency to progression among the LDN193189 treated animals. None of the mice transplanted with the fewest cells (10 cells) and treated with LDN193189 formed tumors, where as 75% of mice receiving vehicle treatment at this cell dose formed palpable tumors (Fig. 7B,left). At the intermediate cell dilution (100 cells), 100% of the mice in the vehicle treatment group developed palpable tumors versus 50% of mice in the LDN193189 treatment group (Fig. 7B). Tumors were measured at the time of harvest, and while no group reached statistical significance, LDN193189 treated animals showed a decreased trend in tumor mass for all three cell doses tested (Supplemental Fig. S7B). The lack of statistical significance is likely attributed to the high variability in tumor mass even within each treatment group. Individual tumor mass data is shown in Supplemental Fig. S7C. Taken together, this data implicate that BMP signaling may be central to regulating epithelial cell plasticity, EMT, and stemness during breast cancer initiation.
Figure 7. Limiting dilution analysis reveals that LDN193189 reduces tumor initiating ability and increases tumor latency.
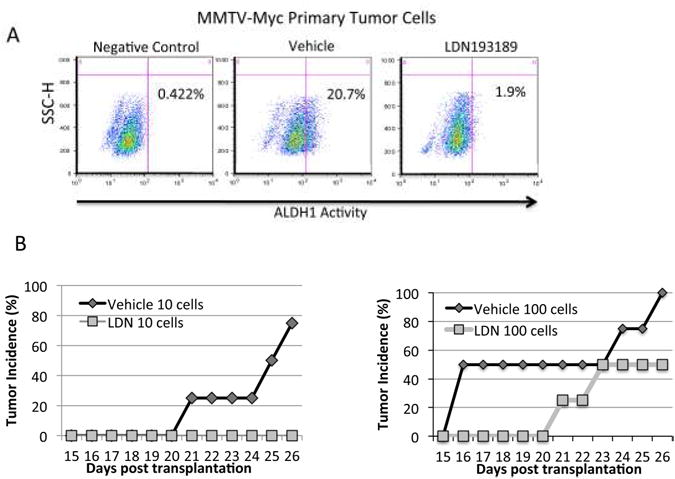
A. Primary MMTV-Myc tumor cells were cultured ex vivo in the presence or absence of 1μM LDN193189 for 48 hours and subjected to FACS-based analysis of ALDH1 activity. Cells were incubated with DEAB, an ALDH1 inhibitor, and used as a negative control. Gates were set according to the negative control. The cells were subsequently transplanted by limiting dilution into the dorsal right flanks of FVB wild-type female recipient mice. Mice were treated two days post transplantation daily for 24 days with 2.5 mg/kg LDN193189 or vehicle control by IP injection. B. Tumor latency is plotted as time to palpitation for each group of mice. N=4 for each group.
Discussion
Breast cancer is the leading cause of cancer-related death in women worldwide, with incidence rates rising globally over the last 25 years (38). In the United States, the lifetime risk of breast cancer for all women is 12.7% (38). Targeted molecular therapies exhibit less toxicity and enhanced tumor selectivity than broad-spectrum chemotherapeutics, and have become important components of current breast cancer treatment modalities. Breast cancer progression and recurrence remains a formidable clinical challenge due to acquired resistance to existing therapies, inability to respond to adjuvant therapies, and a failure of drug combinations to produce durable remissions. This highlights the need for the identification of additional genetic alterations that underlie disease recurrence, and subsequent design of selective inhibitors of these pathways for use in novel combination with conventional cytotoxic agents.
Studies presented here evaluate BMP signaling status by measuring the phosphorylation of SMAD1/5/8, which represents a point of convergence for all BMP signaling. We present evidence that BMP signaling is hyper-activated in human and mouse breast tumors relative to normal tissue controls, and that it is highest in the basal breast cancer subtype. Additionally, we present data indicating that activation of BMP signaling is evident in DCIS and in mouse models of pre-malignancy. Furthermore, numerous reports demonstrate a role for BMP signaling in promoting tumor cell migration and invasion (17, 19-21, 39-41), suggesting that BMP signaling may be targeted for therapeutic benefit. In vitro analysis of LDN193189, a small molecule inhibitor of BMPR1 kinases, revealed that suppressing BMP signaling reduced the stem cell-enriched ALDH1+ population. Here we show that ΔNp63α can induce BMP7 expression and activate canonical BMP signaling, providing a potential mechanism by which ΔNp63α preserves stem cell proliferative potential. Together our data support the conclusion that BMP signaling may be targeted to reduce the activity of breast cancer stem cells.
In vivo studies revealed that biologically active doses of LDN193189 caused reductions in tumor size and volume in mice treated daily with LDN193189. These effects, however, did not reach statistical significance, which we attribute to the very rapid expansion of tumors in the syngeneic allograft model. Longer treatment windows in a model with growth rates that more closely approximate human breast cancers may yield statistically significant effects on tumor volume. An alternative explanation may be that LDN193189 is targeting the rare CSC populations within the tumor causing only modest changes in tumor volume. Administration of LDN193189 with an agent that targets the bulk tumor population may reduce tumor volume and the likelihood of recurrence and metastatic disease.
We observed decreased vimentin expression in tumor tissue from mice treated with LDN193189, suggesting that BMP inhibition causes these tumors to undergo MET. This is consistent with the pro-MET effects we observed with drug treatment in vitro. Limiting dilution analysis demonstrated that LDN193189restricts the tumorigenic capacity of MMTV-Myc allografts. Mice that do form tumors in the presence of LDN193189 administration exhibit an increased latency to palpation. These experiments demonstrate the ability of LDN193189 to repress breast tumor-initiating populations . These initial studies are promising, but much work is needed to understand the efficacy of this drug in cancer treatment and the toxicity to normal stem cell compartments.
Reactivation of normal developmental pathways is a common feature of cancer progression, and has been observed previously with the Hedgehog and Wnt signaling pathways (42). Aberrant activation of these pathways during tumorigenesis is believed to lead to inappropriate specification of cells to a stem-like state. In this study, we demonstrate that BMP signaling is hyper-activated during breast cancer initiation and progression, and that it enhances tumor stem cell populations and EMT, indicating a role for BMP in the pathophysiology of cancer. Overall, this data support a model in which BMP signaling, driven by ΔNp63, governs key aspects of stem cell activity and epithelial plasticity in normal and malignant breast tissue.
Supplementary Material
Acknowledgments
Grant Support: This work was supported in part by grants to J.D.R. from the National Cancer Institute (5RO1CA108539-05), the United States Department of Defense Breast Cancer Research Program (BC095560) and a Prouty Pilot Award from the Friends of the Norris Cotton Cancer Center. This work was also supported in part by National Institutes of Health (NIH) and National Cancer Institute (NCI) grants: R03 CA141564 (L.F.S) and R21 CA141017 (L.F.S). A.L.B. is supported by a training grant from the National Cancer Institute (T32-CA009658).
Footnotes
The authors declare that no conflicts of interest exist.
References
- 1.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer discovery. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–13. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 3.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–8. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 4.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–82. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Molecular cell. 1998;2:305–16. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 6.Nylander K, Vojtesek B, Nenutil R, Lindgren B, Roos G, Zhanxiang W, et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. The Journal of pathology. 2002;198:417–27. doi: 10.1002/path.1231. [DOI] [PubMed] [Google Scholar]
- 7.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. The Journal of clinical investigation. 2007;117:1370–80. doi: 10.1172/JCI30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell stem cell. 2011;8:164–76. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keyes WM, Mills AA. p63: a new link between senescence and aging. Cell Cycle. 2006;5:260–5. doi: 10.4161/cc.5.3.2415. [DOI] [PubMed] [Google Scholar]
- 11.Dutko JA, Mullins MC. SnapShot: BMP signaling in development. Cell. 2011;145:636, e1–2. doi: 10.1016/j.cell.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Buijs JT, Petersen M, van der Horst G, van der Pluijm G. Bone morphogenetic proteins and its receptors; therapeutic targets in cancer progression and bone metastasis? Current pharmaceutical design. 2010;16:1291–300. doi: 10.2174/138161210791033987. [DOI] [PubMed] [Google Scholar]
- 13.Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, et al. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell stem cell. 2010;7:78–89. doi: 10.1016/j.stem.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 14.Tian Q, He XC, Hood L, Li L. Bridging the BMP and Wnt pathways by PI3 kinase/Akt and 14-3-3zeta. Cell Cycle. 2005;4:215–6. [PubMed] [Google Scholar]
- 15.Kobielak K, Stokes N, de la Cruz J, Polak L, Fuchs E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10063–8. doi: 10.1073/pnas.0703004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh-Choudhury N, Woodruff K, Qi W, Celeste A, Abboud SL, Ghosh Choudhury G. Bone morphogenetic protein-2 blocks MDA MB 231 human breast cancer cell proliferation by inhibiting cyclin-dependent kinase-mediated retinoblastoma protein phosphorylation. Biochemical and biophysical research communications. 2000;272:705–11. doi: 10.1006/bbrc.2000.2844. [DOI] [PubMed] [Google Scholar]
- 17.Clement JH, Raida M, Sanger J, Bicknell R, Liu J, Naumann A, et al. Bone morphogenetic protein 2 (BMP-2) induces in vitro invasion and in vivo hormone independent growth of breast carcinoma cells. International journal of oncology. 2005;27:401–7. [PubMed] [Google Scholar]
- 18.Schwalbe M, Sanger J, Eggers R, Naumann A, Schmidt A, Hoffken K, et al. Differential expression and regulation of bone morphogenetic protein 7 in breast cancer. International journal of oncology. 2003;23:89–95. [PubMed] [Google Scholar]
- 19.Helms MW, Packeisen J, August C, Schittek B, Boecker W, Brandt BH, et al. First evidence supporting a potential role for the BMP/SMAD pathway in the progression of oestrogen receptor-positive breast cancer. J Pathol. 2005;206:366–76. doi: 10.1002/path.1785. [DOI] [PubMed] [Google Scholar]
- 20.Pouliot F, Blais A, Labrie C. Overexpression of a dominant negative type II bone morphogenetic protein receptor inhibits the growth of human breast cancer cells. Cancer research. 2003;63:277–81. [PubMed] [Google Scholar]
- 21.Yan W, Chen X. Targeted repression of bone morphogenetic protein 7, a novel target of the p53 family, triggers proliferative defect in p53-deficient breast cancer cells. Cancer research. 2007;67:9117–24. doi: 10.1158/0008-5472.CAN-07-0996. [DOI] [PubMed] [Google Scholar]
- 22.Owens P, Pickup MW, Novitskiy SV, Chytil A, Gorska AE, Aakre ME, et al. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2814–9. doi: 10.1073/pnas.1101139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alarmo EL, Kallioniemi A. Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis? Endocrine-related cancer. 2010;17:R123–39. doi: 10.1677/ERC-09-0273. [DOI] [PubMed] [Google Scholar]
- 24.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer research. 2009;69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiRenzo J, Signoretti S, Nakamura N, Rivera-Gonzalez R, Sellers W, Loda M, et al. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer research. 2002;62:89–98. [PubMed] [Google Scholar]
- 27.Sagorny K, Chapellier M, Laperrousaz B, Maguer-Satta V. BMP and cancer: the Yin and Yang of stem cells. Medecine sciences: M/S. 2012;28:416–22. doi: 10.1051/medsci/2012284020. [DOI] [PubMed] [Google Scholar]
- 28.Laurikkala J, Mikkola ML, James M, Tummers M, Mills AA, Thesleff I. p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development. 2006;133:1553–63. doi: 10.1242/dev.02325. [DOI] [PubMed] [Google Scholar]
- 29.De Rosa L, Antonini D, Ferone G, Russo MT, Yu PB, Han R, et al. p63 Suppresses non-epidermal lineage markers in a bone morphogenetic protein-dependent manner via repression of Smad7. The Journal of biological chemistry. 2009;284:30574–82. doi: 10.1074/jbc.M109.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 31.Jamerson MH, Johnson MD, Furth PA, Dickson RB. Early parity significantly elevates mammary tumor incidence in MMTV-c-myc transgenic mice. Transgenic research. 2003;12:747–50. doi: 10.1023/b:trag.0000005247.69329.ca. [DOI] [PubMed] [Google Scholar]
- 32.Brodie SG, Xu X, Qiao W, Li WM, Cao L, Deng CX. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001;20:7514–23. doi: 10.1038/sj.onc.1204929. [DOI] [PubMed] [Google Scholar]
- 33.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal. 2011;23:1831–42. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 34.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Developmental cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 37.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA: a cancer journal for clinicians. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 39.Peart TM, Correa RJ, Valdes YR, Dimattia GE, Shepherd TG. BMP signalling controls the malignant potential of ascites-derived human epithelial ovarian cancer spheroids via AKT kinase activation. Clinical & experimental metastasis. 2012;29:293–313. doi: 10.1007/s10585-011-9451-3. [DOI] [PubMed] [Google Scholar]
- 40.Lai TH, Fong YC, Fu WM, Yang RS, Tang CH. Osteoblasts-derived BMP-2 enhances the motility of prostate cancer cells via activation of integrins. The Prostate. 2008;68:1341–53. doi: 10.1002/pros.20799. [DOI] [PubMed] [Google Scholar]
- 41.Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene. 2008;27:6322–33. doi: 10.1038/onc.2008.232. [DOI] [PubMed] [Google Scholar]
- 42.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.