

Abstract
Persistent organic pollutants (POPs), including polychlorinated biphenyls (PCBs) and polybrominated diphenylethers (PBDEs) that bioaccumulate in lipid-rich tissues are of concern as developmental neurotoxicants. Epigenetic mechanisms such as DNA methylation act at the interface of genetic and environmental factors implicated in autism-spectrum disorders. The relationship between POP levels and DNA methylation patterns in individuals with and without neurodevelopmental disorders has not been previously investigated. In this study, a total of 107 human frozen post-mortem brain samples were analyzed for 8 PCBs and 7 PBDEs by GC-micro electron capture detector and GC/MS using negative chemical ionization. Human brain samples were grouped as neurotypical controls (n=43), neurodevelopmental disorders with known genetic basis (n=32, including Down, Rett, Prader-Willi, Angelman, and 15q11-q13 duplication syndromes), and autism of unknown etiology (n=32). Unexpectedly, PCB 95 was significantly higher in the genetic neurodevelopmental group, but not idiopathic autism, as compared to neurotypical controls. Interestingly, samples with detectable PCB 95 levels were almost exclusively those with maternal 15q11-q13 duplication (Dup15q) or deletion in Prader-Willi syndrome. When sorted by birth year, Dup15q samples represented five out of six of genetic neurodevelopmental samples born after the 1976 PCB ban exhibiting detectable PCB 95 levels. Dup15q was the strongest predictor of PCB 95 exposure over age, gender, or year of birth. Dup15q brain showed lower levels of repetitive DNA methylation measured by LINE-1 pyrosequencing, but methylation levels were confounded by year of birth. These results demonstrate a novel paradigm by which specific POPs may predispose to genetic copy number variation of 15q11-q13.
Keywords: neurodevelopmental, environmental, copy number variation, autism, epigenetics, DNA methylation
Introduction
Autism spectrum disorders (ASD) are neurodevelopmental disorders characterized by deficits in social interaction, verbal and nonverbal communication, as well as a gain in repetitive stereotyped behaviors (Volkmar and Pauls 2003). Autism affects males more than females with a 4:1 frequency in the total population (Marco and Skuse 2006). A strong genetic component to ASD risk has been presumed based on past studies showing 70-90% concordance in monozygotic twins and 0-10% concordance in dizygotic twins (Bailey et al. 1995; Steffenburg et al. 1989). However, a more recent study of twins born between 1987 and 2004 in California demonstrated a 60% concordance rate in monozygotic twins and 27% concordance in dizygotic twins, suggesting that shared in utero environment is of critical importance in ASDs (Hallmayer et al. 2011). In recent decades, ASD prevalence has apparently increased, with current estimates at 1/88 children or 1/54 boys in the U.S. population (CDC 2012), with similar estimates in South Korea (Kim et al. 2011b; Rice et al. 2007). While changes in interpretation of symptoms and diagnosis are thought to account for 1/3 of the increase in autism rates, the remaining 2/3 increase in autism over recent decades is currently unexplained, therefore suggesting a role for environmental factors (Hertz-Picciotto et al.). Genetic and environmental interactions, as well as the epigenetic interface of genetic and environmental interactions are expected to be important, but are poorly understood for ASDs.
Persistent organic pollutants, especially polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs), that have accumulated in landfills, in the food supply, and human tissues, are of particular concern for human brain development (Gasull et al. 2011; Pessah et al. 2010; Weber et al. 2011). PCBs and PBDEs are known to disrupt neurotransmitter systems, endocrine systems, and intracellular signaling pathways (Dingemans et al. 2011; Eriksson et al. 2001; Guvenius et al. 2003; Pessah et al. 2010).
PCBs are a widely distributed class of environmental pollutants that were used in industrial products until adverse health effects were recognized in the 1970s, resulting in discontinued use after the 1976 Toxic Substances Control Act passage in the US Congress. The developmental neurotoxicity of PCBs became devastatingly apparent after the large-scale consumption of PCB-contaminated rice oil that occurred in 1968 in Japan and in 1979 in Taiwan (Kuratsune et al. 1971; Rogan et al. 1988). PCBs produced adverse effects on the developing brain with exposed offspring displaying behavioral abnormalities as well as significantly lower verbal and full-scale IQ (Chen et al. 1994).
While PCB levels are gradually declining in the environment following discontinued use, exposures to legacy sources remain a major concern to human health (Beyer and Biziuk 2009; Eubig et al. 2010; Pessah et al. 2010), and increasing PBDE body burdens are a growing concern for human exposures (Hertz-Picciotto et al. 2011; Messer 2010). PBDEs are used commercially as a flame retardant for plastics, foams and electronics. DE-71, a commercial penta-PBDE mixture, represents the greatest burden on Western populations. BDE-47 and -99, two specific congeners of DE-71, are the most abundant and found ubiquitously in the environment. Mouse models have demonstrated adverse neurodevelopmental outcomes associated with perinatal exposures to either PCBs or PBDEs, including changes in synaptic plasticity and formation of neuronal networks (Dingemans et al. 2007; Pessah et al. 2010), as well as growth, reproduction, and sociability (Ta et al. 2011; Woods et al. 2012).
PBDEs have been shown to have endocrine disrupting effects. Previous work has shown that these POPs can interact as antagonists or agonists at androgen, progesterone, and estrogen receptors (Branchi et al. 2003; Meerts et al. 2001). PBDEs, like their chemically related nondioxin-like PCBs, disrupt Ca2+ signaling by targeting ryanodine sensitive Ca2+ channels that influence signaling pathways responsible for activity-dependent dendritic growth (Kim et al. 2011a; Wayman et al. 2012a; Wayman et al. 2012b). Once metabolized, hydroxylated PCBs and PBDEs are structurally similar to thyroid hormones (TH) and have been shown to displace TH from thyroxin plasma transporter transthyretin (TTR) and decrease the levels of circulating T3 and T4 (Hamers et al. 2006). Collectively these xenobiotic mechanisms are of particular interest because the convergence of estrogen and thyroid hormone signaling are also mediated through Ca2+-dependent pathways that are important for proper neurodevelopment (Darras 2008; Davies et al. 2008; Eubig et al. 2010; Muchekehu and Harvey 2008; Prossnitz and Maggiolini 2009)
A relatively unexplored area of the effects of POPs on neurodevelopment is at the genetic and epigenetic level. Epigenetic mechanisms, such as DNA methylation, act at the interface of genetic and environmental factors, regulating gene expression and genome stability. Interestingly, an inverse correlation in DNA methylation at repetitive Alu and long interspersed nucleotide element 1 (LINE-1) sequences with high PCB concentrations was observed in blood samples from Greenlandic Inuit population (Rusiecki et al. 2008), and hypomethylation has been observed in PCB exposed rats (Desaulniers et al. 2009). Although PCBs and PBDEs are not known to affect developmental neurotoxicity by direct mutagenesis, epigenetic alterations such as repetitive DNA hypomethylation induced by POP exposures could potentially lead to genome instability and rearrangement, because DNA methylation acts globally on highly repetitive mammalian genomes to suppress retrotransposition. Large-scale chromosomal rearrangements, called copy number variations, occur frequently in the human genome between low copy repeats that are rich in CpG methylation sites. A higher frequency of de novo copy number variations is found in autism and other neurodevelopmental disorders such as schizophrenia, epilepsy, and intellectual disability (Consortium 2008; Helbig et al. 2009; Pinto et al. 2008; Sebat et al. 2007; Stefansson et al. 2008). However, very little is understood about the risk factors that predispose an individual to copy number variations. Even less is understood about the interaction between environmental risk factors and genetics in the etiology of ASDs.
In this study, we performed a comprehensive analysis of POP congener levels on a panel of 107 human postmortem brain samples from idiopathic ASD, known genetic neurodevelopmental disorders, and controls. From these analyses, we discovered an unexpected association between PCB 95 exposure and Dup15q syndrome.
Methods
Human post-mortem brain samples
Frozen cerebral cortex (Broadmann Areas 9 and 19) and cerebellum samples were obtained with assistance by the Autism Tissue Program from the University of Maryland Brain and Tissue bank for Neurodevelopmental Disorders, the Harvard Brain and Tissue Resource Center, and the University of Miami Brain and Tissue Bank for Neurodevelopmental Disorders. Brain samples were chosen from human cadavers where death resulted from non-neurological causes and where the post mortem interval was less than 24 hours. Samples were obtained from three categories: neurotypical controls with no known neurodevelopmental abnormalities, autism of unknown etiology (idiopathic ASD), and neurodevelopmental disorders with known genetic basis (genetic neurodevelopmental disorders). Brain samples were matched for age (range 4-61 years for neurotypical controls, 4-60 years for idiopathic autism, 2-56 years for genetic neurodevelopmental disorders) and sex (M:F ratio 0.86 for neurotypical controls, 2.88 for idiopathic autism, 0.82 for genetic neurodevelopmental disorders) based on sample availability. Genetic analysis of the genetic neurodevelopmental samples were either taken from Autism Tissue Program records or have been published previously (Hogart et al. 2009; Nagarajan et al. 2008; Scoles et al. 2011; Wang et al. 2008).
Persistent organic pollutant analysis
Post-mortem brain samples were stored at -80°C until analyzed. Sample extraction and clean-up for analysis of persistent organic pollutants on human postmortem cortex and cerebellum was performed as described previously (Woods et al. 2012). 100-500 mg of frozen tissue was used per sample, and cortex and cerebellum samples were analyzed separately if available from the same individual. PCBs were analyzed by the methods described previously with minor modifications (Kostyniak et al. 2005). Briefly, two surrogate standards (PCB-46 and -143) and two internal standards (PCB-30 and -204) were added to each sample. After sample extraction and cleanup, each sample was concentrated to 0.2 ml for chromatographic analysis. PCBs were analyzed with an Agilent 6890 GC equipped with a micro electron capture detector, and an HT8 capillary column, (50m × 0.22mm i.d.; 0.25μm film thickness; SGE, Austin, TX). The injection temperature was set at 260°C and helium gas flow rate was 1.2 ml/min. The initial oven temperature was 130°C, and was increased to 200°C at 4°C/min, then to 210°C at 1°C/min, then to 280°C at 2°C/min, and finally increased to 310°C at 20°C/min and held at 310°C for 6 min.
After PCB analysis, the samples were further concentrated to 0.1ml prior to PBDE analysis, as described previously (Woods, et al., 2012). One reagent blank, two sample blanks and three quality spike samples prepared in rendered chicken fat were included in each batch run and concentrations were determined from a standard curve.
Lipid content for each sample was determined by gravimetric analysis. Concentrations were normalized to lipid content in each brain sample.
Pyrosequencing analysis of DNA methylation
Genomic DNA was isolated from cerebral cortex, Brodmann area 19 (BA19) using Gentra Puregene Kit (Qiagen). Bisulfite treatment of 400 ng genomic DNA was performed, using EZ DNA Methylation-Direct conversion kit (Zymo Research). Triplicate PCR amplifications of LINE-1 repetitive elements were performed with Pyromark PCR kit (Qiagen) using the recommended protocol for Pyromark Q24 CpG LINE-1 methylation assay (Qiagen). Pyrosequencing of 6 CpG sites in the human OCA2 locus was performed in duplicate, using Pyromark CpG Assay primers (Qiagen,PM00168168). Published primers for pyrosequencing of p16 (Shaw et al. 2006) were also tested, but preliminary analyses revealed ≥2% methylation at seven assayed CpG positions in all three groups, which is below the level of sensitivity.Following amplification, pyrosequencing was performed on a Pyromark Q24 Pyrosequencer (Qiagen) using the manufacturers recommended protocol. Briefly, samples were purified using Streptavidin Sepharose High Performance beads (GE Healthcare) and prepared for pyrosequencing using the Pyromark Q24 Vacuum Workstation. Samples were sequenced using Pyromark Gold Q24 Reagents (Qiagen) and methylation levels were analyzed using Pyromark Q24 Software. An internal bisulfite conversion control was used in the pyrosequencing assay, which measured methylation at 3 CpG sites.
Statistical analyses
Group comparisons of PCB and PBDE levels were performed as either yes/no (scored for the presence or absence of each congener) or as continuous (using ng/g lipid values or 0 for samples with levels below the level of detection) using exact Wilcoxon non-parametric test with Monte Carlo estimates of exact P-values to account for numerous ties. We constructed multivariate logistic regression models to estimate odds ratios (ORs) adjusted for confounding variables and corresponding 95% confidence intervals (CIs) for each PCB and PBDE congener using SAS, version 9.3 software (SAS Institute Inc., Cary, NC). We examined the following variables as potential confounders of the association between each congener and idiopathic ASD or genetic ND: individual's sex (male = reference, birth period (1976 or before, after 1976 = reference), and brain region (BA 9 = reference, BA 19, cerebellum). Models were adjusted for birth period and brain region, and if numbers allowed, sex. Multivariate linear regression models were also constructed to examine predictors of PCB 95 using SAS, version 9.3 software (SAS Institute Inc., Cary, NC). These models included the same variables described above in addition to diagnostic group indicator variables. Student's t-tests were used for initial comparisons for group means for LINE-1 and OCA2 pyrosequencing, but logistic regression models adjusting for confounding variables were additionally used for methylation analyses.
Results
Comprehensive analysis of POP levels in human postmortem brain samples
To assess the relationship of POP levels in lipid-rich brain tissue with ASD diagnosis, frozen postmortem human brain samples from 107 individuals were chosen from existing tissue banks, with clinical information organized through the Autism Tissue Program (http://atpportal.org). Samples included two different cortical regions (Cx, BA9 or BA19) cerebellum (Cb), or pons, based on remaining availability from the tissue banks. For some individuals, more than one brain region was sampled but only one brain region is shown, for simplicity, in Supplementary Table I as brain region was not found to be a significant confounding variable for exposures. Age of the individual at death, year of birth and sex are also shown when available as these factors were included as variables in statistical tests, in addition to brain region.
Each sample was analyzed for 7 congeners of PBDE by GC/MS using negative chemical ionization and 8 congeners of PCBs by GC-micro electron capture detector, normalized to lipid content, with the results shown in Supplementary Table I. Congeners to be analyzed were selected based on detection in other human tissues and relevancy to neurodevelopmental toxicity.
Samples were grouped in one of three groups: neurotypical controls with no known neurodevelopmental abnormalities, autism of unknown etiology (idiopathic ASD), and neurodevelopmental disorders with known genetic basis, to test the hypothesis that idiopathic autism may correlate with POP exposures. The idiopathic ASD group included samples from 31 individuals diagnosed with autism and one with pervasive developmental disorder, not otherwise specified. Because of the limited availability of tissue for any single genetic neurodevelopmental disorder, the combined genetic neurodevelopmental disorder group (n=32) included a mixture of Angelman syndrome (AS, n=4), Down syndrome (DS, n=4), proximal 15q duplication (Dup15q, n=6), Prader-Willi syndrome (PWS, n=6), and Rett syndrome (RTT, n=12).
Unexpectedly, PCB 95 (2,2′,3,5′,6-pentachlorobiphenyl) was found in significantly higher concentrations in the genetic neurodevelopmental disorder group compared to the neurotypical control group (Figure 1A). Only one control showed detectable levels. PCB 95 detection (yes/no) was significantly associated with the neurodevelopmental disorder group after adjustment for brain region and birth period in regression models (Odds ratio (OR) = 54, 95% confidence interval (CI): 5, 568). In contrast to PCB 95, BDE 153 was significantly lower in the genetic neurodevelopmental disorder group (P = 0.0001) and idiopathic ASD group (P = 0.047), compared to controls (Figure 1B). After adjustment for brain region, birth period, and sex in regression models, BDE 153 detection (yes/no) and continuous levels remained significantly negatively associated with the neurodevelopmental disorder group (odds ratio (OR) = 53.9, 95% confidence interval (CI): 5.1, 568.3, P = 0.0009) and PCB 95 continuous levels associated with borderline significance (OR = 1.7, CI: 0.99, 3.0, P = 0.054). In addition, BDE 153 detection (yes/no) remained significantly negatively associated with idiopathic ASD (OR = 0.8, CI: 0.1, 0.7) but the continuous variable did not (OR = 0.9, CI: 0.8, 1.04). All other findings remained similar (non-significant) after adjustment for brain region, birth period, and sex for both yes/no and continuous detection in regression models.
Figure 1.
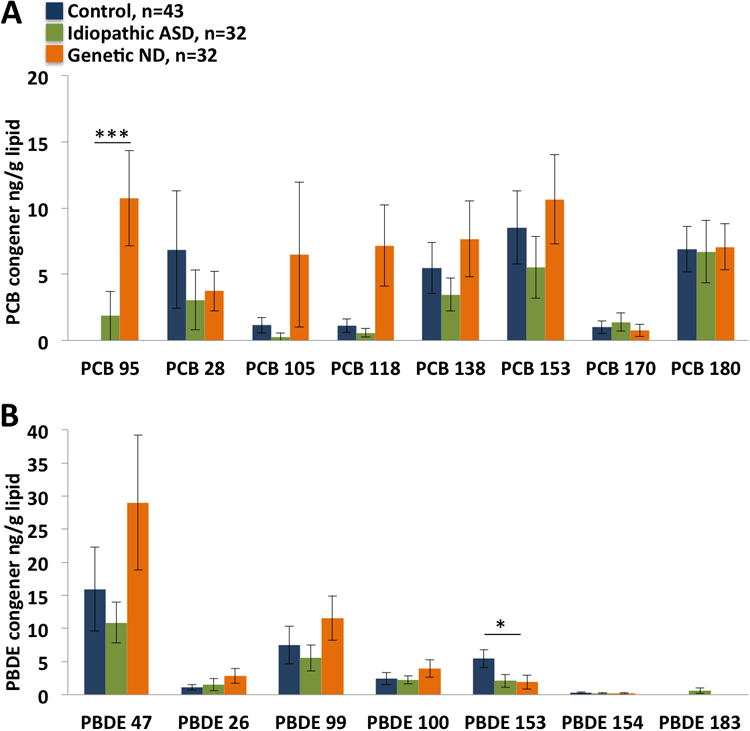
PCB and PBDE congener levels in human postmortem brain samples compared by diagnostic group defined in Supplementary Table I as control, idiopathic ASD, or known genetic neurodevelopmental disorder (ND). A) Average level of each PCB congener is compared between diagnostic groups. The lowest level of detection is 0.1 ng/g lipid for all PCB congeners. Samples having levels below the level of detection were set to a value of 0 and used in the average. B) Average level of each PBDE congener in the sample groups. The lowest level of detection is 0.012 ng/g for PBDE-47, -28, -99, -100, -153, and -154. PBDE-183 lowest level of detection is 0.05 ng/g. Samples with levels below the level of detection were set to a value of 0 and used in the average. Error bars are SEM. *P < 0.05, ***P < 0.001 by exact Wilcoxon non-parametric test with Monte Carlo estimates of exact p-values to account for numerous ties.
Analysis of POP levels by year of birth and genetic ND subgroup
Because of the expected differences in lifetime PCB exposure, depending on generational differences, PCB exposure levels in postmortem brain samples were analyzed by both diagnostic groupings and year of birth. The year 1976 was chosen to divide the samples into two groups, since this was the year of the PCB ban in the U.S. While control samples were roughly equally divided between pre- and post-1976 birth years, idiopathic ASD and genetic neurodevelopmental disorder samples were more frequently represented in the post-1976 grouping (Figure 2A). Figure 2B shows scattergrams of individual samples colored according to diagnostic group and plotted according to year of birth with 1976 as the dividing intersect for four PCB congeners (-95, -118, -138, and -153). Figure 2C shows the means of each diagnostic group divided between pre- and post-1976. While PCB 118 and PCB 153 showed an expected decrease in individual and average levels with increasing year of birth, PCB 95 levels were highest in the genetic neurodevelopmental disorder group, both pre- and post-1976. Both genetic neurodevelopmental disorder diagnosis (OR = 12.6, CI:6.5, 19.7, P <0.0001) and Dup15q (OR = 31.2, CI: 4.6, 6.8, P <0.0001) were significant predictors of PCB 95 by multiple linear regression testing adjusting for brain region, birth year, and sex.
Figure 2. Relationships between PCB exposure and year of birth.
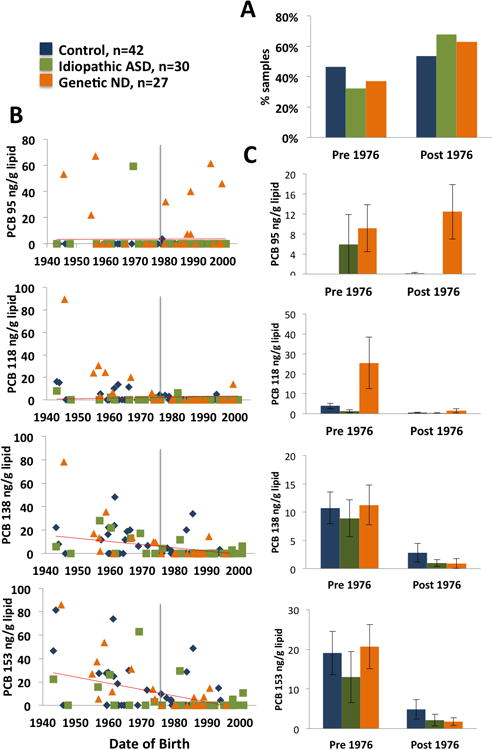
Because of the ban on PCBs in the US in 1976, this year was selected for dividing the samples between pre- and post-1976 (vertical line). A) Distribution of samples by diagnostic group and year of birth group. Controls were evenly distributed between the pre- and post-PCB ban groups, while the idiopathic ASD and genetic ND groups were more frequently born post 1976. B) Each brain sample is graphed individually by PCB congener level (four most common PCB congeners, -95, -118, -138, and -153) and year of birth, with a grey bar set at 1976 and a linear slope shown by the red bar. PCB 138, and -153 (but not PCB 95 or -118) show a trend for decreased levels with increasing year of birth. For PCB 95, six samples from the ND group have detectable levels despite being born post-1976, and five of these samples were from Dup15q syndrome. C) Grouped comparisons of PCB 95, -118, -138, and -153 in samples born before or after 1976.
Strikingly, five samples from individuals born after 1976 with both genetic neurodevelopmental disorder (orange triangles) and detectable PCB 95 levels (Figure 2B, top right) were from the same genetic subgroup of Dup15q, despite only 6 total samples included in this subgroup (Supplementary Table I). Furthermore, the three samples born prior to 1976 from genetic ND (orange triangles) with detectable PCB 95 levels (Figure 2B, top left) were diagnosed with Prader-Willi syndrome (out of 6 total PWS), another 15q genetic copy number variation with large deletions from similar breakpoints as those observed in Dup15q.
Analysis of DNA methylation levels in Dup15q brain samples compared to ASD and controls
Since PCB levels have been associated with reduced levels of repetitive DNA methylation (Rusiecki et al. 2008) and hypomethylation of repetitive DNA has been shown to predispose to genome instability (Kim et al. 2004; Shvachko 2009), we investigated a possible epigenetic link between the chromosomal rearrangements in Dup15q and increased PCB 95 exposure. For methylation analyses, all DNA samples were isolated from the same brain region (cerebral cortex, BA19). DNA isolated from all six Dup15q samples were compared to control and idiopathic ASD samples for repetitive LINE-1 methylation levels by pyrosequencing. Figure 3A demonstrates that Dup15q, but not idiopathic ASD samples showed lower LINE-1 methylation, corresponding to ∼2% decrease in average methylation, which was significant by t-test. In contrast, the promoter of a single gene locus on 15q13.1, OCA2, was not significantly different between control and Dup15q or idiopathic ASD (Fig. 3B). Adjusted analysis for the association between methylation and Dup15q show that age and sex were not confounders (cause less than a 10% change in the beta (and OR) estimate for methylation). Unexpectedly, birth year was a confounder, with earlier year of birth associated with higher methylation for both LINE-1 and OCA2 in controls (Fig. 3C, D). For LINE-1 in Dup15q compared to controls, analysis adjusted for year of birth produced an OR of 0.6 (0.2, 1.6) compared to a crude OR of 0.5 (0.2, 1.1). Therefore, even though Dup15q samples were hypomethylated at LINE-1, we do not know if this difference is due to the genetic, PCB 95 exposure, or year of birth differences in this sample group.
Figure 3. DNA methylation levels of postmortem brain samples.
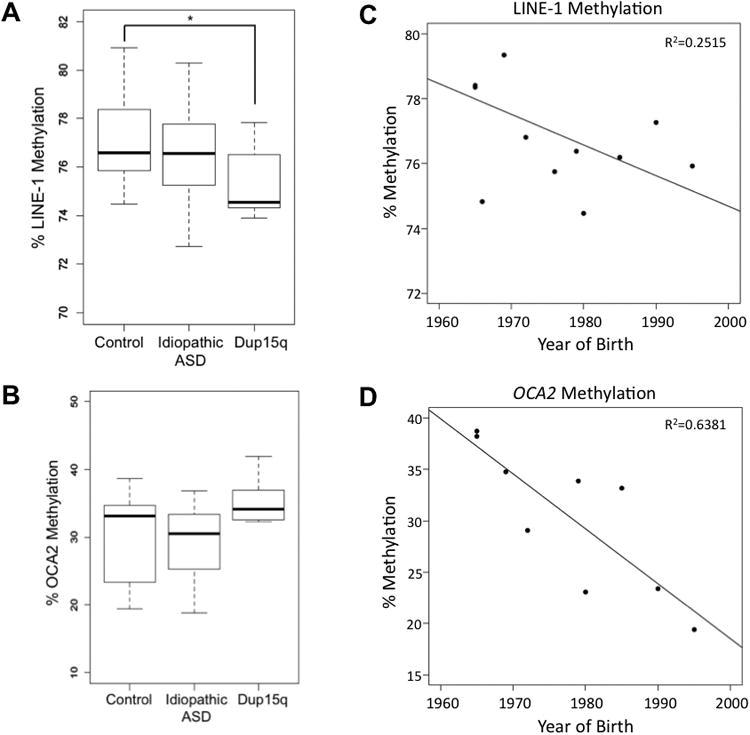
DNA isolated from cerebral cortex Brodmann area 19 (Cx BA19), 12 Control, 12 Idiopathic ASD, 6 Dup15q were assayed for DNA methylation levels by the Pyromark pyrosequencing assay. A) Methylation of repetitive LINE-1. The mean percent methylation of the 3 CpG sites was determined for each individual for the values shown, but results were also compared for each individual CpG site with similar results (data not shown). The CV for the averaged LINE-1 methylation for control, idiopathic autism, and Dup15q were 5.5%, 5.9%, and 4.3%, respectively. Graphs are box and whisker plots representing mean ± SEM of different individuals. * P = 0.05 by t-test. B) Methylation of single copy gene OCA2, averaged from 6 CpG sites. Graphs are box and whisker plots representing mean ± SEM. The CV for the averaged OCA2 methylation for control, idiopathic autism, and Dup15q were 2.5%, 3.3%, and 2.6%, respectively C) Year of birth was the largest confounder, compared to age and sex, with a trend towards lower percent methylation of LINE-1 with increased year of birth in controls (P = 0.06). D) A significant association was observed for decreased methylation with increased year of birth for OCA2 (P = 0.006).
Discussion
This study reaffirms the presence of detectable levels of PCBs in human brain (Caudle et al. 2006; Chu et al. 2003; Dewailly et al. 1999), a tissue that is a target of both the lifetime accumulation and the predominant neurodevelopmental toxicity. In spite of the small sample size analyzed, this investigation revealed some unexpected and provocative results about the relationship between PCB 95 levels and chromosomal rearrangements of 15q11-q13 found in ASD. In this study, we observe that Dup15q genetic diagnosis was the strongest predictor of PCB 95 exposure, over year of birth, brain region, or sex. Furthermore, Dup15q samples showed DNA hypomethylation, suggesting an epigenetic component to the association between Dup15q and PCB 95.
Dup15q is one of the most common copy number variations observed in ASD, at an estimated 1-3% of cases (Hogart et al. 2008). Duplications of proximal 15q occur as either a supernumary isodicentric chromosome (idic15) or interstitial duplications through misalignment errors of low copy repeat rich breakpoint hotspots (BP1-5) in meiosis I. 15q11-q13 deletions that result in Prader-Willi and Angelman syndrome occur between similar breakpoint regions as the duplications. Environmental factors leading to 15q11-q13 deletions or duplications have not been previously reported. However, there are reported unexplained differences in the percentage of PWS cases caused by 15q11-q13 deletion in Taiwan (84%)(Lin et al. 2007) and Japan (80%)(Nakamura et al. 2009) compared to Holland (54%)(Sinnema et al.) and the UK (50%)(Whittington et al. 2007). Our results demonstrated that 3/6 Prader-Willi syndrome and 5/6 Dup 15q brain samples showed detectable levels of PCB 95 suggests that this exposure should be investigated as a potential environmental contributor of the differing copy number variation rates in different geographical regions.
In the emerging area of “environmental epigenetics”, several environmental toxins, such as diethylstilbestrol, bisphenol A, and dioxin, are associated with reduced levels of repetitive DNA methylation (Baccarelli and Bollati 2009). In contrast, increasing the dietary supply of methyl donors through folate supplementation is associated with increased DNA methylation (Dolinoy et al. 2007). These results suggest that PCB congeners like PCB 95 influence both the glutathione synthesis pathway and the one-carbon metabolism pathway, which supplies methyl groups for DNA methylation (Lee et al. 2009). In our previous investigation of perinatal exposure of BDE-47 in a Mecp2 mutant mouse model, defects in sociability corresponded to reduced levels of DNA methylation in brain in female offspring (Woods et al. 2012). High levels of DNA methylation in neurons identified by large-scale genomic mapping identify neurodevelopmental and synaptic genes (Schroeder et al. 2011), so reduced DNA methylation from environmental exposures may compromise gene expression of a large subset of neurodevelopmentally important transcripts.
Developmental exposure of rats to PCB 95 alters cortical networks (Kenet et al. 2007), hippocampal connectivity and long-term potentiation (Kim et al. 2011a; Kim et al. 2009; Schantz et al. 1997), and activity dependent dendritic growth (Lein et al. 2007; Wayman et al. 2012a), apparently by interfering with Ca2+ dependent signaling mediated, at least in part, by direct interactions with ryanodine receptors (Samso et al. 2009). It is important to note that Ca2+-dependent pathways influenced by PCB 95, such as those regulated by Ca2+/CaM kinases (Wayman et al. 2012a; Wayman et al. 2012b), are essential for regulating epigenetic memory. For example, Ca2+/CaM Kinase regulates phosphorylation of the methyl-CpG binding protein MeCP2 (Murgatroyd and Spengler 2011; Zhou et al. 2006), and genes involved in Ca2+-dependent pathways were significantly enriched in neuronal highly methylated domains (Schroeder et al. 2011).
These investigations suggest that an “integrative genetics” approach (LaSalle 2011) of investigating environmental exposures, together with genetic investigations of copy number variations and the dynamic epigenetic landscape in brain, may be most informative for deciphering the complex etiology of autism.
Summary of author contributions
JML, INP, and PJK designed the study and obtained funding. MMM and LHC collected and analyzed the data. RW collected methylation data and analyses. RJS performed statistical analyses. MMM, RW and JML prepared the manuscript draft with important intellectual input from INP, PJK, and RJS. All authors approved the final manuscript.
Supplementary Material
Acknowledgments
This work was supported by NIH R01ES015171, R01ES0210707 (JML), 2R01HD041462 (JML), R01 ES014901 (INP), R01 ES017425 (INP), T32ES002321 (RW), 2K12HD051958-06 (RJS) and the NIEHS/EPA Center for Children's Environmental Health PO1 ES11269, the U.S. Environmental Protection Agency (U.S. EPA) through the Science to Achieve Results (STAR) program award numbers R833292 and R829388. Additional funding was provided by the J.B. Johnson Foundation.
References
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Beyer A, Biziuk M. Environmental fate and global distribution of polychlorinated biphenyls. Rev of Environ Contam Toxicol. 2009;201:137–158. doi: 10.1007/978-1-4419-0032-6_5. [DOI] [PubMed] [Google Scholar]
- Branchi I, Capone F, Alleva E, Costa LG. Polybrominated diphenyl ethers: neurobehavioral effects following developmental exposure. Neurotoxicology. 2003;24:449–462. doi: 10.1016/S0161-813X(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Delea KC, Guillot TS, Wang M, Pennell KD, Miller GW. Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson's disease-associated dopamine toxicity. Toxicol Sci. 2006;92:490–499. doi: 10.1093/toxsci/kfl018. [DOI] [PubMed] [Google Scholar]
- CDC. Prevalence of Autism Spectrum Disorders — Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008. MMWR. 2012;61(SS03):1–19. [PubMed] [Google Scholar]
- Chen YC, Yu ML, Rogan WJ, Gladen BC, Hsu CC. A 6-year follow-up of behavior and activity disorders in the Taiwan Yu-cheng children. Am J Public Health. 1994;84:415–421. doi: 10.2105/ajph.84.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Covaci A, Schepens P. Levels and chiral signatures of persistent organochlorine pollutants in human tissues from Belgium. Environmental Research. 2003;93:167–176. doi: 10.1016/s0013-9351(03)00016-1. [DOI] [PubMed] [Google Scholar]
- Consortium IS. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras VM. Endocrine disrupting polyhalogenated organic pollutants interfere with thyroid hormone signalling in the developing brain. Cerebellum. 2008;7:26–37. doi: 10.1007/s12311-008-0004-5. [DOI] [PubMed] [Google Scholar]
- Davies W, Lynn PM, Relkovic D, Wilkinson LS. Imprinted genes and neuroendocrine function. Frontiers in Neuroendocrinology. 2008;29:413–427. doi: 10.1016/j.yfrne.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Desaulniers D, Xiao GH, Lian H, Feng YL, Zhu J, Nakai J, Bowers WJ. Effects of mixtures of polychlorinated biphenyls, methylmercury, and organochlorine pesticides on hepatic DNA methylation in prepubertal female Sprague-Dawley rats. Int J Toxicol. 2009;28:294–307. doi: 10.1177/1091581809337918. [DOI] [PubMed] [Google Scholar]
- Dewailly E, Mulvad G, Pedersen HS, Ayotte P, Demers A, Weber JP, Hansen JC. Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland. Environ Health Perspect. 1999;107:823–828. doi: 10.1289/ehp.99107823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, Ramakers GM, Gardoni F, van Kleef RG, Bergman A, Di Luca M, van den Berg M, Westerink RH, Vijverberg HP. Neonatal exposure to brominated flame retardant BDE-47 reduces long-term potentiation and postsynaptic protein levels in mouse hippocampus. Environ Health Perspect. 2007;115:865–870. doi: 10.1289/ehp.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, van den Berg M, Westerink RH. Neurotoxicity of brominated flame retardants: (in)direct effects of parent and hydroxylated polybrominated diphenyl ethers on the (developing) nervous system. Environ Health Perspect. 2011;119:900–907. doi: 10.1289/ehp.1003035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Jakobsson E, Fredriksson A. Brominated flame retardants: a novel class of developmental neurotoxicants in our environment? Environ Health Perspect. 2001;109:903–908. doi: 10.1289/ehp.01109903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubig PA, Aguiar A, Schantz SL. Lead and PCBs as risk factors for attention deficit/hyperactivity disorder. Environ Health Perspect. 2010;118:1654–1667. doi: 10.1289/ehp.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasull M, Bosch de Basea M, Puigdomenech E, Pumarega J, Porta M. Empirical analyses of the influence of diet on human concentrations of persistent organic pollutants: a systematic review of all studies conducted in Spain. Environment International. 2011;37:1226–1235. doi: 10.1016/j.envint.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Guvenius DM, Aronsson A, Ekman-Ordeberg G, Bergman A, Noren K. Human prenatal and postnatal exposure to polybrominated diphenyl ethers, polychlorinated biphenyls, polychlorobiphenylols, and pentachlorophenol. Environ Health Perspect. 2003;111:1235–1241. doi: 10.1289/ehp.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers T, Kamstra JH, Sonneveld E, Murk AJ, Kester MH, Andersson PL, Legler J, Brouwer A. In vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol Sci. 2006;92:157–173. doi: 10.1093/toxsci/kfj187. [DOI] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I, Bergman A, Fangstrom B, Rose M, Krakowiak P, Pessah I, Hansen R, Bennett DH. Polybrominated diphenyl ethers in relation to autism and developmental delay: a case-control study. Environ Health. 2011;10:1. doi: 10.1186/1476-069X-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I, Croen LA, Hansen R, Jones CR, van de Water J, Pessah IN. The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ Health Perspect. 2006;114:1119–1125. doi: 10.1289/ehp.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Leung KN, Wang NJ, Wu DJ, Driscoll J, Vallero RO, Schanen NC, LaSalle JM. Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet. 2009;46:86–93. doi: 10.1136/jmg.2008.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Wu D, Lasalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2008 doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenet T, Froemke RC, Schreiner CE, Pessah IN, Merzenich MM. Perinatal exposure to a noncoplanar polychlorinated biphenyl alters tonotopy, receptive fields, and plasticity in rat primary auditory cortex. Proc Nat Acad Sci USA. 2007;104:7646–7651. doi: 10.1073/pnas.0701944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Bose DD, Ghogha A, Riehl J, Zhang R, Barnhart CD, Lein PJ, Pessah IN. Para- and ortho-substitutions are key determinants of polybrominated diphenyl ether activity toward ryanodine receptors and neurotoxicity. Environ Health Perspect. 2011a;119:519–526. doi: 10.1289/ehp.1002728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Inan SY, Berman RF, Pessah IN. Excitatory and inhibitory synaptic transmission is differentially influenced by two ortho-substituted polychlorinated biphenyls in the hippocampal slice preparation. Toxicol Appl Pharm. 2009;237:168–177. doi: 10.1016/j.taap.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Trinh BN, Long TI, Oghamian S, Laird PW. Dnmt1 deficiency leads to enhanced microsatellite instability in mouse embryonic stem cells. Nucleic Acids Res. 2004;32:5742–5749. doi: 10.1093/nar/gkh912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Leventhal BL, Koh YJ, Fombonne E, Laska E, Lim EC, Cheon KA, Kim SJ, Kim YK, Lee H, et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psych. 2011b;168:904–912. doi: 10.1176/appi.ajp.2011.10101532. [DOI] [PubMed] [Google Scholar]
- Kostyniak PJ, Hansen LG, Widholm JJ, Fitzpatrick RD, Olson JR, Helferich JL, Kim KH, Sable HJ, Seegal RF, Pessah IN, et al. Formulation and characterization of an experimental PCB mixture designed to mimic human exposure from contaminated fish. Toxicol Sci. 2005;88:400–411. doi: 10.1093/toxsci/kfi338. [DOI] [PubMed] [Google Scholar]
- Kuratsune M, Yoshimura T, Matsuzaka J, Yamaguchi A. Yusho, a poisoning caused by rice oil contaminated with polychlorinated biphenyls. HSMHA Health Rep. 1971;86:1083–1091. [PMC free article] [PubMed] [Google Scholar]
- LaSalle JM. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics. 2011;6:862–869. doi: 10.4161/epi.6.7.16353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Jacobs DR, Jr, Porta M. Hypothesis: a unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ Health Perspect. 2009;117:1799–1802. doi: 10.1289/ehp.0900741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein PJ, Yang D, Bachstetter AD, Tilson HA, Harry GJ, Mervis RF, Kodavanti PR. Ontogenetic alterations in molecular and structural correlates of dendritic growth after developmental exposure to polychlorinated biphenyls. Environ Health Perspect. 2007;115:556–563. doi: 10.1289/ehp.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY, Lin SP, Chuang CK, Chen MR, Yen JL, Lee YJ, Huang CY, Tsai LP, Niu DM, Chao MC, et al. Genotype and phenotype in patients with Prader-Willi syndrome in Taiwan. Acta Paediatr. 2007;96:902–905. doi: 10.1111/j.1651-2227.2007.00284.x. [DOI] [PubMed] [Google Scholar]
- Marco EJ, Skuse DH. Autism-lessons from the X chromosome. Social Cognitive and Affective Neuroscience. 2006;1:183–193. doi: 10.1093/scan/nsl028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerts IA, Letcher RJ, Hoving S, Marsh G, Bergman A, Lemmen JG, van der Burg B, Brouwer A. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol A compounds. Environ Health Perspect. 2001;109:399–407. doi: 10.1289/ehp.01109399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer A. Mini-review: polybrominated diphenyl ether (PBDE) flame retardants as potential autism risk factors. Physiol Behav. 2010;100:245–249. doi: 10.1016/j.physbeh.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Muchekehu RW, Harvey BJ. 17beta-estradiol rapidly mobilizes intracellular calcium from ryanodine-receptor-gated stores via a PKC-PKA-Erk-dependent pathway in the human eccrine sweat gland cell line NCL-SG3. Cell Calcium. 2008;44:276–288. doi: 10.1016/j.ceca.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Murgatroyd C, Spengler D. Epigenetic programming of the HPA axis: early life decides. Stress. 2011;14:581–589. doi: 10.3109/10253890.2011.602146. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz-Picciotto I, Hansen RL, Van de Water J, Pessah IN, Jiang R, et al. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 2008;1:169–178. doi: 10.1002/aur.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Nagai T, Iida T, Ozeki S, Nohara Y. Epidemiological aspects of scoliosis in a cohort of Japanese patients with Prader-Willi syndrome. Spine J. 2009;9:809–816. doi: 10.1016/j.spinee.2009.06.017. [DOI] [PubMed] [Google Scholar]
- Pessah IN, Cherednichenko G, Lein PJ. Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacology & Therapeutics. 2010;125:260–285. doi: 10.1016/j.pharmthera.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2008;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Moland Cell Endocrinology. 2009;308:32–38. doi: 10.1016/j.mce.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CE, Baio J, Van Naarden Braun K, Doernberg N, Meaney FJ, Kirby RS. A public health collaboration for the surveillance of autism spectrum disorders. Paediatric and Perinatal Epidemiology. 2007;21:179–190. doi: 10.1111/j.1365-3016.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- Rogan WJ, Gladen BC, Hung KL, Koong SL, Shih LY, Taylor JS, Wu YC, Yang D, Ragan NB, Hsu CC. Congenital poisoning by polychlorinated biphenyls and their contaminants in Taiwan. Science. 1988;241:334–336. doi: 10.1126/science.3133768. [DOI] [PubMed] [Google Scholar]
- Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect. 2008;116:1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samso M, Feng W, Pessah IN, Allen PD. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS biology. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schantz SL, Seo BW, Wong PW, Pessah IN. Long-term effects of developmental exposure to 2,2′,3,5′,6-pentachlorobiphenyl (PCB 95) on locomotor activity, spatial learning and memory and brain ryanodine binding. Neurotoxicology. 1997;18:457–467. [PubMed] [Google Scholar]
- Schroeder DI, Lott P, Korf I, LaSalle JM. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Research. 2011;21:1583–1591. doi: 10.1101/gr.119131.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoles HA, Urraca N, Chadwick SW, Reiter LT, Lasalle JM. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Molecular Autism. 2011;2:19. doi: 10.1186/2040-2392-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, Field JK, Risk JM. Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006;94:561–568. doi: 10.1038/sj.bjc.6602972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvachko LP. DNA hypomethylation as Achilles' heel of tumorigenesis: a working hypothesis. Cell Biol Int. 2009;33:904–910. doi: 10.1016/j.cellbi.2009.02.018. [DOI] [PubMed] [Google Scholar]
- Sinnema M, van Roozendaal KE, Maaskant MA, Smeets HJ, Engelen JJ, Jonker-Houben N, Schrander-Stumpel CT, Curfs LM. Different distribution of the genetic subtypes of the Prader-Willi syndrome in the elderly. Eur J Hum Genet. 18:993–998. doi: 10.1038/ejhg.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Ta TA, Koenig CM, Golub MS, Pessah IN, Qi L, Aronov PA, Berman RF. Bioaccumulation and behavioral effects of 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47) in perinatally exposed mice. Neurotoxicol Teratol. 2011;33:393–404. doi: 10.1016/j.ntt.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmar FR, Pauls D. Autism. Lancet. 2003;362:1133–1141. doi: 10.1016/S0140-6736(03)14471-6. [DOI] [PubMed] [Google Scholar]
- Wang NJ, Parokonny AS, Thatcher KN, Driscoll J, Malone BM, Dorrani N, Sigman M, LaSalle JM, Schanen NC. Multiple forms of atypical rearrangements generating supernumerary derivative chromosome 15. BMC Genet. 2008;9:2. doi: 10.1186/1471-2156-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Bose DD, Yang D, Lesiak A, Bruun D, Impey S, Ledoux V, Pessah IN, Lein PJ. PCB 95 Modulates Calcium-Dependent Signaling Pathway Responsible for Activity-Dependent Dendritic Growth. Env Health Perspect. 2012a doi: 10.1289/ehp.1104833. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Yang D, Bose DD, Lesiak A, Ledoux V, Bruun D, Pessah IN, Lein PJ. PCB 95 Promotes Dendritic Growth via Ryanodine Receptor-Dependent Mechanisms. Env Health Perspect. 2012b doi: 10.1289/ehp.1104832. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber R, Watson A, Forter M, Oliaei F. Review Article: Persistent organic pollutants and landfills - a review of past experiences and future challenges. Waste management&research: the journal of the International Solid Wastes and Public Cleansing Association, ISWA. 2011;29:107–121. doi: 10.1177/0734242X10390730. [DOI] [PubMed] [Google Scholar]
- Whittington JE, Butler JV, Holland AJ. Changing rates of genetic subtypes of Prader-Willi syndrome in the UK. Eur J Hum Genet. 2007;15:127–130. doi: 10.1038/sj.ejhg.5201716. [DOI] [PubMed] [Google Scholar]
- Woods R, Vallero RO, Golub MS, Suarez JK, Ta TA, Yasui DH, Chi LH, Kostyniak PJ, Pessah IN, Berman RF, et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Human Molecular Genetics. 2012 doi: 10.1093/hmg/dds046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, et al. Brain-Specific Phosphorylation of MeCP2 Regulates Activity-Dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.