

Abstract
The present study investigated the effects of the multikinase inhibitor sorafenib on androgen-independent cancer cells viability and intracellular signaling. Human androgen-independent PC-3 prostate cancer cells were treated with sorafenib. At concentration that suppresses extracellular signal-regulated kinase phosphorylation, sorafenib treatment reduced the mitochondrial transmembrane potential. Sorafenib also down-modulated the levels of myeloid cell leukemia 1, survivin and cellular inhibitor of apoptosis protein 2. Sorafenib induced caspase-3 cleavage and the mitochondrial release of cytochrome c. However, no nuclear translocation of apoptosis inducing factor was detected after treatment and the pan-caspase inhibitor Z-VAD-FMK had an obvious protective effect against the drug. In conclusion, sorafenib induces apoptosis through a caspase-dependent mechanism with down-regulated anti-apoptotic proteins in androgen-independent prostate cancer cells in vitro.
Keywords: apoptosis, PC-3 prostate cancer cells, prostate cancer, sorafenib
Introduction
Prostate cancer is the most common malignancy and the second leading cause of cancer-related death in men 1. In addition, the overall incidence of prostate cancer is increasing in China 2. Inhibition of androgen receptor (AR) signalling through hormone deprivation therapy, a common treatment, is initially effective but ultimately leads to a highly aggressive and frequently lethal hormone-insensitive form of the disease. Although advances in chemotherapy have improved patient outcomes 3, 4, 5, 6, there remains a clear need for development of novel therapeutic approaches to control this disease and improve the outcomes for patients.
One of the most important intracellular pathways involved in controlling cell proliferation, cell differentiation and cell death is the canonical extracellular signal regulated protein kinases (ERK) signalling cascade. This signalling pathway appears to be involved in prostate cancer drug resistance 7, 8. Furthermore, previous research has shown that inhibition of just one of the signalling proteins in this pathway can induce apoptosis through down-regulation of myeloid cell leukaemia 1 (MCL-1), an anti-apoptotic member of the B-cell lymphoma 2 (BCL-2) gene family 9, 10. MCL-1 is expressed in a fairly high percentage of prostate tumours 11, 12, and the inhibition of ERK pathway-mediated signals, and consequently expression of MCL-1, might be a key target for treatment of advanced prostate cancer cells, as suggested by Cavarretta et al 13. These observations form the basis for our hypothesis that targeting p42/p44 mitogen-activated protein kinase signalling pathways may inhibit tumour growth and progression in prostate cancer.
Sorafenib (commercial name Nexavar), a novel bi-aryl urea, has been shown to inhibit the kinase activities of C-Raf, and both wild-type and mutant V600E B-Raf in vitro. Sorafenib treatment also diminishes mitogen extracellular kinase MEK/ERK activation in various tumour cell lines 14, 15. Sorafenib has recently been approved by the US Food and Drug Administration for treatment of renal cancer and is currently being investigated in over 30 clinical trials for use against a wide variety of human cancers, including melanoma, prostate, ovarian, pancreatic, lung cancer and others (http://www.clinicaltrials.gov).
Although sorafenib is undergoing phase I/II clinical evaluation for treatment of prostate cancer 16, 17, 18, 19, 20, the apoptotic pathways and the changes in biomarker expression resulting from sorafenib treatment have not been studied. Thus, the aim of this study was to investigate the effects of sorafenib on androgen-independent prostate cancer cell viability in vitro.
Materials and methods
Cell lines
The human prostate cancer cell lines PC-3, DU145 and LNCaP were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in RPMI 1640 containing 10% foetal bovine serum (GibcoBRL, LifeTechnologies, Carlsbad, CA, USA), 2 mmol L−1 glutamine, 100 IU mL−1 ampicillin and 100 ng mL−1 streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Reagents and antibodies
Sorafenib was purchased from the Bayer Corporation (West Haven, CT, USA). The pan-caspase inhibitor, Z-VAD-FMK, was purchased from Merck Chemical Ltd. (Nottingham, UK). Compounds were dissolved in 100% DMSO (Amresco Inc., Solon, OH, USA). The following antibodies were used for immunoblotting at the indicated dilutions: p-ERK (rabbit monoclonal, 1:500; Cell Signaling Technology Inc., Beverly, MA, USA), ERK (mouse monoclonal, 1:1 500; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), MCL-1 (mouse monoclonal, 1:500; Santa Cruz), survivin (rabbit monoclonal, 1:1 500; R&D Systems Inc., Minneapolis, MN, USA), cellular inhibitor of apoptosis protein 2 (c-IAP2) (rabbit polyclonal, 1:1 000; Santa Cruz), cytochrome c (rabbit polyclonal, 1:600; Santa Cruz), apoptosis-inducing factor (AIF; rabbit monoclonal, 1:500; Epitomics Inc., Burlingame, CA, USA), tubulin (rabbit polyclonal, 1:1 000; Santa Cruz) and HSP60 (mouse monoclonal, 1:1 000; Kangcheng, Shanghai, China). A cleaved caspase-3 antibody was used for immunochemistry (mouse monoclonal, 1:200; Cell Signaling Technology Inc.).
Western blot analysis
Whole-cell extracts were prepared by lysing cells in buffer containing 50 mmol L−1 Tris-HCl (pH 7.5), 150 mmol L−1 NaCl, 2 mmol L−1 EDTA, 2% NP40, 1 mmol L−1 DTT, 100 mg L−1 aprotinin, 100 mg L−1 leupeptin, 100 mg L−1 pepstatin and 100 mg L−1 PMSF. Protein concentrations were measured with the BCA protein assay kit (Pierce, Rockford, IL, USA). In all, 30 μg of protein was separated by 10% or 15% SDA-PAGE and electroblotted onto PVDF membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA). After blocking non-specific binding sites with 5% skim milk in TBS-T for 2 h, the membranes were incubated at 37°C for 1 h, followed by 4°C overnight, with primary antibodies. After three washes in TBS-T, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Kangcheng) for 2 h at 37°C. Signals were detected by exposure to X-ray films after treatment with the super-signal enhanced chemiluminescence detection kit (Roche Diagnostics GmbH, Mannheim, Germany).
Apoptosis and mitochondrial potential assays
Cells were seeded at approximately 5 × 105 mL−1 per well in six-well plates and allowed to reach exponential growth for 24 h before 20 h of treatment with different concentrations of sorafenib. Adherent and floating cells were then harvested together, washed twice with PBS and incubated with a reagent containing Annexin V conjugated to fluorescence isothiocyanate (FITC, 2.5 μg mL−1; BD Pharmingen, San Diego, CA, USA) and propidium iodide (PI, 5 μg mL−1; BD Pharmingen) for 15 min at room temperature. Cells were analyzed using a flow cytometer (FASCAria, BD Biosciences, San Jose, CA, USA). To measure change in mitochondrial membrane potential, cells were incubated with 10 μmol L−1 Rhodamine 123 (Amresco) for 30 min at 37°C, 5% CO2. Next, both adherent and floating cells were harvested, washed twice with PBS and analysed by means of flow cytometry (FASCAria). Samples were run in triplicate, and 10 000 events were collected for each replicate. The data are presented as the average of three experiments with error under 5%.
Immunocytochemistry for detection of cleaved caspase-3
PC-3 cells were seeded at approximately 5 × 105 mL−1 per well on coverslips in six-well plates and allowed to reach exponential growth for 24 h. After 20 h of treatment with 10 μmol L−1 sorafenib, the cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for 30 min at 4°C. Primary anti-cleaved caspase-3 antibody, biotinylated secondary antibody (1:200; Zymed Laboratories Inc., San Francisco, CA, USA) and horseradish peroxidase-labelled streptavidin (1:200; Zymed) were added sequentially and the cells were then counterstained with 3,3′-diaminobenzidine for analysis.
Detecting the contribution of caspase activation to the lethality induced by sorafenib
To assess the contribution of caspase activation to the lethality of sorafenib, approximately 6 × 103 PC-3 cells per well were grown in 96-well plates and incubated overnight in 100 μL of culture medium. Cells were then treated with 10 μmol L−1 sorafenib for 20 h at 37°C with 5% CO2, with or without 20 μmol L−1 of the pan-caspase inhibitor, Z-VAD-FMK incubated for 30 min before, then 20 μL MTT (5 mg mL−1, Sigma; St. Louis, MO, USA) was added to each well and the cells were incubated for another 4h at 37°C. The supernatants were removed, and 50 μL of DMSO was added to each well. A micro ELISA reader (BIO-TEK FL600A, Hercules, CA, USA) was used to measure absorbance at a wavelength of 590 nm. Cells treated with DMSO (the same concentration as used with sorafenib) as the negative control.
Detecting cytochrome c and AIF translocation
Mitochondrial and cytosolic fractionation was performed by washing 1 × 107 PC-3 cells treated with 10 μmol L−1 sorafenib for 20 h in PBS followed by resuspension in mitochondrial isolation buffer (20 mmol L−1 HEPES [pH 7.5], 250 mmol L−1 sucrose, 10 mmol L−1 KCl, 1.5 mmol L−1 MgCl2, 1 mmol L−1 DTT, 1 mmol L−1 EDTA, 1 mmol L−1 EGTA, 2 mmol L−1 PMSF, 1 μg mL−1 aprotinin, 1 μg mL−1 leupeptin and 1 μg mL−1 pepstatin) on ice for 20 min and by homogenization with 20 passages through a 26-gauge needle. Large cellular debris was removed from the homogenate by centrifugation at 4°C for 10 min at 700 × g. The supernatant was again centrifuged at 4°C at 700 × g for 10 min, collected and centrifuged at 4°C, 10 000 × g for 25 min, generating a pellet containing mitochondrial proteins and a supernatant containing cytosolic proteins. The mitochondrial fraction was resuspended in mitochondrial storage buffer (10 mmol L−1 HEPES-KOH [pH 7.5], 250 mmol L−1 sucrose, 1 mmol L−1 ATP, 5 mmol L−1 sodium succinate, 0.08 mmol L−1 ADP, 2 mmol L−1 K2HPO4) and all samples were stored at −80°C. Cytosolic and nuclear fractions were prepared with a nuclear-cytosolic protein isolation kit (Beyotime Institute of Biotechnology, Hangzhou, China).
Lysates from the mitochondrial and cytosolic fractions were analysed for cytochrome c by Western blot. Lysates from the mitochondrial and nuclear fractions were assayed to analyse AIF translocation.
Statistical analysis
All experiments were set up in triplicate, and the results were expressed as mean ± SD. The significance of differences between experimental conditions was determined using one-way ANOVA and differences were considered significant if P < 0.05.
Results
Exposure of PC-3 cells to sorafenib is associated with a decrease in ERK phosphorylation
Dose–response studies revealed that exposure of PC-3 cells to sorafenib at concentrations as low as 5 μmol L−1 resulted in a discernable decrease in ERK phosphorylation after 4-h treatment (Figure 1A). Exposure to 10 and 20 μmol L−1 sorafenib produced even more pronounced reductions in ERK phosphorylation. Total levels of ERK remained unchanged in all cases. Furthermore, ERK phosphorylation was completely suppressed after treatment with 10 μmol L−1 sorafenib in LNCap cells (Figure 1B), while DU145 cells required 20 μmol L−1 (Figure 1C).
Figure 1.
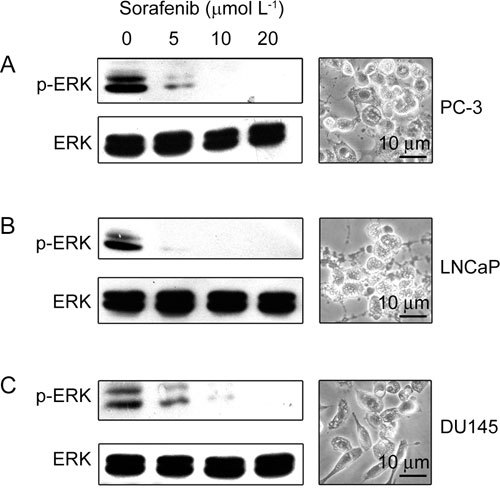
Inhibition of the canonical extracellular signal regulated protein kinases (ERK) phosphorylation by sorafenib treatment. Prostate cancer cells were exposed to the drug at the indicated concentrations for 4 h. The cells were then lysed and the lysates were analyzed by Western blot for phosphorylated-ERK (p-ERK), using total ERK levels as an internal control. The morphological pictures represent PC-3, LNCap, and DU145 cells treated with 10 μmol L−1 sorafenib for 24 h.
Sorafenib induces apoptosis and mitochondrial injury in PC-3 cells
Dose–response studies were performed to characterize the apoptotic effect of sorafenib treatment on PC-3 cells. As shown in Figure 2A, 20 h of exposure of PC-3 cells to increasing concentrations of sorafenib revealed a moderate induction of apoptosis at concentrations as low as 5 μmol L−1, indicated by Annexin V analysis. Higher concentrations of sorafenib resulted in greater amounts of cell death (27.7% at 10 μmol L−1).
Figure 2.
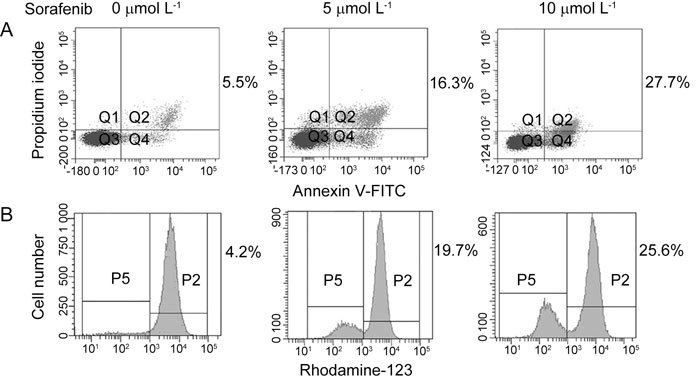
Induction of apoptosis and mitochondrial depolarization in PC-3 cells in response to sorafenib treatment. (A): PC-3 cells were exposed to the drug at the indicated concentration for 20 h, stained with Annexin-FITC V and propidium iodide (PI) and analyzed by flow cytometry. Numbers represent the percentage of cells stained with Annexin V alone (Q4) and double stained with Annexin-V and PI (Q2). (B): PC-3 cells stained with Rhodamine-123 and analyzed by flow cytometry. Numbers represent the percentage of cells displaying diminution of mitochondrial transmembrane potential. P, percentage. Q, quantitative.
To assess the effect of sorafenib treatment on mitochondrial membrane potential, PC-3 cells were incubated with different concentrations of sorafenib for 20 h and assayed by Rhodamine-123 staining. As shown in Figure 2B, sorafenib treatment induced decay of Rhodamine-123 fluorescence in a dose-dependent manner, indicating a diminution of mitochondrial uptake and transmembrane potential.
Sorafenib induces cytochrome c, but not AIF, release from mitochondria in PC-3 cells
To determine whether the mitochondrial depolarization induced by sorafenib treatment was associated with the release of pro-apoptotic proteins from the mitochondria, PC-3 cells were exposed to the drug (10 μmol L−1 for 20 h) and separated into cytosolic and mitochondrial fractions. The individual fractions were analyzed for the presence of cytochrome c by Western blot. As shown in Figure 3A, sorafenib induced leakage of cytochrome c from the mitochondria into the cytosol.
Figure 3.
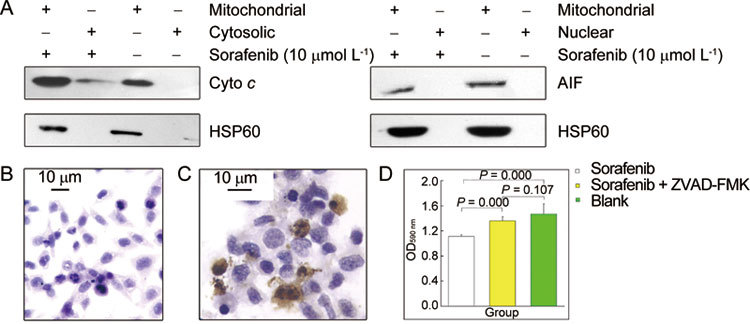
(A): Induction of cytochrome c (Cyto c), but not apoptosis-inducing factor (AIF) release, from mitochondria in response to sorafenib treatment using HSP60 as a mitochondrial marker. (B) and (C): Untreated and sorafenib-treated PC-3 cells assayed for caspase-3 cleavage by immunocytochemical staining. (D): The contribution of caspase activation to the lethality of sorafenib treatment with or without the pan-caspase inhibitor Z-VAD-FMK (MTT assay).
To determine whether sorafenib treatment induced AIF translocation from the mitochondria to the nucleus, PC-3 cells were exposed to the drug (10 μmol L−1 for 20 h), separated into mitochondrial and nuclear fractions, and analysed for the presence of AIF by Western blot. As shown in Figure 3A, no AIF was detected in the nuclear fraction following sorafenib treatment, indicating an absence of AIF translocation into the nucleus in response to treatment.
Sorafenib induces caspase-3 cleavage in PC-3 cells
To measure caspase-3 activation in response to sorafenib treatment of prostate cancer cells, PC-3 cells were plated on coverslips and incubated with the drug (10 μmol L−1 for 20 h). Cleaved caspase-3 was then detected by immunocytochemistry. As shown in Figure 3C, sorafenib-treated cells showed positive staining, which was always accompanied by nuclear condensation or nuclear fragmentation. These morphological changes indicate sorafenib-induced apoptosis. In contrast, no positive staining was observed in PC-3 cells lacking sorafenib treatment (Figure 3B). A pan-caspase inhibitor, Z-VAD-FMK, was used to demonstrate the involvement of caspase in sorafenib-induced apoptosis. PC-3 cells were treated with 10 μmol L−1 sorafenib for 20 h in the presence or absence of the caspase inhibitor. Sorafenib treatment significantly inhibited cell growth by 24.5% compared with DMSO-treated controls (P = 0.000). When Z-VAD-FMK was added to sorafenib treatment, growth was only inhibited by 7.5% compared with DMSO-treated controls (P = 0.000). As shown in Figure 3D, the percentage of viable cells increased significantly following Z-VAD-FMK treatment compared with sorafenib treatment alone (P = 0.107), indicating that caspase-3 has an important role in sorafenib-induced apoptosis.
Treatment of PC-3 cells with sorafenib results in a decrease in MCL-1, survivin and c-IAP2 protein levels
In view of the critical role that MCL-1, survivin and c-IAP2 have in the regulation of apoptosis, expression of these proteins was monitored in PC-3 cells after treatment with different concentrations of sorafenib for 6 and 24 h. As shown in Figure 4, sorafenib treatment reduced the levels of these proteins after 6 h. This decrease in the levels of these proteins was more pronounced after 24 h of treatment.
Figure 4.
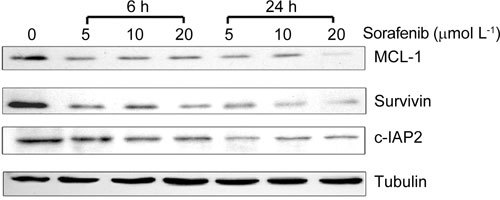
The effect of sorafenib treatment on down-regulation of anti-apoptotic proteins. PC-3 cells were exposed to the drug at the indicated concentrations for 6 and 24 h. Decreased levels of the MCL-1, survivin and c-IAP2 proteins were confirmed by Western blot.
Discussion
It has been well-established that drug-induced apoptosis in most cancer cells is mediated through the mitochondrial (intrinsic) pathway and/or the death receptor (extrinsic) pathway of caspase activation 21. One of the central control steps in the induction of apoptosis through the mitochondrial pathway is the disruption of the mitochondrial membrane potential, which leads to the release of cytochrome c and triggers caspase-9 activation. This critical step is controlled and mediated by the BCL-2 family of proteins 22. MCL-1, one of the BCL-2 family members that inhibits cytochrome c release and caspase activation, has been shown to be down-regulated following sorafenib treatment in a variety of human tumour cell lines 23, 24, 25, 26.
The activity of caspases-3, -7, and -9 is often further regulated by inhibitor of apoptosis protein (IAP) family members (i.e., c-IAP1 [cellular inhibitor of apoptosis protein 1], c-IAP2, XIAP [X-linked inhibitor of apoptosis protein] and survivin) 27, 28. To date, most studies of this family of proteins have focussed on the expression of survivin. This molecule has been identified in a wide variety of cancers 27 and is associated with adverse features in many malignant tumours, including breast carcinoma, gastric carcinoma, primary and recurrent colorectal carcinoma, neuroblastoma and non-small-cell lung cancer 29. Transfection with anti-sense oligonucleotides against survivin results in increased levels of apoptosis and prevents both cytokinesis and anchorage-dependent cell growth 30. Similar anti-sense studies, or use of dominant-negative mutants (T34A), have confirmed a role for survivin in conferring apoptotic resistance in prostate cancer 31, 32. In the case of c-IAP2, reports have indicated an association between higher expression levels of c-IAP2 and more aggressive disease, consistent with the documented function of this anti-apoptotic protein as a suppressor of caspases 28. In this study, we demonstrated that sorafenib treatment reduces constitutive expression of the anti-apoptotic proteins MCL-1, survivin and c-IAP2 in PC-3 cells.
AIF is a flavoprotein with both oxidoreductase and DNA-binding domains but no intrinsic DNase activity. It is involved in initiating a caspase-independent pathway of apoptosis (positive intrinsic regulator of apoptosis) 33, 34. It also participates in the regulation of apoptotic mitochondrial membrane permeabilization and exhibits an NADH oxidase activity 35. Under normal circumstances, AIF is sequestered behind the outer mitochondrial membrane. However, upon induction of apoptosis, AIF translocates to the cytosol and the nucleus and binds to the DNase Endo G, another mitochondrial constituent that is released in response to apoptotic stimuli 34. This recruitment of Endo G results in DNA fragmentation and cell death 36, 37.
We have demonstrated that sorafenib treatment results in the release of cytochrome c from the mitochondria and induction of caspase-3 activation in PC-3 cells. Immunocytochemistry showed positive staining of active caspase-3 in cells with nuclear fragmentation and condensation, coincident with morphological changes that are characteristic of apoptotic cells. However, release of AIF from the mitochondria and subsequent translocation of AIF to the nucleus were not observed in sorafenib-treated PC-3 cells. In addition, sorafenib-induced apoptosis can be inhibited by treatment with Z-VAD-FMK. Together, these findings indicate that sorafenib treatment results in the induction of mitochondrial injury and caspase-dependent apoptosis in PC-3 cells.
In more sensitive cell lines, such as Jurkat leukaemia cells or SKMEL5 melanoma cells, sorafenib-induced apoptosis is largely caspase independent because Z-VAD-FMK is not inhibitory and AIF translocation has an essential role in the apoptotic process 23, 38. A previous study observed a higher rate of sorafenib-induced apoptosis than that observed in this study: ∼80% cell death for Jurkat cells at 15 μmol L−1 and 80.9% cell death for SKEML5 cells at 20 μmol L−1 (27.7% cell death was observed for PC-3 cells at 10 μmol L−1 in the current study). It is possible that this cell-type difference explains why the efficacy of sorafenib is not as obvious in clinical phase II trials as for other tumour types 20.
Rahmani et al. 39 found that constitutive activation of MEK1 failed to protect leukaemia cells from sorafenib-induced lethality. Their data suggested that endoplasmic reticulum (ER) stress represents a central component of an MEK1/2-ERK1/2-independent cell death program that is triggered by sorafenib treatment. In a previously published phase II clinical trial using sorafenib for castration-resistant prostate cancer, ERK levels were inconclusively down-regulated and did not correlate clinically with response or PSA levels 19. These findings indicate that mechanisms of action of sorafenib other than the ERK pathway need to be studied in androgen-independent prostate cancer.
This study found that sorafenib-induced apoptosis is caspase-dependent in PC-3 cells. However, additional research will be required to elucidate the mechanism of sorafenib-induced lethality in androgen-independent prostate cancer.
Conflicts of interest
All authors declare that they have no financial conflicts of interest with Bayer Corporation (West Haven, CT, USA).
Acknowledgments
We thank Mr Wen-Tong Meng and Mr Ji-Long Gou (Stem Cell Research Laboratory, West China Hospital, Sichuan University, Chengdu, China) for technical assistance with the flow cytometry. We also thank BioMed Proofreading for their editing work. This work was supported by grants to Prof Hao Zeng and Dr Rui Huang from the National Natural Science Foundation of China (NSFC 30700977 and 30600153).
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, et al. Cancer statistics. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wu S, Guo LR, Zhao XJ. Diagnostic strategies and the incidence of prostate cancer: reasons for the low reported incidence of prostate cancer in China. Asian J Androl. 2009;11:9–13. doi: 10.1038/aja.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrylak DP.New paradigms for advanced prostate cancer Rev Urol 20079Suppl 2): S3–12. [PMC free article] [PubMed] [Google Scholar]
- Pienta KJ, Smith DC.Advances in prostate cancer chemotherapy: a new era begins CA Cancer J Clin 200555300–18.quiz 23–5. [DOI] [PubMed] [Google Scholar]
- Berry WR. The evolving role of chemotherapy in androgen-independent (hormone-refractory) prostate cancer. Urology. 2005;65:2–7. doi: 10.1016/j.urology.2005.03.080. [DOI] [PubMed] [Google Scholar]
- Gao QZ, Lu JJ, Liu ZD, Zhang H, Wang SM, et al. Dexamethasone suppresses DU145 cell proliferation and cell cycle through inhibition of the extracellular signal-regulated kinase 1/2 pathway and cyclin D1 expression. Asian J Androl. 2008;10:635–41. doi: 10.1111/j.1745-7262.2008.00352.x. [DOI] [PubMed] [Google Scholar]
- Carey AM, Pramanik R, Nicholson LJ, Dew TK, Martin FL, et al. Ras-MEK-ERK signaling cascade regulates androgen receptor element-inducible gene transcription and DNA synthesis in prostate cancer cells. Int J Cancer. 2007;121:520–7. doi: 10.1002/ijc.22715. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, et al. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13:4934–42. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, et al. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–69. [PubMed] [Google Scholar]
- Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, et al. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. 1996;148:1567–76. [PMC free article] [PubMed] [Google Scholar]
- Royuela M, Arenas MI, Bethencourt FR, Sanchez-Chapado M, Fraile B, et al. Immunoexpressions of p21, Rb, mcl-1 and bad gene products in normal, hyperplastic and carcinomatous human prostates. Eur Cytokine Netw. 2001;12:654–63. [PubMed] [Google Scholar]
- Cavarretta IT, Neuwirt H, Untergasser G, Moser PL, Zaki MH, et al. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene. 2007;26:2822–32. doi: 10.1038/sj.onc.1210097. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, et al. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–21. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23:965–72. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- Steinbild S, Mross K, Frost A, Morant R, Gillessen S, et al. A clinical phase II study with sorafenib in patients with progressive hormone-refractory prostate cancer: a study of the CESAR Central European Society for Anticancer Drug Research-EWIV. Br J Cancer. 2007;97:1480–5. doi: 10.1038/sj.bjc.6604064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi KN, Ellard SL, Hotte SJ, Czaykowski P, Moore M, et al. A phase II study of sorafenib in patients with chemo-naive castration-resistant prostate cancer. Ann Oncol. 2008;19:746–51. doi: 10.1093/annonc/mdm554. [DOI] [PubMed] [Google Scholar]
- Dahut WL, Scripture C, Posadas E, Jain L, Gulley JL, et al. A phase II clinical trial of sorafenib in androgen-independent prostate cancer. Clin Cancer Res. 2008;14:209–14. doi: 10.1158/1078-0432.CCR-07-1355. [DOI] [PubMed] [Google Scholar]
- Aragon-Ching JB, Jain L, Gulley JL, Arlen PM, Wright JJ, et al. Final analysis of a phase II trial using sorafenib for metastatic castration-resistant prostate cancer. BJU Int. 2009;103:1636–40. doi: 10.1111/j.1464-410X.2008.08327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Green DR. At the gates of death. Cancer Cell. 2006;9:328–30. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–27. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- Ding Q, Huo L, Yang JY, Xia W, Wei Y, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68:6109–17. doi: 10.1158/0008-5472.CAN-08-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12:66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, et al. The role of Mcl-1 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. 2005;24:6861–9. doi: 10.1038/sj.onc.1208841. [DOI] [PubMed] [Google Scholar]
- Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. Embo J. 1997;16:6914–25. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Survivin study: what is the next wave. J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, et al. Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia. 2000;2:235–41. doi: 10.1038/sj.neo.7900091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi N, Asano K, Suzuki H, Yamamoto T, Tanigawa N, et al. Adenoviral infection of survivin antisense sensitizes prostate cancer cells to etoposide in vivo. Prostate. 2005;65:10–9. doi: 10.1002/pros.20232. [DOI] [PubMed] [Google Scholar]
- Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005;24:2474–82. doi: 10.1038/sj.onc.1208490. [DOI] [PubMed] [Google Scholar]
- Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–23. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–96. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111:147–50. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death. J Cell Sci. 2002;115:4727–34. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- Panka DJ, Wang W, Atkins MB, Mier JW. The Raf inhibitor BAY 43-9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells. Cancer Res. 2006;66:1611–9. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]