

Abstract
Gonadotropin-releasing hormone (GnRH) is secreted from neurons within the hypothalamus and is necessary for reproductive function in all vertebrates. GnRH is also found in organs outside of the brain and plays an important role in Leydig cell steroidogenesis in the testis. However, the signalling pathways mediating this function remain largely unknown. In this study, we investigated whether components of the mitogen-activated protein kinase (MAPK) pathways are involved in GnRH agonist (GnRHa)-induced testis steroidogenesis in rat Leydig cells. Primary cultures of rat Leydig cells were established. The expression of 3β-hydroxysteroid dehydrogenase (3β-HSD) and the production of testosterone in response to GnRHa were examined at different doses and for different durations by RT-PCR, Western blot analysis and radioimmunoassay (RIA). The effects of GnRHa on ERK1/2, JNK and p38 kinase activation were also investigated in the presence or absence of the MAPK inhibitor PD-98059 by Western blot analysis. GnRHa induced testosterone production and upregulated 3β-HSD expression at both the mRNA and protein levels; it also activated ERK1/2, but not JNK and p38 kinase. Although the maximum effects of GnRHa were observed at a concentration of 100 nmnol L−1 after 24 h, activation of ERK1/2 by GnRHa reached peak at 5 min and it returned to the basal level within 60 min. PD-98059 completely blocked the activation of ERK1/2, the upregulation of 3β-HSD and testosterone production. Our data show that GnRH positively regulates steroidogenesis via ERK signalling in rat Leydig cells. ERK1/2 activation by GnRH may be responsible for the induction of 3β-HSD gene expression and enzyme production, which may ultimately modulate steroidogenesis in rat Leydig cells.
Keywords: 3β-HSD, ERK1/2, GnRH, JNK, Leydig cell, p38
Introduction
Gonadotropin-releasing hormone (GnRH) is a hypothalamic neuronal decapeptide that is secreted in a pulsatile manner. Due to its pivotal role in vertebrate reproduction, GnRH and its analogues have been used extensively to treat hormone-dependent diseases.1 Results from previous studies have shown that GnRH agonists stimulate testosterone production in Leydig cells, and the stimulatory effect is dependent on calcium but not on cAMP or cGMP.2, 3, 4
3β-hydroxysteroid dehydrogenase (3β-HSD) is a steroid-metabolising enzyme that converts pregnenolone to progesterone.5 This enzyme is an obligatory and regulatory intermediate in testosterone biosynthesis and is used as a histochemical marker for testosterone-producing Leydig cells. 3β-HSD has a wide tissue distribution with cell-specific expression in the placenta, adrenal gland, ovary and testis, and is regulated by multiple signal transduction pathways in response to the stimulation of several growth factors, steroids, and cytokines.6 In prepubertal murine Leydig cells, the 3β-HSD enzyme is upregulated during GnRH-induced steroidogenesis.7
GnRH-induced signalling pathways have been studied in a number of cell systems. GnRH is known to activate ERK, JNK and p38 mitogen-activated protein kinase (MAPK) in both pituitary cells and GnRH receptor-transfected cells.8, 9, 10, 11 A panel of signalling molecules, including protein kinase C, ERK, JNK, p38 and c-Src, but not Ca2+, have been shown to be involved in GnRH-stimulated ovine follicle-stimulating hormone-β promoter activity in LβT-2 cells,9 and ERK1/2 has been shown to be involved in the expression of the gonadotropin α-subunit in pituitary cells.12, 13 The ERK and JNK MAPK pathways are involved in both basal and GnRH-stimulated luteinizing hormone-β (LH-β) transcription.14 Although the MAPK pathway has been implicated in many of the functions of GnRH, the signalling pathway mediating GnRH-induced steroidogenesis is still largely unknown.
Recently, several groups have reported that the ERK1/2, JNK and p38 MAP kinases are involved in the regulation of testosterone production15, 16, 17 in response to a variety of extracellular factors, including LH,18 interleukin-1α15 and human chorionic gonadotropin.19 Although these studies demonstrate an important role for ERK1/2 in the acute stimulation of steroidogenesis in immature Leydig cells, the contribution of MAPK signalling pathways to GnRH-induced steroidogenesis in rat Leydig cells remains unclear. In our study, we examined the effects of GnRH on 3β-HSD expression and steroidogenesis and investigated whether the activation of the MAPK is involved in these events in rat Leydig cells.
Materials and methods
Materials
The GnRH agonist (GnRHa) ‘alarelin acetate' was obtained from Anhui Fengyuan (Ma'anshan, China). Dulbecco's modified Eagle's medium/nutrient mixture F12 was purchased from Invitrogen (Grand Island, NY, USA). Percoll, HEPES and collagenase type I were purchased from Sigma (St. Louis, MO, USA). Hanks' balanced salt solution, without Ca2+ and Mg2+, and penicillin-streptomycin were purchased from Life Technologies (Paisley, Scotland, UK). The MEK1 inhibitor PD-98059 and the Reverse Transcription System were purchased from Promega (Madison, WI, USA). Rabbit monoclonal antibodies against phospho-p44/42 (p-ERK1/2, doubly phosphorylated), phospho-SAPK/JNK (doubly phosphorylated) and SAPK/JNK were purchased from Cell Signalling Technology (Beverly, MA, USA). Polyclonal ERK1/2 and phospho-p38 antibodies were purchased from Santa Cruz Biotechnology (Eugene, OR, USA). The antibody against β-actin as well as the secondary horseradish peroxidase-conjugated goat anti-mouse antibodies and goat anti-rabbit antibodies were purchased from Boster (Wuhan, China). The polyclonal antibody against 3β-HSD was produced in our laboratory, and its reactivity and specificity were confirmed by Western blot analysis with a testicular extract as a positive control (data not shown). All other chemicals were commercially available products of the highest grade.
Animals
Male Sprague–Dawley rats (9–10 weeks old) were bred in our laboratory. The vivarium was maintained at 22–24 °C under a constant 12-h light:12-h dark cycle. The animals were fed a standard pellet diet and given water ad libitum. The animal experiments were carried out following the guidelines for animal treatment of the Nanjing Jinling Hospital (approved by the local ethics committee), which are in accordance with the principles and procedures of the NIH guidelines for the care and use of laboratory animals.
Isolation and culturing of Leydig cells
Leydig cells were prepared from immature rat testes and cultured in Dulbecco's modified Eagle's medium/nutrient mixture F12 supplemented with 15 mmol l−1 HEPES (pH 7.4), 1 mg ml−1 bovine serum albumin, 365 mg l−1 glutamine, 100 IU ml−1 penicillin and 100 µg ml−1 streptomycin (complete medium), as described previously.20 Briefly, the testes were isolated and decapsulated. They were then incubated with collagenase type I (0.25 mg ml−1) for 20 min at 37 °C. The crude interstitial cells were collected by centrifugation at 1000g for 10 min and then washed twice in Hanks' balanced salt solution containing 0.1% (w/v) bovine serum albumin. To obtain purified Leydig cells, the crude cell suspension was loaded on top of a discontinuous Percoll gradient (20%, 40%, 60% and 90% Percoll in Hanks' balanced salt solution) and subsequently centrifuged at 800g for 20 min. The fractions with enriched Leydig cells were collected and centrifuged in a continuous, self-generating density gradient starting with 60% Percoll at 20 000g for 30 min at 4 °C. The purified cells were then collected into a new tube.
The purity of the Leydig cells in the final preparation was 90%, as determined by histochemical staining for 3β-HSD.21 The cell viability, as assessed by trypan blue exclusion, was greater than 90%. The purified Leydig cells were washed twice with Dulbecco's modified Eagle's medium/nutrient mixture F12 and resuspended to a concentration of 106 cells ml−1 in complete medium. The cell suspension was plated into six-well plates (Costar, New York, USA) at 2 ml/well and then cultured at 34 °C in a humidified atmosphere consisting of 5% CO2 and 95% air.
Experimental groups and treatment
Twenty-four hours after plating, all of the cells were changed to fresh media and subjected to the experimental conditions described below.
In the first experiment, the cells in each well were treated either with GnRHa at different concentrations (10 nmol l−1, 100 nmol l−1, 1 µmol l−1 or 10 µmol l−1) for 24 h or with 100 nmol l−1 GnRHa for different durations (6, 12, 24, 36 or 48 h). The GnRHa was replaced every 12 h when the medium was changed. The culture medium was collected and stored at −20 °C for radioimmunoassay (RIA) determination of testosterone levels, and the cells were harvested for RNA and protein extractions.
In another experiment, the cells were washed once with fresh medium and incubated in serum-free medium for 6 h. They were then treated with 100 nmol l−1 GnRHa and harvested at different time points (0, 5, 10, 30, 60 or 90 min) for Western blot analysis to determine the levels of phosphorylated MAPK (ERK1/2, JNK and p38). In parallel, some cells were pre-treated with PD-98059 (50 µmol l−1) for 20 min and then with GnRHa (100 nmol l−1) for another 5 min, 24 h prior to harvesting. Cell viability was confirmed based on an analysis of cell morphology using a light microscope. The culture medium was then collected for RIA determination of testosterone levels, and the cells were harvested for RNA and protein extractions.
Western blotting analysis
The cells were washed twice with ice-cold phosphate-buffered saline and lysed in 200 µl of ice-cold radioimmunoprecipitation assay lysis buffer (150 mmol l−1 NaCl, 1% NP-40, 0.25% deoxycholate, 0.1% sodium dodecyl sulphate, 50 mmol l−1 Tris (pH 7.4), 1 mmol l−1 phenylmethylsulphonyl fluoride, 1 mmol l−1 Na3VO4 and 1 mmol l−1 NaF). The cell lysates were harvested and centrifuged at 10 000g for 20 min at 4 °C. The supernatants were added to new tubes, and the protein concentrations were determined using the Bradford method. Thirty micrograms of total protein was mixed with 6× loading buffer and boiled for 5 min. The sample mixtures were run on 10% SDS-PAGE gels in 1× running buffer at 25 mA for 2 h. The proteins were then transferred to polyvinylidene difluoride membranes (Millipore Corporation, Bedford, MA, USA) in transfer buffer at 70 V for 3.5 h. After the transfer, the membranes were incubated in blocking buffer (TBST (0.5% Tween 20 in Tris-buffered saline) containing 5% (w/v) non-fat dry milk) at 37 °C for 1.5 h and washed three times with TBST for 30 min. The membranes were then incubated with the primary antibodies against phosphorylated ERK1/2 MAPK, total ERK1/2 MAPK, 3β-HSD or β-actin for 16–18 h at 4 °C. After washing, the blots were incubated with a horseradish peroxidase-conjugated secondary antibody, and the protein bands were visualized with an ECL reaction (Promega), as described by the manufacturer, and exposed to X-ray film. The immunoblot assays for JNK and p38 were conducted in a similar manner. The antibodies against activated JNK and ERK recognize the dually phosphorylated proteins. The intensity of each protein band was quantified using Quantity-One software (Bio-Rad Laboratories, Hercules, CA, USA).
RNA extraction and real-time quantitative RT-PCR
Total RNA was extracted from rat Leydig cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instruction. The quality and concentration of the RNA were determined using an Eppendorf Biophotometer (Eppendorf, Hamburg, Germany). Total RNA was reverse-transcribed in a 25-µl reaction containing 5× avian myeloblastosis virus (AMV) buffer, 2.5 mmol l−1 dNTPs, OligdT, 10 U µl−1 AMV and RNA at 42 °C for 60 min and then heat-inactivated at 95 °C for 5 min.
After reverse transcription, real-time quantitative PCR was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The PCR reactions were carried out for 40 cycles at 94 °C for 30 s, 57 °C for 30 s and 72 °C for 30 s in 25-µl reactions containing 12.5 µl of 2× PCR mix, 1.5 µl of each 10 pmol µl−1 gene-specific forward and reverse primer, 6.5 µl of ddH2O, 1 µl of 10 pmol µl−1 TaqMan probe and 2 µl of cDNA template. The primers and TaqMan probes (Invitrogen, Shanghai, China) specific to 3β-HSD and Rp119 cDNA were designed using Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA). The sequences of the primers and TaqMan probes are listed in Table 1. Each RT-PCR experiment was repeated three times. The average threshold cycles were used to calculate the relative levels of mRNA expression. The quantity of specific mRNA was normalized as a ratio to the amount of Rp119 mRNA.
Table 1. Sequences of primers and TaqMan probes used in quantitative RT-PCR for 3β-HSD and Rpl19.
| Gene | Primer/probe | Sequence (5′ to 3′) | Product length (bp) |
|---|---|---|---|
| 3β-HSD | Forward | AGCAAAAAGATGGCCGAGAA | |
| Reverse | GGCACAAGTATGCAATGTGCC | 80 | |
| TaqMan | (FAM)TGGCAGCCAATGGGAGCATCCT(TAMRA) | ||
| Rpl19 | Forward | CATGAGTATGCTTAGGCTACAGAAG | |
| Reverse | GCGATTTCGTTGGTTTCATT | 96 | |
| TaqMan | (FAM)CGCTGTGGTAAAAAGAAGGTGTGGTTG(TAMRA) |
Determination of testosterone levels
The levels of testosterone secreted into the media were measured using an RIA with the Access testosterone assay kit, according to the manufacturer's instructions (Beckman Coulter Inc., Fullerton, CA, USA).
Statistic analysis
All data are expressed as mean±s.e.m. of the values obtained from at least three independent experiments. Differences between the means were analysed by one-way analysis of variance and the least significance difference method. P<0.05 was considered to be significant.
Results
GnRHa stimulates testosterone production
Primary cultures of rat Leydig cells were treated with GnRHa either at different concentrations or for different durations, and the amount of testosterone secreted into the media was examined by RIA. During a 24-h incubation, GnRHa significantly stimulated testosterone production over the baseline by 33% and 51% at 10 and 100 nmol l−1, respectively (P<0.05). GnRHa treatment also caused an insignificant increase in testosterone levels (P>0.05) at 1 µmol l−1 but showed no effect at 10 µmol l−1 (Figure 1a). When the cells were treated with GnRHa at the optimal concentration of 100 nmol l−1, the mean testosterone levels increased steadily 6–24 h post-treatment and gradually returned to baseline after 48 h (Figure 1b). Although the increases compared to the baseline levels were not statistically significant at 6 or 36 h (P>0.05), they were 44% and 60% higher (P<0.05) after 12 and 24 h, respectively.
Figure 1.
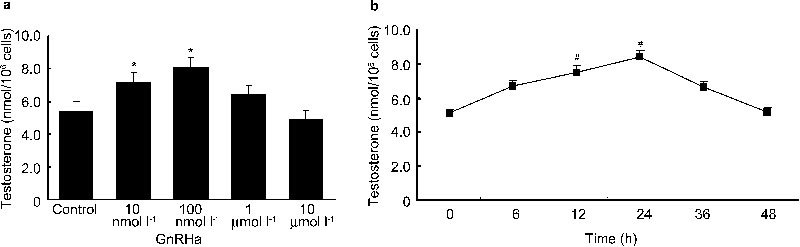
The effects of gonadotropin-releasing hormone agonist (GnRHa) on testosterone production at various doses and time points. Rat Leydig cells were treated with GnRHa at various concentrations for 24 h (a) or with a concentration 100 nmol l−1 for various time courses (b). GnRHa was replaced along with the medium every 12 h. The culture media were collected, and the levels of testosterone production were measured by radioimmunoassay (RIA). Data are presented as mean±s.e.m. of three separate experiments. * P < 0.05 compared with the control; #, P < 0.05 compared with the testosterone level at 0 h.
GnRHa stimulates 3β-HSD expression
When cells were treated with GnRHa in a manner similar to the above experiments, 3β-HSD expression increased at both the mRNA and protein levels. GnRHa treatment for 24 h significantly increased 3β-HSD mRNA expression by 160%, 198% and 72% compared to baseline at 10 nmol l−1, 100 nmol l−1 and 1 µmol l−1, respectively (P<0.05). However, the treatment had no significant effect at 10 µmol l−1 (Figure 2a). 3β-HSD protein levels significantly increased by 28% and 41% (P<0.05) in response to GnRHa at 10 and 100 nmol l−1 concentrations, respectively, but the increase was insignificant at two higher concentrations (Figure 2c).
Figure 2.
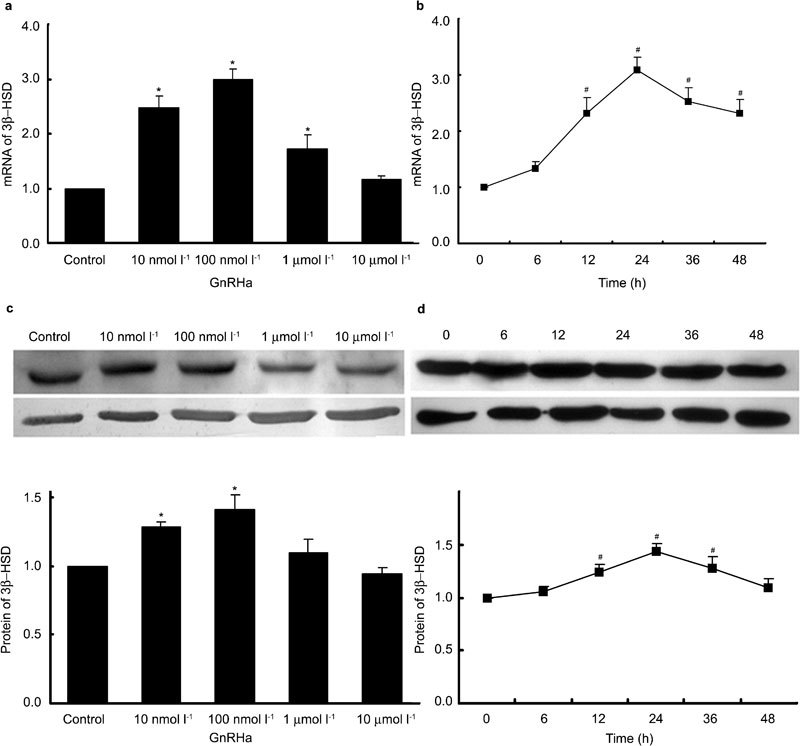
Dose-dependent effects and time course of the effects of gonadotropin-releasing hormone agonist (GnRHa) on 3β-hydroxysteroid dehydrogenase (3β-HSD) expression. Rat Leydig cells were treated with GnRHa at various concentrations for 24 h or at a concentration of 100 nmol l−1 for various time courses. GnRHa was replaced along with the medium every 12 h. Cells were harvested for total RNA or protein extractions, and 3β-HSD expression was detected by real-time PCR (a, b) and Western blot analysis (c, d). The quantity of 3β-HSD mRNA was normalized as a ratio to the amount of Rp119 mRNA. The intensities of the protein bands in the Western blots were quantified, and 3β-HSD protein levels were normalized to that of β-actin. Data are presented as mean±s.e.m. of three separate experiments. * P < 0.05 compared with the control; #, P < 0.05 compared with 0 h.
Treatment with 100 nmol l−1 GnRHa significantly induced 3β-HSD mRNA expression by 131%, 209%, 152% and 133% after 12, 24, 36 and 48 h, respectively (P<0.05) (Figure 2b). The protein levels also increased steadily 6–24 h after administration of GnRHa and then gradually returned to baseline after 48 h. The increase was insignificant (P>0.05) after 6 h; however, the protein levels were significantly increased by 21%, 43% and 29% (P<0.05) after 12, 24 and 36 h, respectively (Figure 2d).
Effect of GnRHa on activation of ERK1/2, JNK and p38 MAPK
To investigate whether GnRHa activates any of the MAPK signalling pathways in rat Leydig cells, we measured the levels of ERK1/2, JNK and p38 MAPK phosphorylation by Western blot analysis using antibodies that specifically recognize the phosphorylated form of these MAPK proteins. When cells were treated with GnRHa at a concentration of 100 nmol l−1 for 90 min, the levels of ERK1/2 phosphorylation rapidly increased by 258% as early as 5 min after administration and remained elevated by 140% over baseline after 10 min (P<0.05) (Figure 3a). However, the levels of unphosphorylated total ERK1/2 did not change during the treatment, and the levels of JNK and p38 MAPK phosphorylation were not altered by GnRHa treatment either (Figure 3b). However, pre-treatment with PD-98059 at a concentration of 50 µmol l−1 for 20 min completely abolished the effect of GnRHa (Figure 4) on ERK1/2 and repressed the basal level of ERK1/2 MAPK activation (P<0.05).
Figure 3.
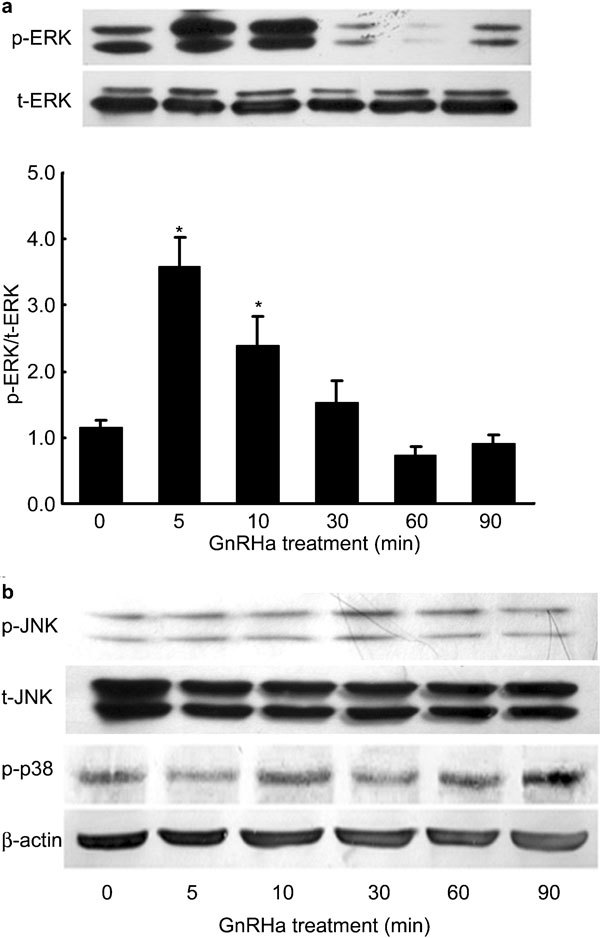
Time course of the effects of gonadotropin-releasing hormone agonist (GnRHa) on ERK1/2 MAPK activation. Rat Leydig cells were cultured for 24 h and then treated with GnRHa (100 nmol l−1) for up to 90 min. The whole-cell lysates were collected at different time points as indicated, and total protein (30 µg) samples were subjected to Western blot analysis of total ERK1/2 (t-ERK1/2) and phosphorylated ERK1/2 (p-ERK1/2) (a), or total SAPK/JNK (t-JNK), phosphorylated SAPK/JNK (p-JNK1/2) phosphorylated p38 (p-p38) and β-actin (b). The intensities of the protein bands in the western blot were quantified, and the p-MAPK protein levels were normalized to those of t-MAPK. Data are presented as mean±s.e.m. of three separate experiments. * P < 0.05 compared with 0 min.
Figure 4.
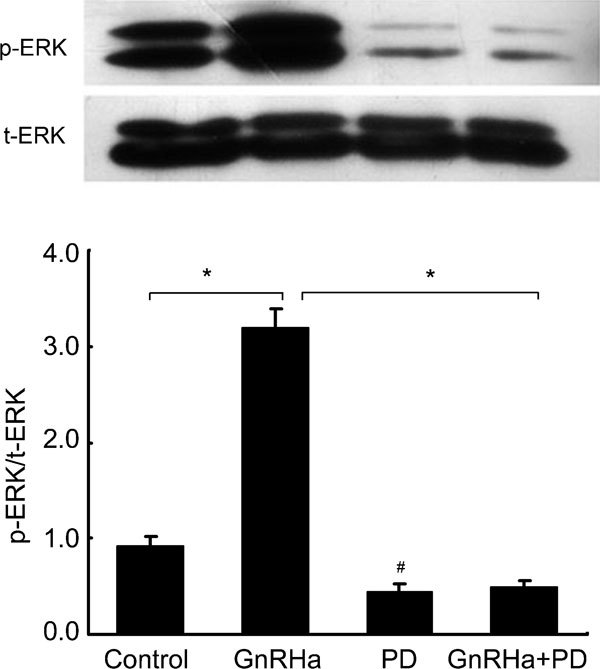
Inhibitory effects of PD-98059 on gonadotropin-releasing hormone agonist (GnRHa)-induced ERK1/2 MAPK activation. Rat Leydig cells were pre-treated with PD-98059 (50 µmol l−1) for 20 min and then stimulated with 100 nmol l−1 GnRHa for 5 min. The whole-cell lysates were collected, and total protein (30 µg) samples were subjected to Western blot analysis of total ERK1/2 (t-ERK1/2) and phosphorylated ERK1/2 (p-ERK1/2). The intensities of the protein bands in the blot were quantified, and the p-MAPK protein levels were normalized to those of t-ERK1/2. Data are presented as mean±s.e.m. of three separate experiments. *, P < 0.05, compared with GnRHa treatment alone; #, P < 0.05, compared with the control.
Role of the MAPK pathway in GnRHa-stimulated 3β-HSD expression
To investigate whether the ERK1/2 MAPK signalling pathway mediates the GnRHa-induced expression of 3β-HSD in rat Leydig cells, we examined the ability of PD-98059 to block the effects of GnRHa. In the absence of PD-98059 and similar to the observations described earlier, GnRHa treatment at a concentration of 100 nmol l−1 for 24 h resulted in a 200% increase in 3β-HSD mRNA levels (Figure 5a) and a 42% increase in protein levels (Figure 5b) compared to the untreated cells (P<0.05). However, pre-treatment with 50 µmol l−1 of PD-98059 for 20 min markedly blocked the increases in 3β-HSD expression at both the mRNA and protein levels (P<0.05). PD-98059 also significantly repressed basal 3β-HSD expression in Leydig cells (P<0.05).
Figure 5.
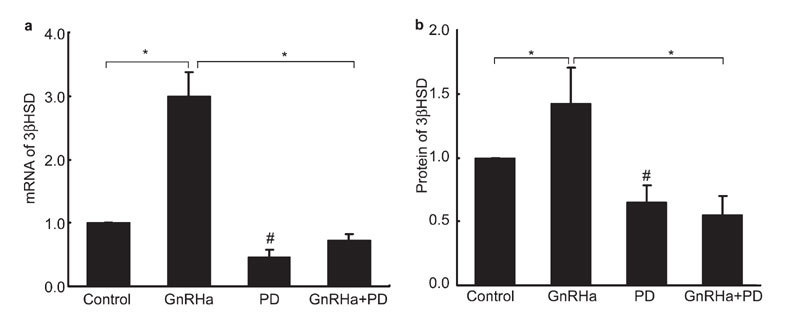
Inhibitory effects of PD-98059 on gonadotropin-releasing hormone agonist (GnRHa)-induced 3β-hydroxysteroid dehydrogenase (3β-HSD) expression. Leydig cells were pretreated with PD-98059 (50 µmol l−1) for 20 min and then stimulated with 100 nmol l−1 GnRHa for 24 h. Cells were harvested, and total RNA and protein were extracted. 3β-HSD expression was analysed by real-time PCR (a) and Western blot analysis (b). 3β-HSD mRNA was normalized as a ratio to the amount of Rp119 mRNA. The intensities of the protein bands in the western blot were quantified, and 3β-HSD protein levels were normalized to that of β-actin. Data are presented as mean±s.e.m. of three separate experiments. *, P < 0.05, compared with GnRHa; #, P < 0.05, compared with the control.
Role of the MAPK pathway in GnRHa-stimulated testosterone production
We also examined the effects of PD-98059 on GnRHa-induced steroidogenesis in rat Leydig cells. As shown in Figure 6, treatment with 100 nmol l−1 of GnRHa for 24 h significantly increased testosterone production in the cells (P<0.05), which was similar to our earlier observation. Pre-treatment with 50 µmol l−1 of PD-98059 for 20 min completely blocked the stimulatory effect of GnRHa. The basal level of testosterone production was also significantly reduced (P<0.05) (Figure 6).
Figure 6.
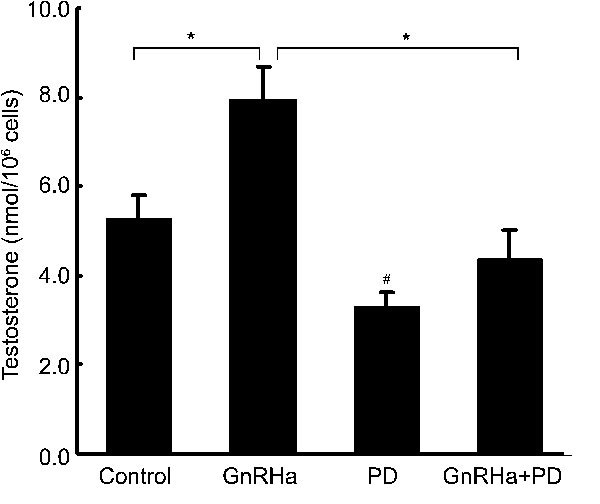
Inhibitory effects of PD-98059 on gonadotropin-releasing hormone agonist (GnRHa)-stimulated testosterone production. Rat Leydig cells were pre-treated with PD-98059 (50 µmol l−1) for 20 min and then stimulated with 100 nmol l−1 GnRHa for another 24 h. The amount of testosterone in the culture media was measured by radioimmunoassay (RIA). Data are presented as mean±s.e.m. of three separate experiments. *, P < 0.05, compared with GnRHa; #, P < 0.05, compared with the control.
Discussion
Many recent studies have suggested the existence of important local regulatory mechanisms that control the secretion and synthesis of testosterone from Leydig cells. Under in vitro conditions, Sertoli cells release an array of cytokines, such as insulin-like growth factor-1, inhibin, transforming growth factor-β and GnRH, in response to follicle-stimulating hormone stimulation.22 These secreted cytokines can further stimulate Leydig cells to produce testosterone via an autocrine pathway. Sertoli cells also release oestrogen and transforming growth factor-α, both of which inhibit testosterone production. Testicular macrophages regulate testosterone synthesis in Leydig cells, as well as the development and proliferation of pubertal Leydig cells in response to follicle-stimulating hormone. Macrophages can inhibit the synthesis and secretion of testosterone from Leydig cells by releasing nitric oxide.23 Leydig cells themselves can also secrete growth factors and bioactive substances to regulate, through autocrine signalling and direct contact, the expression and activity of the Leydig cell enzymes related to testosterone synthesis.24 The discovery of new mechanisms involved in the regulation of testosterone synthesis may help us understand the pathogenesis of clinical diseases, such as late-onset hypogonadism, and thus identify more effective approaches to their prevention and treatment.
Cell culturing provides a simplified in vitro system to study the effects of stimulators and inhibitors on a particular type of cells. MA-10 Leydig tumour cells have been used extensively to study many of the functions of Leydig cells.25, 26, 27, 28. However, the primary culture system employed in this study more closely resembles the cells in vivo. Using these cells, we showed that the GnRHa stimulates the production of testosterone and the expression of 3β-HSD, both following similar bell-shaped dose–response curves and time courses in response to the hormone treatment that peaked at the 100 nmol l−1 dose and at the 24-h time point, respectively. These results show that GnRH stimulates testosterone secretion at low concentrations, but high concentrations of GnRH may lead to receptor desensitisation, reducing the stimulation of the production of enzymes for steroid hormone synthesis and testosterone production. More importantly, we demonstrated for the first time the involvement of the ERK1/2 MAPK signal transduction pathway in the GnRH-induced upregulation of 3β-HSD expression and in testosterone biosynthesis.
Similar stimulatory effects of GnRHa on 3β-HSD protein expression and testosterone production have been reported in murine Leydig cells,7 but the signalling mediators remain elusive. Consistent with the results observed in murine Leydig cells, GnRHa increased testosterone production in rat Leydig cells more efficiently at low doses in the range of 10–100 nmol l−1 in the first 24 h. However, high doses or prolonged treatment reduced these effects. These functional characteristics of GnRHa suggest that these effects may be mediated through specific GnRH receptors on the Leydig cells. Based on their coregulation by GnRHa, either 3β-HSD or its enzymatic activity may be necessary for GnRH-induced steroidogenesis in Leydig cells, although other pathways may also be important.29
LH is known to stimulate Leydig cells to produce testosterone. In light of the present findings, it is important to clarify the relationship between GnRH and LH and in the production of testosterone in Leydig cells. Our preliminary study revealed that the effects of LH on testosterone production could be blocked by a GnRH antagonist (Cetrorelix, Baxter Oncology GmbH, Halle, Germany) and could be augmented by a GnRHa (data shown in Supplementary Figures 1 and 2). These results imply that, in Leydig cells, LH-induced stimulation of testosterone production may be mediated through the function of GnRH. The details of this mechanism are currently being investigated in our lab.
In this study, GnRHa (100 nmol l−1) caused a rapid and transient increase in the level of phosphorylated ERK1/2 (2.58-fold increase over the baseline) as early as 5 min after treatment, and activated ERK1/2 declined to the basal level within 60 min. EEK1/2 activation by GnRHa followed a much shorter time course (in minutes) compared with the GnRHa-induced effects on 3β-HSD protein expression and testosterone production (in hours), which is consistent with the results from a previous study examining MLTC-1 mouse Leydig cells.30 Furthermore, the GnRHa-induced activation of ERK1/2 in this study also occurred more rapidly than the previous reports of ERK1/2 activation by other stimuli. For example, ERK1/2 MAPK activation by interleukin-1α takes 15 minutes in the same cells.15 Similarly, human chorionic gonadotropin-induced ERK phosphorylation occurs after 60 min, peaks at 180 min and declines to basal levels within 360 min.19 The discrepancies in ERK temporal regulation may represent the specific responses of the cells to different extracellular signals in their specific environment. The LH/human chorionic gonadotropin receptor predominantly couples to Gs upon binding to its ligands,18, 19 whereas the GnRH receptor mainly couples to Gq/11. It has been reported that the GnRH receptor does not directly couple to Gs and Gi/o.31 Gs mainly activates adenylyl cyclase and enhances the levels of cAMP to exert downstream effects, whereas Gq/11 principally exerts its effects via activation of phospholipase C but not cAMP. This activation then leads to several signalling processes, including enhanced phosphoinositide turnover, production of diacylglycerol, activation of protein kinase C and MAPK cascades. The temporal relationship of MAPK activation, followed by 3β-HSD expression and testosterone production, also delineates an intercellular pathway connecting these three events, from the binding of the GnRHa receptor to the activation of ERK1/2, then the upregulation of 3β-HSD and ultimately testosterone production.
GnRH has also been shown to activate other components of the MAPK pathways. For example, it activates ERK1/2 and JNK, and subsequentially protein kinase C, c-Src and CDC4210 in both αT3-1 cells32 and LβT-2 cells.14 GnRH-II also induces p38 MAPK in the apoptotic pathway through the activation of activator protein-1 in ovarian cancer cells.33 In this study, JNK and p38 MAPK were not significantly activated by GnRHa in the rat Leydig cells. Therefore, ERK1/2 may be the only MAP kinase mediating GnRHa-induced testosterone biosynthesis in these cells. We also demonstrated that GnRHa increases phosphorylated ERK1/2 levels but exerts no effect on the total non-phosphorylated form of ERK1/2. In addition, the activation of ERK1/2 MAPK was completely blocked by PD-98059. This ERK1/2 inhibitor also blocked subsequent 3β-HSD induction and ultimately blocked testosterone production. These results suggest an essential role for ERK1/2 in the regulation of 3β-HSD and the mediation of GnRHa-induced testes steroidogenesis. PD-98059 treatment alone also reduced the basal levels of ERK1/2 activation, 3β-HSD expression and testosterone production, suggesting a role for the ERK pathway in the maintenance of the basic functions of Leydig cells. Although it is difficult to exclude the influence of non-specific effects of PD-98059, this inhibitor is well recognized as a specific antagonist of ERK1/2 and has been employed in many previous studies involving many cell types, including Leydig cells.34, 35
While our findings are mostly consistent with the results from a recent study reporting that inhibition of LH-activated ERK1/2 MAPK decreases steroidogenesis in Leydig cells,18 the mechanisms of steroidogenesis may vary in different experimental systems. Although both PKA and MAPK are activated by LH in mLTC-1 Leydig cells, only cAMP-mediated PKA activation is essential, as the MAPK pathway is only involved in the early stage (30–60 min)30 of steroidogenesis.
As one of the Sertoli cell paracrine factors mediating the function of Leydig cells,36 GnRH is produced in testicular cells and secreted outside of the cell to stimulate testosterone production in Leydig cells via a paracrine loop. Rat Leydig cells contain high-affinity receptors for GnRH.37 In regard to the events that occur downstream of GnRH signalling, the stimulatory effect of GnRH has been shown to be independent on the cAMP-protein kinase system. However, this effect is calcium-dependent2, 3, 4 and involves protein kinase C in rat Leydig cells.38 Nevertheless, we show here that the activation of ERK1/2 is a key downstream event that is modulated by GnRH and may serve as a signal transduction pathway mediating GnRH-induced steroidogenesis.
Our data demonstrate the critical role of ERK1/2 in GnRH-induced 3β-HSD expression and testosterone production in primary cultures of Leydig cells. We provide the first line of evidence to suggest that ERK1/2 activation by GnRH may be responsible for the induction of 3β-HSD gene expression and enzyme production, which then ultimately modulates steroidogenesis in rat Leydig cells. These findings may help define the mechanisms of many of the clinical diseases that result from the abnormal synthesis and secretion of testosterone.
Author contributions
BY and HYL conceived and designed the study, and carried out Leydig cells culture and the molecular genetic studies, and drafted the manuscript. YCG performed the statistical analysis and drafted the manuscript. SSS and XQT carried out the immunoassays. XJL participated in the statistical analysis. YFG and YXC participated in the design of the study and revising the manuscript. GBY conceived of the study and coordination and helped to draft the manuscript, and gave final approval of the version to be published. All authors read and approved the final manuscript.
Acknowledgments
This work was supported in part by the Natural Scientific Foundation of China (No. 30770801 and No. 81070480).
The authors declare no competing financial interests.
Footnotes
Note: Supplementary information is available on the Asian Journal of Andrology website(http://www.nature.com/aja/).
Supplementary Information
References
- Millar RP, Pawson AJ, Morgan K, Rissman EF, Lu ZL. Diversity of actions of GnRHs mediated by ligand-induced selective signaling. Front Neuroendocrinol. 2008;29:17–35. doi: 10.1016/j.yfrne.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. Mechanism of action of gonadotropin-releasing hormone stimulated Leydig cell steroidogenesis. III. The role of arachidonic acid and calcium/phospholipid dependent protein kinase. Life Sci. 1985;36:1255–64. doi: 10.1016/0024-3205(85)90270-x. [DOI] [PubMed] [Google Scholar]
- Lin T. Mechanism of action of gonadotropin-releasing hormone-stimulated Leydig cell steroidogenesis. I. The stimulatory effect is calcium dependent and not mediated by cyclic nucleotides. J Androl. 1984;5:193–200. doi: 10.1002/j.1939-4640.1984.tb02392.x. [DOI] [PubMed] [Google Scholar]
- Lin T. Mechanism of action of gonadotropin-releasing hormone-stimulated Leydig cell steroidogenesis. II. Gonadotropin-releasing hormone stimulates phospholipid labeling. J Androl. 1984;5:201–5. doi: 10.1002/j.1939-4640.1984.tb02393.x. [DOI] [PubMed] [Google Scholar]
- Lorence MC, Naville D, Graham-Lorence SE, Mack SO, Murry BA, et al. 3 beta-hydroxysteroid dehydrogenase/delta 5–4-isomerase expression in rat and characterization of the testis isoform. Mol Cell Endocrinol. 1991;80:21–31. doi: 10.1016/0303-7207(91)90139-j. [DOI] [PubMed] [Google Scholar]
- Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, et al. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5–delta4 isomerase gene family. Endocr Rev. 2005;26:525–82. doi: 10.1210/er.2002-0050. [DOI] [PubMed] [Google Scholar]
- Lin YM, Liu MY, Poon SL, Leu SF, Huang BM. Gonadotrophin-releasing hormone-I and -II stimulate steroidogenesis in prepubertal murine Leydig cells in vitro. Asian J Androl. 2008;10:929–36. doi: 10.1111/j.1745-7262.2008.00434.x. [DOI] [PubMed] [Google Scholar]
- Gur G, Bonfil D, Safarian H, Naor Z, Yaron Z. GnRH receptor signaling in tilapia pituitary cells: role of mitogen-activated protein kinase (MAPK) Comp Biochem Physiol B Biochem Mol Biol. 2001;129:517–24. doi: 10.1016/s1096-4959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, et al. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology. 2004;145:2228–44. doi: 10.1210/en.2003-1418. [DOI] [PubMed] [Google Scholar]
- Levi NL, Hanoch T, Benard O, Rozenblat M, Harris D, et al. Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary alpha T3-1 cell line is mediated by protein kinase C, c-Src, and CDC42. Mol Endocrinol. 1998;12:815–24. doi: 10.1210/mend.12.6.0120. [DOI] [PubMed] [Google Scholar]
- Burger LL, Haisenleder DJ, Aylor KW, Marshall JC. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol Reprod. 2009;81:1206–15. doi: 10.1095/biolreprod.109.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone alpha-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–9. doi: 10.1128/mcb.15.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor Z, Harris D, Shacham S. Mechanism of GnRH receptor signaling: combinatorial cross-talk of Ca2+ and protein kinase C. Front Neuroendocrinol. 1998;19:1–19. doi: 10.1006/frne.1997.0162. [DOI] [PubMed] [Google Scholar]
- Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, et al. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology. 2002;143:1018–25. doi: 10.1210/endo.143.3.8675. [DOI] [PubMed] [Google Scholar]
- Renlund N, Jo Y, Svechnikova I, Holst M, Stocco DM, et al. Induction of steroidogenesis in immature rat Leydig cells by interleukin-1alpha is dependent on extracellular signal-regulated kinases. J Mol Endocrinol. 2006;36:327–36. doi: 10.1677/jme.1.01963. [DOI] [PubMed] [Google Scholar]
- Svechnikov K, Stocco DM, Soder O. Interleukin-1alpha stimulates steroidogenic acute regulatory protein expression via p38 MAP kinase in immature rat Leydig cells. J Mol Endocrinol. 2003;30:59–67. doi: 10.1677/jme.0.0300059. [DOI] [PubMed] [Google Scholar]
- Manna PR, Stocco DM. The role of JUN in the regulation of PRKCC-mediated STAR expression and steroidogenesis in mouse Leydig cells. J Mol Endocrinol. 2008;41:329–41. doi: 10.1677/JME-08-0077. [DOI] [PubMed] [Google Scholar]
- Martinat N, Crepieux P, Reiter E, Guillou F. Extracellular signal-regulated kinases (ERK) 1, 2 are required for luteinizing hormone (LH)-induced steroidogenesis in primary Leydig cells and control steroidogenic acute regulatory (StAR) expression. Reprod Nutr Dev. 2005;45:101–8. doi: 10.1051/rnd:2005007. [DOI] [PubMed] [Google Scholar]
- Martinelle N, Holst M, Soder O, Svechnikov K. Extracellular signal-regulated kinases are involved in the acute activation of steroidogenesis in immature rat Leydig cells by human chorionic gonadotropin. Endocrinology. 2004;145:4629–34. doi: 10.1210/en.2004-0496. [DOI] [PubMed] [Google Scholar]
- Svechnikov K, Sultana T, Soder O. Age-dependent stimulation of Leydig cell steroidogenesis by interleukin-1 isoforms. Mol Cell Endocrinol. 2001;182:193–201. doi: 10.1016/s0303-7207(01)00554-8. [DOI] [PubMed] [Google Scholar]
- Payne AH, Downing JR, Wong KL. Luteinizing hormone receptors and testosterone synthesis in two distinct populations of Leydig cells. Endocrinology. 1980;106:1424–9. doi: 10.1210/endo-106-5-1424. [DOI] [PubMed] [Google Scholar]
- Fujisawa M. Cell-to-cell cross talk in the testis. Urol Res. 2001;29:144–51. doi: 10.1007/s002400100179. [DOI] [PubMed] [Google Scholar]
- Weissman BA, Niu E, Ge R, Sottas CM, Holmes M, et al. Paracrine modulation of androgen synthesis in rat leydig cells by nitric oxide. J Androl. 2005;26:369–78. doi: 10.2164/jandrol.04178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JM. Leydig cells: endocrine, paracrine, and autocrine regulation. Endocr Rev. 1994;15:574–626. doi: 10.1210/edrv-15-5-574. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim JU, Kim C, Min CK. Inhibitory actions of mibefradil on steroidogenesis in mouse Leydig cells: involvement of Ca2+ entry via the T-type Ca2+ channel. Asian J Androl. 2010;12:807–13. doi: 10.1038/aja.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore MW, Rommerts FF, Wirtz KW, Stocco DM. Regulation of steroidogenesis in subclones of the MA-10 mouse Leydig tumor cell line. Mol Cell Endocrinol. 1990;69:9–16. doi: 10.1016/0303-7207(90)90083-k. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, D'Souza B, Conti M, Ascoli M. Attenuation of cAMP-mediated responses in MA-10 Leydig tumor cells by genetic manipulation of a cAMP-phosphodiesterase. J Biol Chem. 1991;266:14383–9. [PubMed] [Google Scholar]
- Levine SL, Han Z, Liu J, Farmer DR, Papadopoulos V. Disrupting mitochondrial function with surfactants inhibits MA-10 Leydig cell steroidogenesis. Cell Biol Toxicol. 2007;23:385–400. doi: 10.1007/s10565-007-9001-6. [DOI] [PubMed] [Google Scholar]
- Feek CM, Tuzi NL, Edwards CR. The adrenal gland and progesterone stimulates testicular steroidogenesis in the rat in vivo. J Steroid Biochem. 1989;32:573–9. doi: 10.1016/0022-4731(89)90392-0. [DOI] [PubMed] [Google Scholar]
- Evaul K, Hammes SR. Cross-talk between G protein-coupled and epidermal growth factor receptors regulates gonadotropin-mediated steroidogenesis in Leydig cells. J Biol Chem. 2008;283:27525–33. doi: 10.1074/jbc.M803867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CD, Coetsee M, Morgan K, Flanagan CA, Millar RP, et al. A crucial role for Galphaq/11, but not Galphai/o or Galphas, in gonadotropin-releasing hormone receptor-mediated cell growth inhibition. Mol Endocrinol. 2008;22:2520–30. doi: 10.1210/me.2008-0122. [DOI] [PubMed] [Google Scholar]
- Zhang T, Roberson MS. Role of MAP kinase phosphatases in GnRH-dependent activation of MAP kinases. J Mol Endocrinol. 2006;36:41–50. doi: 10.1677/jme.1.01881. [DOI] [PubMed] [Google Scholar]
- Kim KY, Choi KC, Park SH, Chou CS, Auersperg N, et al. Type II gonadotropin-releasing hormone stimulates p38 mitogen-activated protein kinase and apoptosis in ovarian cancer cells. J Clin Endocrinol Metab. 2004;89:3020–6. doi: 10.1210/jc.2003-031871. [DOI] [PubMed] [Google Scholar]
- Manna PR, Chandrala SP, King SR, Jo Y, Counis R, et al. Molecular mechanisms of insulin-like growth factor-I mediated regulation of the steroidogenic acute regulatory protein in mouse Leydig cells. Mol Endocrinol. 2006;20:362–78. doi: 10.1210/me.2004-0526. [DOI] [PubMed] [Google Scholar]
- Diaz ES, Pellizzari E, Casanova M, Cigorraga SB, Denduchis B. Type IV collagen induces down-regulation of steroidogenic response to gonadotropins in adult rat Leydig cells involving mitogen-activated protein kinase. Mol Reprod Dev. 2005;72:208–15. doi: 10.1002/mrd.20259. [DOI] [PubMed] [Google Scholar]
- Clayton RN, Katikineni M, Chan V, Dufau ML, Catt KJ. Direct inhibition of testicular function by gonadotropin-releasing hormone: mediation by specific gonadotropin-releasing hormone receptors in interstitial cells. Proc Natl Acad Sci USA. 1980;77:4459–63. doi: 10.1073/pnas.77.8.4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe RM, Fraser HM. Leydig cell receptors for luteinizing hormone releasing hormone and its agonists and their modulation by administration or deprivation of the releasing hormone. Biochem Biophys Res Commun. 1980;95:256–62. doi: 10.1016/0006-291x(80)90732-9. [DOI] [PubMed] [Google Scholar]
- Nikula H, Huhtaniemi I. Gonadotropin-releasing hormone agonist activates protein kinase C in rat Leydig cells. Mol Cell Endocrinol. 1988;55:53–9. doi: 10.1016/0303-7207(88)90090-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.