

Abstract
Partial androgen insensitivity syndrome (PAIS) is the milder variant of androgen receptor (AR) defects. The subtle effects of AR mutations present in a patient with micropenis, peno-scrotal hypospadias, infertility, clitoromegaly and posterior labial fusion. We studied the association of isolated micropenis with the genetic defects resulting in androgen resistance, that is, AR gene defects and 5-α reductase type 2 (SRD5A2) deficiency. We describe two cases of isolated micropenis: one in a 14-year-old boy and the other in a 3-year-old boy who was followed until he was 10 years old. There were no findings of hypospadias, cryptorchidism or gynecomastia in either of these patients. Serum gonadotrophin and androgen levels were obtained and karyotyping was done. Human chorionic gonadotropin (hCG) stimulation testing assessed the functional capacity of the testes. DNA was extracted from peripheral leukocytes, and all exons of the SRD5A2 and AR genes were amplified by polymerase chain reaction and sequenced. In both patients, baseline testosterone (T) level was low and the values were elevated after hCG testing. The sequence of the SRD5A2 gene was normal in patient 1, and a heterozygous polymorphism, V89L, was found in patient 2. Two known mutations, P390S and A870V, were identified in patients 1 and 2, respectively. Mutations in the AR gene can be associated with isolated micropenis without other features of PAIS, such as hypospadias or gynecomastia. This underlines the importance of including AR gene analysis in the evaluation of isolated micropenis with normal plasma T to ensure proper management of the patient and appropriate genetic counseling for the family.
Keywords: androgen receptor, gynecomastia, isolated micropenis, male infertility
Introduction
Micropenis can be a manifestation of underlying growth hormone deficiency, hypogonadotrophic hypogonadism, or defects in androgen biosynthesis and action. It has been associated with abnormalities in the androgen receptor (AR) leading to partial androgen insensitivity syndrome (PAIS) 1, 2, 3, 4. However, in addition to micropenis, these patients had other features of PAIS, such as scant pubic hair development, gynecomastia during adolescence and penile chordae during infancy 1, 2, 3, 4.
In a study from the UK, the association of gonadotrophin deficiency, testosterone biosynthesis and AR binding was studied in 18 boys with isolated micropenis. No evidence of abnormal AR binding was found in these patients 5. The length of the CAG trinucleotide on exon 1 that encodes for the polyglutamine tract (with a polyglutamine length of 28 or more) has been associated with male infertility 6, but no association between the CAG trinucleotide repeat and micropenis has been observed 7. Severe micropenis has also been described in association with a polymorphism in the steroidogenic factor-1 gene 8. The Gly146Ala polymorphism was found to reduce the function by approximately 20% 8. In this study, a higher percentage of patients with severe micropenis carried this polymorphism compared with control subjects and patients with mild micropenis 8. A polymorphism causing androgen production defects might also explain severe isolated micropenis, but the number of cases showing this association is unknown.
PAIS with evidence of AR gene defects has not been reported in the context of isolated micropenis 4. In this study, we sought a link between isolated micropenis and an AR defect. We describe two patients with isolated micropenis and AR gene mutations, P390S and A870V. The P390S mutation has been described in relation to infertility and the A870V mutant has been related to hypospadias and cryptorchidism 9, 10. This is the first report of these mutations in association with isolated micropenis.
Materials and methods
Hormonal testing
Human chorionic gonadotropin (hCG) stimulation tests were performed to assess the functional capacity of the testes, as previously described 11. Steroids were analyzed by radioimmunoassay after selective solvent extraction, column chromatography, hydrolysis and high-performance liquid chromatography tandem mass spectrometry.
DNA isolation and sequencing
DNA was extracted from peripheral leukocytes and all exons of the AR genes were amplified by polymerase chain reaction using primer pairs and standard conditions. The SRD5A2 gene was also analyzed in both patients. DNAs from the mothers of both patients were also obtained and extracted and the AR gene was analyzed using standard conditions. Written informed consent and assent to perform the mutational analysis was obtained from the parents and subjects, respectively. The study was approved by the Institutional Review Board of Maimonides Medical Center, Brooklyn, and Hôpital Lapeyronie, Centre Hospitalier Universitaire, Montpellier.
AR gene mutation prediction model
Amino-acid substitutions were studied in silico to predict the effects using PolyPhen (Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph/index.html) 12 and SIFT 13 software (http://sift.jcvi.org/ ). The PolyPhen algorithm predicts the functional effects of amino-acid changes by considering evolutionary conservation, physicochemical differences, and the proximity of the substitution to the predicted functional domains and/or structural features. The SIFT algorithm predicts the functional importance of the amino-acid substitutions based on the alignment of orthologous and/or paralogous protein sequences. Original sequences of proteins were obtained from the Ensembl and UniProt/Swiss, Prot databases.
Patient description
Patient 1: A 14-year-old boy was seen in a pediatric endocrinology clinic for evaluation of micropenis. He had been born full term and the micropenis had been noted at the time of birth; however, the exact measurement was not available for our review. Both parents were healthy and the family had no history of delayed puberty or infertility. Physical examination revealed axillary hair at Tanner stage 2 and pubic hair at Tanner 3. The testes were descended bilaterally and 8 cm3 in volume, the scrotum was rugated normally, and the stretched penile length was 2.0 cm (below −2 SD). No gynecomastia or hypospadias was noted. The patient's height was 163 cm (50%, +0.05 SD). His karyotype was a normal 46,XY complement. The anti-Mullerian hormone and inhibin-B levels were in the normal range (Table 1). The ultrasensitive luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels were in the range of early puberty (Table 1). The baseline testosterone (T) concentration was low and rose to 15.29 nmol L−1 (442 ng dL−1) after the hCG stimulation test, the T:dihydrotestosterone (DHT) ratio was elevated to 31.85 and the T:Δ4 (androstenedione) ratio was normal (Table 1). The penis was carefully measured during the examination by a pediatric endocrinologist. After assessment and diagnosis, the patient was treated with T replacement. Within 6 months, his penile length increased from 2 to 4 cm (−2 SD to −1 SD); T replacement was continued until 16 years of age.
Table 1. Hormonal data and hCG stimulation test in patients 1 and 2.
| Test | Patient 1 (14 years old) |
Patient 2 (3 years old) |
||
|---|---|---|---|---|
| Pre-stimulation | Post-stimulation | Pre-stimulation | Post-stimulation | |
| Δ4, nmol L−1 (ng dL−1) | 1.19 (34) | 2.84 (81) | ||
| T, nmol L−1 (ng dL−1) | 0.52 (15) | 15.29 (442) | 0.35 (10) | 22.84 (660) |
| DHT, nmol L−1 (ng dL−1) | 0.08 (2.2) | 0.48 (14) | ||
| T: DHT ratio | 6.50 | 31.85 | ||
| T: Δ4 ratio | 0.44 | 5.38 | ||
| LH, IU L−1 | 0.72 | 0.10 | ||
| FSH, IU L−1 | 3.9 | 0.6 | ||
| Inhibin-B, pg mL−1 | 86 | |||
| AMH, ng dL−1 | 20 | |||
Abbreviations: AMH, anti-Mullerian hormone; Δ4, androstenedione; DHT, dihydrotestosterone; FSH, follicle-stimulating hormone; hCG, Human chorionic gonadotropin; LH, luteinizing hormone; T, testosterone.
Patient 2: At 3 years of age, this boy was referred to a pediatric endocrinology clinic because of isolated micropenis. He had been born to non-consanguineous parents at 39 weeks of gestation, after an uncomplicated pregnancy and delivery (birth weight: 3.6 kg, birth length: 51 cm). His height was 101 cm (+2 SD) and his weight was 15 kg (+1 SD). His stretched penile length was 2.5 cm (−3.3 SD), and the testes were descended bilaterally and 2 cm3 in volume with pubic hair development at Tanner stage 1. The urethral meatus was located at the top of the glans. Bone age was 2.6 years. Basal LH and FSH were in the normal prepubertal range. T was 0.35 nmol L−1 (10 ng dL−1) and increased to 22.84 nmol L−1 (660 ng dL−1) after hCG stimulation testing. The stretched penile length after hCG testing was 4 cm (−1.7 SD), which indicated a good clinical response. This penile length was conserved 6 months later. Nevertheless, a decrease in penile length was observed at 10 years, to 3.5 cm (−2.5 SD). We thus decided to treat the patient with T enanthate (Androtardyl®, Bayer Santé, Puteaux cdx, France) at 100 mg m−2 (bodily area), 100 mg every month, for 3 months. This yielded a very good clinical response, with penile length increasing to 5 cm (+0.9 SD) after T injections. No history of micropenis, cryptorchidism, hypospadias or infertility was found in the mother's family.
Results
Patient 1: The SRD5A2 gene was analyzed and was found to be normal. However, a Pro390Ser mutation on exon 1 of the AR gene was identified. The mother carried the same mutation, which is consistent with an X-linked pattern of PAIS (Figure 1A).
Figure 1.
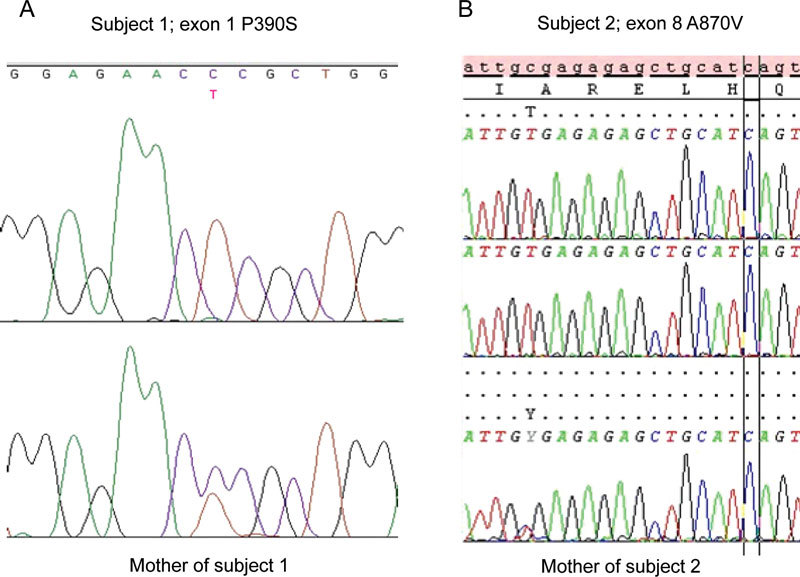
(A): The sequence of the patient with P390S exon 1 mutation and, below, the sequence of the mother of subject 1 carrying the same mutation. (B): The sequence of the patient with A870V exon 8 mutation and, below, the sequence of the mother of subject 2.
Patient 2: An A870V mutation on exon 8 of the AR gene was found. The mother was found to be a carrier for the A→V substitution identified in the propositus (Figure 1B). The SRD5A2 gene was analyzed and a V89L polymorphism on exon 1 was found in a heterozygous state. The same polymorphism has previously been described 14.
Prediction of mutation effects
To assess the potential deleterious effect of the amino-acid change, we used two types of software to predict the functional effects. First, we performed testing with PolyPhen, a tool that predicts the possible impact of an amino-acid substitution on the structure and function of a human protein using straightforward physical and comparative analysis. The mutations were evaluated as 'Probably damaging'.
The same assessment was performed with SIFT software. SIFT uses protein sequence conservation data to calculate the probability of a mutation being deleterious. Scores less than 0.05 suggest potential pathogenicity. Similar to the PolyPhen method, SIFT placed the mutations in the 'Affects protein function' class, with a score of 0.03, and confirmed the first evaluation with the PolyPhen algorithm.
Discussion
Mutations in the AR gene are associated with a wide spectrum of phenotypes. To date, hundreds of mutations have been described, with clinical phenotypes from externally normal females with no androgenic effects to partial androgen insensitivity characterized by genital ambiguity and males with undervirilization at one end of the spectrum. Moreover, the same AR mutation can result in different clinical presentations within the same family and even among siblings 3, 15. In one report, for example, a sibling with clitoromegaly and labial fusion was raised as a girl while the other sibling with micropenis and peno-scrotal hypospadias was raised as a boy 15. In another report of an extended Chinese family with an AR mutation, some of the family members were infertile with gynecomastia and/or hypospadias, whereas other members were fertile 3.
Patient 1 was evaluated for isolated micropenis. The patient's normal stature and the detection of pubertal levels of LH and FSH ruled out growth hormone deficiency and hypogonadotrophic hypogonadism as the cause of isolated micropenis. The patient's T levels were low at baseline and increased normally after the hCG stimulation test. The T:DHT ratio was high (> 20:1) and consistent with a 5-α reductase type 2 enzyme deficiency 16. However, it was shown in previous studies that the T:DHT ratio is not sensitive for 5-α reductase type 2 deficiency 17. The T:Δ4 ratio was normal (> 0.8) and is a very sensitive marker for 17 βHSD-3 enzyme deficiency 18. Upon hCG stimulation, the T:DHT ratio was high and initially the diagnosis of 5-α reductase deficiency was proposed. The entire coding region of the SRD5A2 gene was analyzed and found to be normal. We further investigated whether AR defects could have led to micropenis. We found a mutation on exon 1 of the AR gene, which led to the Pro390Ser change. This proline to serine change at position 390 was previously reported to be associated with decreased spermatogenesis and infertility in men 9. At the time patient 1 was evaluated, he was at Tanner 3 and only had micropenis; gynecomastia was not noted on examination. The testicular size was also compatible with Tanner 3 development. At this point, we cannot exclude the possibility of abnormal breast development later on in puberty.
Patient 2 was evaluated at 3 years of age for micropenis. The patient had a good response to hCG stimulation testing as the T levels were high, and we observed a subsequent increase in the penile length from 2.5 cm (−3.3 SD) to 4 cm (−1.7 SD). A similar response to T replacement was obtained later on. It has been reported that androgen resistance can be overcome by exogenous androgen therapy. Therefore, despite the good response to exogenous androgens, we decided to analyze the AR gene and found that patient 2 carried an A870V mutation on exon 8, which is situated in the hormone-binding domain of the receptor.
There have been some reports of micropenis with AR mutations, but a direct association has not been reported 3, 4. In a large Chinese family with the Arg840Cys mutation, some of the family members were infertile with gynecomastia and/or hypospadias, whereas others were fertile but had a 'small penis', according to their wives 3. These patients, reportedly with micropenis, were not examined by clinicians, nor was their DNA analyzed for AR mutations. There was no report of gynecomastia or hypospadias in these patients. As shown in table 2, another family member was found to have a 'slightly diminished penile length' and gynecomastia, but the patient's DNA was not studied for AR gene mutation 3. In another report, a 16-year-old boy was described with a 'slightly diminished penile length', normal scrotal size and Tanner stage 3–4 gynecomastia 2. Unfortunately, the measurements and SD of penile length were not provided. This patient had elevated T and LH levels with normal levels of inhibin-B. This patient was fertile but had a low seminal volume, low sperm concentration and highly immotile sperms 2. Another report was on infants with micropenis and penile chordae. This patient was the brother of a newborn boy with PAIS and the L712F mutation of the AR gene 4. Of note, these patients were examined in infancy and may or may not develop gynecomastia in puberty.
Table 2. Androgen receptor gene mutations leading to micropenis.
| Phenotype | Mutation | Exon | Functional affect | References |
|---|---|---|---|---|
| Isolated micropenis | P390S | 1 | Transactivation defect | This report |
| Isolated micropenis | A870V | 8 | Hormone binding | This report |
| Reported slightly diminished penile size | A840C (presumed mutation) | 7 | Hormone binding | 3 |
| Reported slightly diminished penile size, gynecomastia | G824L | 7 | Hormone binding | 2 |
| Micropenis, penile chordae | L712F | 4 | Transactivation defect | 4 |
The Pro390Ser mutation on exon 1 of the AR gene in patient 1 has also been reported to be associated with infertility and seminoma 9, 19, 20. The variation lies within the first exon encoding for the transactivation domain of the receptor 9. Interestingly, this mutation is located within a region of the AR gene that is important for transcriptional activity of the receptor 21. In the in vitro model, the Ser390 AR mutant did not lead to gross alterations in transcriptional activity in the presence of the combination of T and DHT, compared with the wild-type AR 9. The authors suggested that the in vitro model was oversimplified and would not reflect the changes induced in vivo leading to defective spermatogenesis 9. This might explain our patient's mild presentation of isolated micropenis without other features of PAIS. The A870V mutation in patient 2 has already been reported in association with peno-scrotal hypospadias and bilateral cryptorchidism 10. Although the phenotype of the patient in this earlier study was more severe than that of our patient, he was assigned male gender and responded very well to T treatment, presenting normal prepubertal male genitalia after the treatment and surgical correction. Other mutations affecting this amino acid in the hormone-binding domain have been reported and lead to a wide range of phenotypes, from isolated gynecomastia to ambiguous genitalia to complete sex reversal 22, 23, 24.
We conclude that AR gene mutations may be linked with the phenotype of isolated micropenis without other features of PAIS, such as hypospadias or gynecomastia. This underlines the importance of evaluating the AR gene sequence in cases of isolated micropenis, as the identification of a mutation will have an impact on patient management and will orient appropriate genetic counseling for the family.
References
- Grino PB, Griffin JE, Cushard WG, Jr, Wilson JD. A mutation of the androgen receptor associated with partial androgen resistance, familial gynecomastia, and fertility. J Clin Endocrinol Metab. 1988;66:754–61. doi: 10.1210/jcem-66-4-754. [DOI] [PubMed] [Google Scholar]
- Giwercman A, Kledal T, Schwartz M, Giwercman YL, Leffers H, et al. Preserved male fertility despite decreased androgen sensitivity caused by a mutation in the ligand-binding domain of the androgen receptor gene. J Clin Endocrinol Metab. 2000;85:2253–9. doi: 10.1210/jcem.85.6.6626. [DOI] [PubMed] [Google Scholar]
- Chu J, Zhang R, Zhao Z, Zou W, Han Y, et al. Male fertility is compatible with an Arg(840)Cys substitution in the AR in a large Chinese family affected with divergent phenotypes of AR insensitivity syndrome. J Clin Endocrinol Metab. 2002;87:347–51. doi: 10.1210/jcem.87.1.8167. [DOI] [PubMed] [Google Scholar]
- Holterhus PM, Sinnecker GH, Hiort O. Phenotypic diversity and testosterone-induced normalization of mutant L712F androgen receptor function in a kindred with androgen insensitivity. J Clin Endocrinol Metab. 2000;85:3245–50. doi: 10.1210/jcem.85.9.6812. [DOI] [PubMed] [Google Scholar]
- Evans BA, Williams DM, Hughes IA. Normal postnatal androgen production and action in isolated micropenis and isolated hypospadias. Arch Dis Child. 1991;66:1033–6. doi: 10.1136/adc.66.9.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tut TG, Ghadessy FJ, Trifiro MA, Pinsky L, Yong EL. Long polyglutamine tracts in the androgen receptor are associated with reduced trans-activation, impaired sperm production, and male infertility. J Clin Endocrinol Metab. 1997;82:3777–82. doi: 10.1210/jcem.82.11.4385. [DOI] [PubMed] [Google Scholar]
- Ishii T, Sato S, Kosaki K, Sasaki G, Muroya K, et al. Micropenis and the AR gene: mutation and CAG repeat-length analysis. J Clin Endocrinol Metab. 2001;86:5372–8. doi: 10.1210/jcem.86.11.7999. [DOI] [PubMed] [Google Scholar]
- Wada Y, Okada M, Hasegawa T, Ogata T. Association of severe micropenis with Gly146Ala polymorphism in the gene for steroidogenic factor-1. Endocr J. 2005;52:445–8. doi: 10.1507/endocrj.52.445. [DOI] [PubMed] [Google Scholar]
- Hiort O, Holterhus PM, Horter T, Schulze W, Kremke B, et al. Significance of mutations in the androgen receptor gene in males with idiopathic infertility. J Clin Endocrinol Metab. 2000;85:2810–5. doi: 10.1210/jcem.85.8.6713. [DOI] [PubMed] [Google Scholar]
- Hiort O, Klauber G, Cendron M, Sinnecker GH, Keim L, et al. Molecular characterization of the androgen receptor gene in boys with hypospadias. Eur J Pediatr. 1994;153:317–21. doi: 10.1007/BF01956409. [DOI] [PubMed] [Google Scholar]
- Scurry MT, Bruton J, Barry KG. The effect of chorionic gonadotropin on steroid excretion. Arch Intern Med. 1971;128:561–5. [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki G, Ogata T, Ishii T, Kosaki K, Sato S, et al. Micropenis and the 5alpha-reductase-2 (SRD5A2) gene: mutation and V89L polymorphism analysis in 81 Japanese patients. J Clin Endocrinol Metab. 2003;88:3431–6. doi: 10.1210/jc.2002-021415. [DOI] [PubMed] [Google Scholar]
- Evans BA, Hughes IA, Bevan CL, Patterson MN, Gregory JW. Phenotypic diversity in siblings with partial androgen insensitivity syndrome. Arch Dis Child. 1997;76:529–31. doi: 10.1136/adc.76.6.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato-McGinley J, Gautier T, Pichardo M, Shackleton C. The diagnosis of 5 alpha-reductase deficiency in infancy. J Clin Endocrinol Metab. 1986;63:1313–8. doi: 10.1210/jcem-63-6-1313. [DOI] [PubMed] [Google Scholar]
- Boudon C, Lumbroso S, Lobaccaro JM, Szarras-Czapnik M, Romer TE, et al. Molecular study of the 5 alpha-reductase type 2 gene in three European families with 5 alpha-reductase deficiency. J Clin Endocrinol Metab. 1995;80:2149–53. doi: 10.1210/jcem.80.7.7608269. [DOI] [PubMed] [Google Scholar]
- Bertelloni S, Dati E, Hiort O. Diagnosis of 17 β-hydroxysteroid dehydrogenase-3 deficiency. Expert Rev Endocrinol Metab. 2009;4:53–65. [Google Scholar]
- Ferlin A, Vinanzi C, Garolla A, Selice R, Zuccarello D, et al. Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 2006;65:606–10. doi: 10.1111/j.1365-2265.2006.02635.x. [DOI] [PubMed] [Google Scholar]
- Garolla A, Ferlin A, Vinanzi C, Roverato A, Sotti G, et al. Molecular analysis of the androgen receptor gene in testicular cancer. Endocr Relat Cancer. 2005;12:645–55. doi: 10.1677/erc.1.00954. [DOI] [PubMed] [Google Scholar]
- Berrevoets CA, Doesburg P, Steketee K, Trapman J, Brinkmann AO. Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2) Mol Endocrinol. 1998;12:1172–83. doi: 10.1210/mend.12.8.0153. [DOI] [PubMed] [Google Scholar]
- Albers N, Ulrichs C, Gluer S, Hiort O, Sinnecker GH, et al. Etiologic classification of severe hypospadias: implications for prognosis and management. J Pediatr. 1997;131:386–92. doi: 10.1016/s0022-3476(97)80063-7. [DOI] [PubMed] [Google Scholar]
- Hiort O, Sinnecker GH, Holterhus PM, Nitsche EM, Kruse K. Inherited and de novo androgen receptor gene mutations: investigation of single-case families. J Pediatr. 1998;132:939–43. doi: 10.1016/s0022-3476(98)70387-7. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Chavez B, Vilchis F, Kofman-Alfaro S. Phenotypic heterogeneity associated with identical mutations in residue 870 of the androgen receptor. Horm Res. 2002;57:90–3. doi: 10.1159/000057958. [DOI] [PubMed] [Google Scholar]