

Abstract
Limited knowledge of the genetic causes of male infertility has resulted in few treatment and targeted therapeutic options. Although the ideal approach to identify infertility causing mutations is to conduct studies in the human population, this approach has progressed slowly due to the limitations described herein. Given the complexity of male fertility, the entire process cannot be modeled in vitro. As such, animal models, in particular mouse models, provide a valuable alternative for gene identification and experimentation. Since the introduction of molecular biology and recent advances in animal model production, there has been a substantial acceleration in the identification and characterization of genes associated with many diseases, including infertility. Three major types of mouse models are commonly used in biomedical research, including knockout/knockin/gene-trapped, transgenic and chemical-induced point mutant mice. Using these mouse models, over 400 genes essential for male fertility have been revealed. It has, however, been estimated that thousands of genes are involved in the regulation of the complex process of male fertility, as many such genes remain to be characterized. The current review is by no means a comprehensive list of these mouse models, rather it contains examples of how mouse models have advanced our knowledge of post-natal germ cell development and male fertility regulation.
Keywords: infertility, male fertility, mouse models, spermatogenesis
Introduction
Infertility is a major medical problem worldwide. In the Western societies, one in eight couples of reproductive age is infertile (http://www.endotext.org/male/male7/maleframe7.htm). Of these, male factors are the sole or a contributing cause of the infertility in >40% of cases.1 Causes of male infertility may be broadly classified into four categories: (i) defective sperm production; (ii) reproductive tract obstruction; (iii) inflammation; and (iv) sexual disorders, for example, erectile dysfunction and ejaculatory failure.2, 3 Approximately half of the causes of male infertility result from defective sperm production, for example, complete blockade of spermatogenesis, low sperm counts, abnormal sperm motility, morphology or function.3 Although the etiology of most cases remains unknown, there is increasing evidence that a significant percentage of male infertility is caused by genetic defects including chromosome aberrations, gene mutations and single-nucleotide polymorphisms.4, 5, 6, 7, 8, 9, 10, 11
The complexity of mammalian male fertility regulation
Following the crucial process of in utero sex determination, the establishment of male fertility comprises a series of complex and highly structured steps involving cell division, differentiation and cell–cell interactions. The production of fully functional spermatozoa in mammals takes place within the seminiferous tubules of the testis and is under strict hormonal regulation. Several key hormones secreted by the hypothalamus (gonadotrophin-releasing hormone), pituitary gland (luteinizing hormone and follicle-stimulating hormone) and hormones secreted by the testis (for example, androgens, oestrogens, inhibins and follistatin) are involved in the regulation of spermatogenesis.12, 13, 14, 15 Deregulation of hormonal regulation has been shown to contribute to male/female infertility in human15 and mouse models.4, 16
Mammalian spermatogenesis can be separated into three categories of cellular events: the proliferative phase (spermatogonia), the meiotic phase (spermatocytes) and the differentiation phase or spermiogenesis (spermatids), followed by a series of post-testicular maturation processes required for fully functional spermatozoa (capable of motility and fertilization). In addition to germ cells, the testis contains two other specialized cell types, Sertoli and Leydig cells, which support spermatogenesis.12 Sertoli cells form the blood–testis barrier through the presence of intercellular tight junctions, which provides the isolated environment necessary for development of spermatocytes and spermatids. Sertoli cells also provide paracrine support to all adult germ cell types.17 Leydig cells in the interstitial tissue of the testis are uniquely positioned to provide testosterone to the seminiferous tubules to drive spermatogenesis.
In simple eukaryotes, such as flies and worms, it is estimated that over 1000 genes are involved in the regulation of male fertility.18, 19 In mice, it has been calculated that the male germ cell transcriptome comprises more than 30 000 transcripts.20 The somatic cells of the testis and those involved in endocrine regulation of spermatogenesis will contain many others. Theoretically, defects in any one of these genes could lead to infertility.
Challenges in defining male infertility genes in humans
Two common approaches used to evaluate the function of a gene are reverse genetic and forward genetic approaches (Figure 1). In a reverse genetic approach, the functional analysis of a gene begins by altering gene expression (that is, complete or partial inactivation or ectopic expression), followed by an assessment of the phenotypic consequences. In contrast, a forward genetic approach, or phenotypic-driven approach, starts by the relatively undirected generation of and identification of phenotypic variants of interest. The underlying mutation is identified subsequently. The latter approach is a powerful method for revealing completely novel genes that function in a particular process as it does not require fore knowledge of a gene's function.
Figure 1.
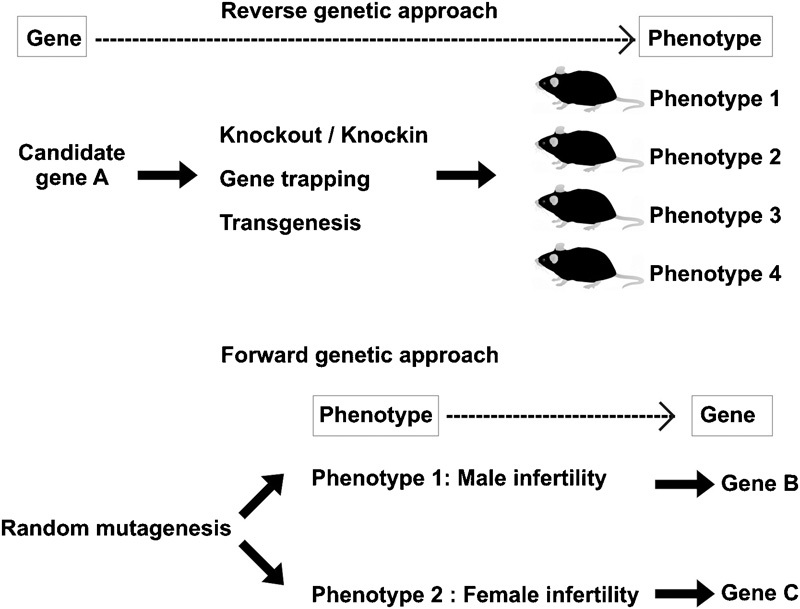
Reverse and forward genetic approaches for in vivo gene functional analyses in mouse models. The reverse approach begins by selecting a candidate gene of interest followed by evaluating its in vivo function using gene manipulation techniques and animal model production. Once generated, animal models are used for phenotypic characterization to define pathological abnormalities. In many cases, candidate genes have multiple roles in different tissues/organs; thus, the animal models often give rise to more than one phenotypic defect. By contrast, the forward genetic approach is initiated by the creation of animal models exhibiting a phenotypic defect of interest followed by defining the genetic alteration responsible for the phenotypic defects.
Cytogenetic and molecular studies have revealed that chromosome abnormalities including deletions (for example, the azoospermia factor region on the long arm of the Y chromosome), translocations and chromosome aneuploidies, occur with an increased incidence in infertile men compared with fertile men.4, 5, 6, 7, 8, 9, 10, 11 Although recent advances in molecular biology and the completion of human genome sequence have enabled the prediction of candidate genes within the deleted or translocated regions, the precise functions of the majority of these genes have yet to be determined. In addition, infertility-causing genes and risk factors are identified via mutation screening of candidate genes in infertile cases compared with fertile control men. However, the progress of this approach has been slow for a number of reasons, including: (i) inadequate numbers of patient and control groups for case–control studies; (ii) insufficiently detailed clinical/phenotypic information; (iii) the large genetic diversity in human populations that may contribute to different phenotypic outcomes (effects of modifier genes); (iv) the design of the mutation screens, for example, the majority of mutation screens focus on protein-coding regions of candidate genes that can eliminate or bias against the identification of intronic mutations; and (v) follow-up in vivo by gene functional analyses in humans remains problematic due to the limited availability of human samples and the associated ethical restrictions. The use of mouse models, however, does not have such problems and, as such, a substantial amount of fertility research is performed in the mouse.
The identification of genes required for spermatogenesis, post-testicular sperm maturation, capacitation and fertilization is imperative if we hope to obtain a complete understanding of male fertility. The outcomes of these studies have ramifications for the diagnosis and treatment of human male infertility as well as the development of the much needed male-based contraceptives for humans and animals. Male-based contraceptives, using both hormonal and immunological approaches, have been under trial with limited success.15, 21 Recent focus has been on identifying targets for non-hormonal contraception by taking advantage of the specific cellular and physiological processes unique to the reproductive organs. The main goal is to interfere in a highly specific manner in key developmental or functional processes. This requires the identification/characterization of many of the unknown male fertility regulators. With the exception of the spermatogonial stem cells, the temporary disruption of testicular germ cells is attractive as upon cessation of treatment it should require only one wave of spermatogenesis to restore germ cell production and fertility.
Different types of mouse models
Given the complexity of male fertility, the entire process cannot be fully modeled in vitro. As such, animal models provide a viable alternative for experimentation. Mice are the most commonly used animal models in biomedical research, including reproductive biology, because of their short reproductive cycle with a large litter size and relatively cheap housing conditions. Most importantly, mice are genetically very similar to humans22 and their embryos are relatively easy to manipulate at the genetic level. Moreover, mouse spermatogenesis and oogenesis are comparable to humans. Over several decades, many types of mouse models have been made available for biomedical research, including knockout/knockin and transgenic models (using the reverse genetic or candidate gene approach). More recently, chemical-mutagenized mutant mouse models (using the forward genetic approach) have been increasingly used to reveal disease-associated genes, including causes of male infertility.23, 24, 25, 26, 27, 28
Knockout/knockin mice and gene-trapped mice
The most common approach used to define a gene function in vivo is gene ablation by homologous recombination in embryonic stem (ES) cells, known as ‘knockout' or ‘gene targeting'.29, 30 This strategy is designed for the evaluation of a gene function on the basis of the complete elimination (null allele) or partially elimination of a candidate gene function (that is, deletion of particular domain(s) of the encoded protein).
Generally, targeting constructs are designed to carry a selectable marker to enable the selection of correctly targeted ES clones (for example, kanamycin/neomycin resistance gene), flanked by 5′ and 3′ homology regions of the target gene (Figure 2). Targeted ES cells are injected into blastocysts of a different mouse strain to produce chimeric progeny, which are subsequently mated with wild-type mice to generate heterozygous knockout mice, that is, carrying one deleted allele and one wild-type allele. Homozygous knockout mice, that is, carrying two deleted alleles, are generated by heterozygous intercrosses.31, 32 Once generated, the phenotypic characterization is performed to define pathological defects due to the complete or partial loss of the targeted gene function.
Figure 2.
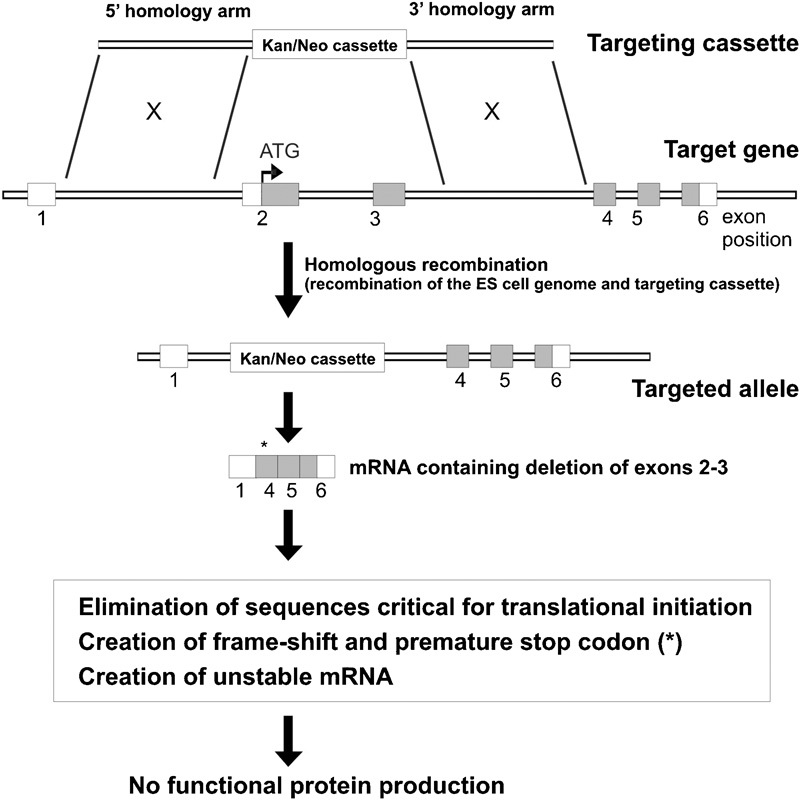
Gene targeting strategy for the generation knockout mice. A targeting cassette is designed to contain homology arms flanking a drug selectable marker (for example, kanamycin/neomycin) to facilitate the identification of targeted ES clones. Depending upon the design of the targeting cassette, a null allele can be generated by: (i) elimination of sequences critical for translational initiation; (ii) creation of a frame shift in the coding region and premature stop codon (indicated by *); and (iii) creation of unstable mRNA. Untranslated regions are depicted by unfilled boxes; protein-coding regions are depicted by filled boxes; ATG: initiation codon. ES, embryonic stem.
In addition to the generation of null alleles, specific disease-causing mutations can be precisely introduced into the mouse genome by a ‘knockin' approach. The production of knockin mice requires two rounds of homologous recombination in ES cells in order to eliminate the remaining sequence of targeting vectors from the targeted locus. Although the generation of knockin mice is more complicated than knockout mice, this approach enables the creation of mouse models carrying specific disease-causing mutations rather than a gene deletion that completely eliminates the protein function. For example, such alleles could carry mutations observed within the human population.
The production of null alleles by which the targeted genes are inactivated in every cell in the animal throughout its life can be very valuable in defining in vivo gene function. This approach, however, has a limitation due to a large percentage of knockout mice exhibiting unexpected embryonic/neonatal lethality, which limits the analysis of the gene function in later stages of life. In some instances, the gene may contribute to different aspects of health at different stages, for example, different functions in embryo stage compared with neonate and adult stages. In the case of infertility and contraceptive research, this is a serious limitation. To overcome this limitation, the ablation of a target gene at a precise time and/or specific tissue can be achieved by using the conditional knockout approach, which allows the mouse to develop up to a certain stage of development prior to inactivation of the gene. This approach utilizes the site-specific recombination systems, Cre/LoxP or Flp/FRT.33
In addition to knockout and knockin approaches, a high throughput method for random gene disruption, known as ‘gene trapping', has been developed.34 This approach is based on the use of a gene-trapping vector containing a conserved 3′ acceptor splice site, a eukaryotic drug-selectable marker (and/or markers for gene expression analysis) and transcriptional termination (poly A) signal to randomly insert and interrupt gene transcription in the ES cell genome (Figure 3). If the insertion of a gene trap cassette occurs within an intron of a gene, it will result in alternative splicing via the use of the 3′ acceptor splice site within the cassette. The transcription of the trapped gene will terminate at the poly A signal and result in the production of truncated protein or no protein depending upon where the cassette is inserted. Insertion in an intron prior to the ATG start site is likely to generate null alleles.34
Figure 3.
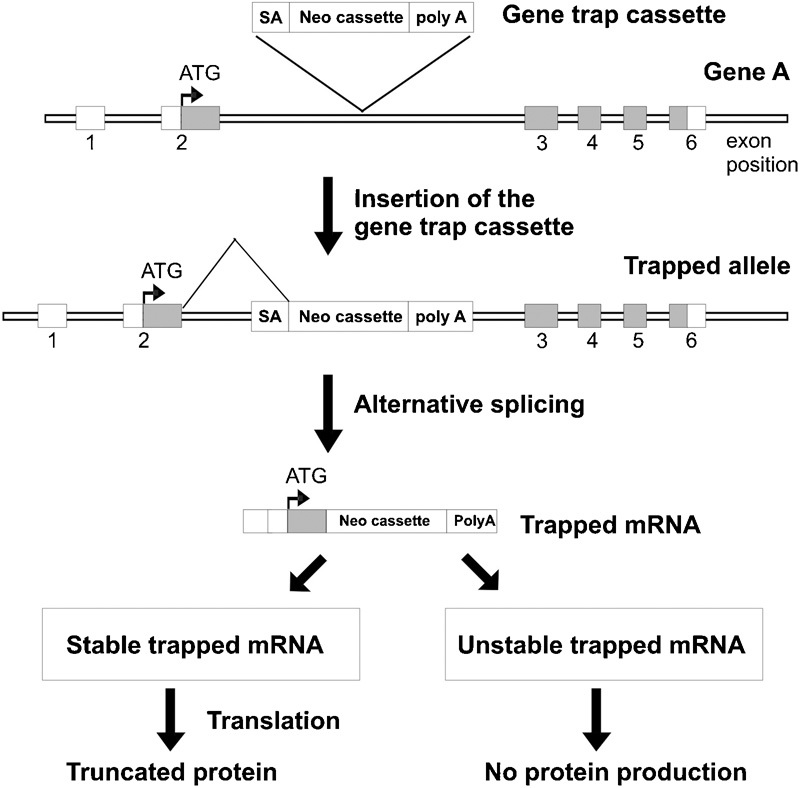
A high-throughput gene trapping approach. A gene trap cassette is designed to contain the features necessary for its integration into the ES cell genome and termination of transcription of the trapped allele. These features include SA site and poly A signal. If inserted within an intron of a gene, the native splicing pattern is affected by the SA, resulting in fusion of the upstream exons with the trapped cassette. Transcription of the trapped mRNA is subsequently terminated at the poly A site. If inserted within the 5′ UTR, the trapped mRNA will produce no functional protein. If insertion occurs within an intron downstream of the translational start site of a gene, a truncated protein (fused with the trapped cassette) may be produced depending upon the stability of the trapped mRNA. ES, embryonic stem; poly A, polyadenylation; SA, slicing acceptor; UTR, untranslated region.
Through the use of high-throughput technologies, ES clones containing thousands of different trapped genes have been produced. The collection of ES gene trap cell lines has been made publically available and centralized through the International Gene Trap Consortium, which aims to generate a library of mouse mutant ES cells covering most of the genes in the mouse genome.35, 36, 37 Furthermore, researchers should be aware of the existence of the International Knockout Mouse Consortium (http://www.knockoutmouse.org), which aims to mutate all protein-coding genes in the mouse genome using a combination of gene trapping and gene targeting in C57BL/6 mouse ES cells.38 The International Knockout Mouse Consortium includes the following programs: the Knockout Mouse Project (http://www.knockoutmouse.org/about/komp) (USA), the European Conditional Mouse Mutagenesis Program (http://www.eucomm.org),39 the North American Conditional Mouse Mutagenesis Project (http://www.norcomm.org/index.htm) (Canada) and the Texas A&M Institute for Genomic Medicine (http://www.tigm.org) (USA).
Transgenic mice
A transgenic organism refers to an organism that has had its genome modified to carry a copy (or more than one copy) of a piece of DNA of foreign origin. These models enable researchers to evaluate in vivo phenotypic defects as a result of ectopic expression of a gene, for example, overexpression of a wild-type gene and expression of a dominant-negative form to diminish the normal function of a gene. The production of transgenic mice is achieved by direct injection of a purified fragment of a transgene of interest into fertilized oocytes (Figure 4). Following injection, the transgene is inserted into the genome of fertilized oocytes, which are subsequently used to generate transgenic mice. The fact that transgene integration usually occurs randomly, and the expression level of transgenes can be influenced by positional effect and transgene copy number.
Figure 4.
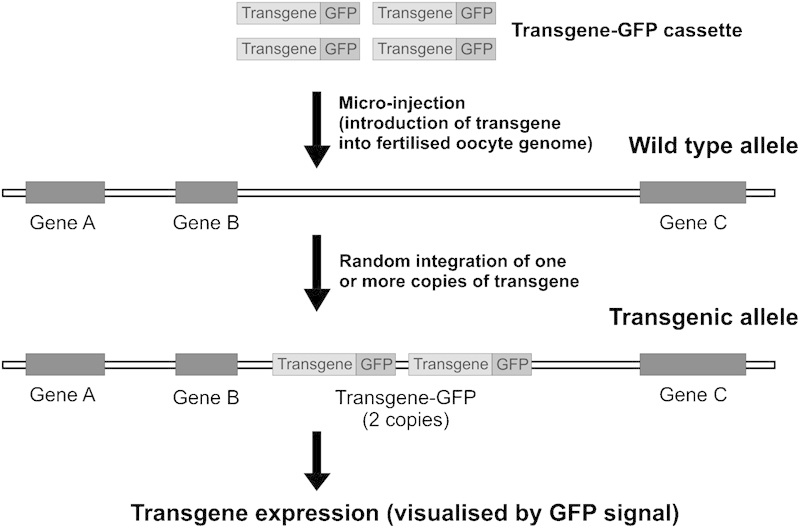
Transgenesis strategy. A transgene cassette is designed to contain features necessary for gene expression, for example, promoter and polyadenylation signal. Additional markers such as fluorescent protein tags (for example, GFP and YFP) can be included to facilitate the detection of transgene expression. The transgene is introduced into the genome of fertilized mouse oocytes. Integration of the transgene occurs randomly and multiple copies of the transgene may be integrated in the same chromosomal location or on different chromosomes. GFP, green fluorescent protein; YFP, yellow fluorescent protein.
Chemical-induced point mutant mice
To facilitate unbiased gene discovery in the mouse, a forward genetic-based approach using the potent mutagen, N-ethyl-N-nitrosourea (ENU) has been developed.40, 41, 42 ENU primarily acts as an alkylating agent, transferring its ethyl group to any of a number of nucleophilic nitrogen or oxygen sites on each of the four deoxyribonucleotides.40, 41, 42 The transferred ethyl group constitutes a DNA adduct that, during cell proliferation and DNA replication, results in heritable mutations. ENU induces the highest mutation rate of any agent tested in mice (on average 0.0015 point mutations per locus per gamete in the C57BL/6 strain). An ENU mutagenesis program involves injecting adult male mice with a series of ENU intraperitoneal injections. Following a transient period of sterility (10–12 weeks, depending on mouse strain and doses of ENU used), owing to the depletion of differentiated spermatogonia, mice are subjected to controlled breeding schemes to propagate the induced mutations. Ultimately descendents of these mice are screened for a phenotype of interest and linkage analysis used to identify the underlying mutation. Mutations are often introduced onto one mouse strain and offspring are outcrossed to a different strain to enable mapping of the mutated chromosomal region. Outbreeding can occur either during the generation of mice for phenotypic screening (Figure 5) or once a phenotype of interest has been identified. Linkage analysis (the localization of the causal mutation to a relatively small region in the whole genome) can be done using a number of techniques. The majority, however, exploit small differences in genomic DNA sequence between individual strains of mice. Once the linkage region is defined, the mutation is identified through the sequencing of candidate genes. Candidate genes within the linkage interval are generally selected based on gene expression profile, predicted protein function and available mouse/human disease phenotypes. Most recently, mutation identification without the need for candidate gene selection has been achieved by whole genome sequencing technologies (O'Bryan MK, pers. commun.). For more details on an ENU mutagenesis approach used to discover male fertility regulators, readers are referred to Refs 23–28.
Figure 5.
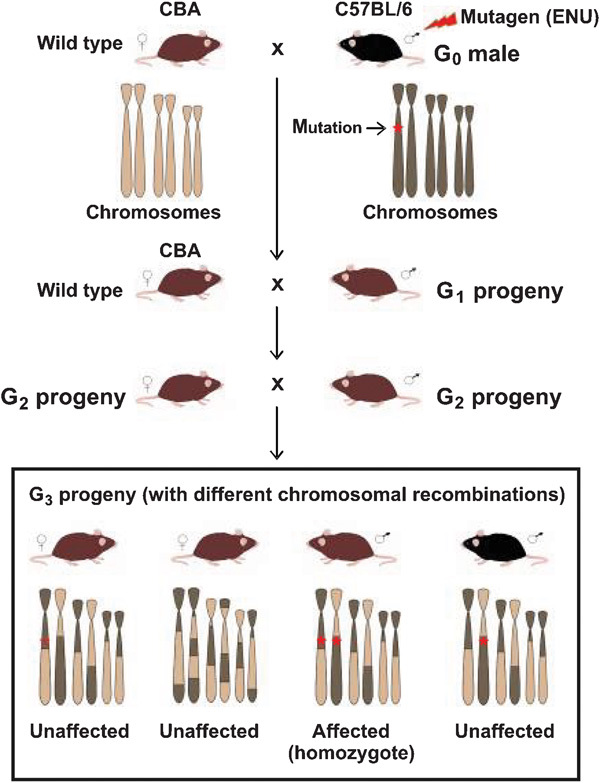
Example of a three-generation breeding scheme used to identify recessive mutations. To screen for recessive mutations, a three-generation breeding scheme is required. Founder male mouse (referred to as generation 0 (G0)) of an inbred strain (for example, C57BL/6) is injected with ENU. The ENU-treated male is subsequently mated with wild-type females of a different inbred strain (for example, CBA) to produce G1 offspring. G2 progeny can be produced from G1 littermate intercrosses or from G1×wild-type CBA crosses (as shown here). Finally, G3 progeny are generated from G2 littermate intercrosses and/or G2 females×G1 fathers. During each step of crossing, each progeny will have different combinations of chromosomes from the ENU-treated mouse strain and the strain used for subsequent outcrossing. These differences enable researchers to map the region containing the ENU-induced mutation causing phenotypic defects of interest (indicated by *). In this case, mutations are introduced into the C57BL/6 genome. CBA, cytometric bead array; ENU, N-ethyl-N-nitrosourea.
In contrast to a candidate gene approach (knockout/knockin and transgenesis), ENU mutagenesis provides a powerful tool for the discovery of previously unsuspected genes within biological processes. In addition to defining gene function, critical domains/regions of the mutated protein can be identified. The first large-scale ENU mutagenesis began in 1997.43, 44 Currently, there are at least 20 ENU consortiums around the world.45 Such programs have revealed many critical genes for human diseases, including male infertility.46, 47, 48, 49
Mouse models for male infertility
In recent years, the identification of infertility genes spanning all aspects of spermatogenesis, sperm maturation, capacitation and fertilization has been accelerated by mouse model studies.4, 50, 51, 52 At least 400 fertility essential genes have been modeled in mice. The majority of these genes were identified using knockout models and to a lesser extent, knockin, transgenic and ENU-mutagenized models. The current review is by no means a comprehensive list of these mouse models, rather it contains examples of how mouse models have advanced our knowledge of male fertility regulation. Herein, we focus on single-gene defects that affect the process of spermatogenesis and post-testicular maturation and fertilization. Defects in genes involved in hormonal regulation of spermatogenesis are not included in the review. Readers are referred to a comprehensive list of these genes in Refs 4 and 16.
Premeiotic defects
In mammals, although spermatogenesis begins in fetal life, germ cells undergo mitotic arrest prior to regaining their proliferative and differentiative capacity at puberty. The stem cells involved in the process of spermatogenesis are called spermatogonia. Spermatogonia differentiate and undergo numerous cycles of mitosis at the basement of seminiferous tubules. In mice, spermatogonia begin to proliferate (as well as self-renew) and differentiate at ∼4 days after birth and divide continuously through mitosis to give rise to spermatocytes. Premeiotic defects can lead to the complete disruption of spermatogenesis and result in a phenotype equivalent to that of the ‘Sertoli cell-only' syndrome seen in ∼15.7% of men with no sperm in their ejaculates.53 Several genes involving mouse spermatogonia self-renewal, apoptosis and cell cycle regulation have been shown to contribute to male infertility. Examples of such genes are listed in Table 1.
Table 1. Examples of genes essential for premeiotic germ cell development implicated by mouse model studies.
| Gene | Proposed function | Knockout phenotype | Fertility status | Reference |
|---|---|---|---|---|
| Etv5 (Erm) (Ets variant gene 5) | Transcription factor | Azoospermia; failed to maintain spermatogonia | Male infertility | 54 |
| Bax (BCL2-associated X protein) | Regulation of apoptosis | Premeiotic germ cell arrest | Male infertility | 55 |
| Pi3k (phosphoinositide-3-kinase) | Phosphatidylinositol 3′-kinase signaling pathway | Impaired spermatogonia proliferation and increased apoptosis of spermatogonia | Male infertility | 56 |
| Nanos2 (Nanos homolog 2 (Drosophila)) | Germ cell differentiation | Germ cell apoptosis and complete loss of spermatogonia | Male infertility | 57a, 58 |
| Ddx4 (Vasa) (DEAD (Asp–Glu–Ala–Asp) box polypeptide 4) | Germ cell proliferation and differentiation | Impaired premeiotic germ cells differentiation and increased apoptosis of spermatogonia | Male infertility | 59 |
| Dazl (Deleted in azoospermia-like) | Germ cell proliferation and differentiation | Azoospermia; few spermatogonia enter meiosis, and those that do fail to proceed beyond pachytene | Male/female infertility | 60 |
Transgenic mouse model.
Meiotic defects
Meiosis is a special cell division whereby diploid parental cells produce genetically diverse haploid sperm (or eggs). The reduction in chromosome number is achieved by one round of DNA replication followed by two successive rounds of chromosome segregation (meiosis I and II). Meiosis I involves the segregation/separation of homologous chromosomes from each other, whereas meiosis II involves the segregation of sister chromatids and therefore resembles mitosis.61 Male germ cells in the mouse testis enter meiosis in the second week of life. During meiosis, germ cells are termed spermatocytes. Spermatocytes pass through G1 and S phase and subsequently enter the meiotic prophase, during which time chromosome condensation and the formation of DNA double-strand breaks (DSBs) (leptotene spermatocytes), followed by the initiation of pairing (synapsis) between homologous chromosomes (zygotene spermatocytes) occur. The completion of synapsis of homologous chromosomes and the repair of DSBs, using homologous chromosomes as templates, occur from the midzygotene through the pachytene spermatocyte periods. The culmination of this process results in the reciprocal exchange of genetic information between homologues, and is completed through the midpachytene to diplotene periods.62 Genetic exchange takes place through the formation of DSBs followed by a crossover (synapsis) of genetic material between homologous chromosome pairs. This leads to the reassortment of maternal and paternal alleles and the production of genetically diverged haploid sperm. The formation and repair of meiotic DSBs is a pivotal process that drives genetic diversity.
Due to the complexity of the meiotic process, a large number of genes are proposed to be involved in its regulation. Defects in this process can lead to meiosis failure, the production of aneuploid gametes and infertility. Furthermore, gamete aneuploidy can result in embryonic death or developmental defects in the offspring.62 A list of some crucial meiotic genes identified by the use of mouse models is shown in Table 2. Many of these genes are involved in the initiation of programmed DSB formation, meiotic recombination, DSB repair and cell cycle regulation, and play a pivotal role in both male and female meiosis.
Table 2. Examples of genes essential for meiosis implicated by mouse model studies.
| Gene | Proposed function | Knockout phenotype | Fertility status | Reference |
|---|---|---|---|---|
| Spo11 (Sporulation protein, meiosis-specific, SPO11 homolog (S. cerevisiae)) | Programmed DSB formation | Impaired DSB formation and recombination initiation | Male/female infertility | 63 |
| Mei1 (Meiosis defective 1) | Programmed DSB formation | Impaired DSB formation and recombination initiation | Male/female infertility | 64a |
| Atm (Ataxia telangiectasia mutated homolog (human)) | DSB sensing and repairing | Impaired chromosomal synapsis and chromosome fragmentation | Male/female infertility | 65 |
| Dmc1 (Dosage suppressor of mck1 homolog, meiosis-specific homologous recombination (yeast)) | Meiotic recombination | Impaired chromosomal synapsis | Male/female infertility | 66 |
| H2afx (H2A histone family, member X) | Assembly of specific DSB repair complexes | Meiosis arrested at pachytene stage | Male infertility/female subfertility | 67 |
| Trip13 (Thyroid hormone receptor interactor 13) | Meiotic recombination and DSB repair | Impaired DSB repair | Male/female infertility | 68 |
| Mlh1 (MutL homolog 1 (E. coli)) | Mismatch repair | Impaired meiotic recombination | Male/female infertility | 69 |
| Mlh3 (MutL homolog 3 (E. coli)) | Mismatch repair | Impaired chromosomal synapsis | Male/female infertility | 70 |
| Pms2 (Postmeiotic segregation increased 2 (S. cerevisiae)) | Mismatch repair | Impaired chromosomal synapsis | Male infertility | 71 |
| Msh4 (MutS homolog 4 (E. coli)) | Mismatch repair | Impaired chromosomal synapsis | Male/female infertility | 72 |
| Msh5 (MutS homolog 5 (E. coli)) | Mismatch repair | Impaired chromosomal synapsis and synaptonemal complexes formation | Male/female infertility | 73 |
| Exo1 (Exonuclease 1) | Mismatch repair | Dynamic loss of chiasmata and apoptosis | Male/female infertility | 74 |
| Cdk2 (Cyclin-dependent kinase 2) | Cell cycle regulation | Meiosis arrest at prophase I and atrophy of the testes and ovaries | Male/female infertility | 75 |
| Ccna1 (Cyclin A1) | Cell cycle regulation | Meiosis prophase I arrest and increased germ cell apoptosis | Male infertility | 76 |
| Fkbp6 (FK506-binding protein 6) | Homologous chromosomes synapsis | Meiosis arrest at pachytene stage | Male infertility | 77 |
| Psmc3ip (Hop2, Tbpip) (Proteasome (prosome, macropain) 26S subunit, ATPase 3, interacting protein) | Homologous chromosomes synapsis | Meiosis arrest | Male/female infertility | 78 |
| Dnmt3l (DNA (cytosine-5-)-methyltransferase 3-like) | DNA methylation | Meiosis arrest and epigenetic defects | Male infertility; heterozygous progeny of homozygous females die by midgestation | 79, 80 |
| Siah1 (Seven in absentia 1A) | Protein ubiquitination and degradation | Meiosis prophase I arrest | Male infertility/female subfertility | 81 |
| Prdm9 (Meisetz) (PR domain containing 9) | Histone H3 methyltransferase that controls epigenetic events required for meiotic prophase | Impaired chromosomal synapsis and sex body formation | Male/female infertility | 82 |
| Rec8 (Rec8 homolog (yeast)) | Synaptonemal complex formation | Abnormal synaptonemal complexes | Male/female infertility | 83 |
| Sycp1 (Synaptonemal complex protein 1) | Synaptonemal complex assembly, meiotic recombination, and XY body formation | Impaired chromosomal synapsis | Male/female infertility | 84 |
| Sycp2 (Synaptonemal complex protein 2) | Synaptonemal complex assembly and chromosomal synapsis | Impaired axial element formation | Male infertility/female subfertility | 85 |
| Sycp3 (Synaptonemal complex protein 3) | Axial/lateral elements and synaptonemal complex formation and chromosomal synapsis | Impaired axial/lateral elements and synaptonemal complexes formation | Male infertility/female subfertility | 86 |
| Syce1 (Synaptonemal complex central element protein 1) | DSB repair | Impaired DSB repair | Male/female infertility | 87 |
| Syce2 (Synaptonemal complex central element protein 2) | Synaptonemal complex formation, homologous recombination and DSB repair | Impaired chromosomal synapsis and sex body formation | Male/female infertility | 88 |
| Smc1b (Structural maintenance of chromosomes 1B) | Segregation of chromosomes | Impaired sister chromatid cohesion formation and chromosomal synapsis | Male/female infertility | 89 |
| Eif4g3 (Eukaryotic translation initiation factor 4γ, 3) | Translational regulation | Meiotic arrest; spermatocytes failed to exit prophase via G2/MI transition | Male infertility | 49a |
Chemical-mutagenized point mutant mouse models.
Abbreviations: DSB, double-strand break; E. coli, Escherichia coli; S. cerevisiae, Saccharomyces cerevisiae.
Postmeiotic or spermiogenesis defects
Upon the completion of meiosis, haploid germ cells termed ‘round spermatids' are produced and subsequently undergo a series of differentiation processes collectively known as ‘spermiogenesis' to give rise to highly polarized spermatozoa. Spermiogenesis involves dramatic morphological alterations, which include chromatin reorganization and condensation, acrosome formation, cytoplasmic removal, sperm tail assembly and spermiation.
During sperm head formation, the characteristic shape of the sperm in each species is formed through the coordinated repackaging of the chromatin and the sculpting of the sperm head through processing involving the acroplaxome–manchette complex. Specifically, histones that package DNA in the majority of cell types are removed and replaced first with transition proteins (TPs) and then protamines (Prms) to form a highly condensed and transcriptionally inactive nucleus. As evidence in both TP (Tp1 and Tp2) and Prm (Prm1 and Prm2) knockout models, the attainment of the correct ratio of these proteins is essential for the formation of ‘normal' looking sperm and for fertility.90, 91, 92, 93 In agreement with mouse model studies, clinical association data suggest that attainment of the correct ratio of Prms is essential for human male fertility.94, 95
In parallel with the changes in nuclear packaging, mounting data suggest that the elongating spermatid nucleus is shaped through processes involving the acroplaxome–manchette.96 The acroplaxome is an F-actin-containing series of loop-like structures that form around the elongating spermatid and appear to be involved in the progressive extrusion of the head, as well as in the anchoring of the growing acrosome.97, 98 In addition, the acroplaxome is coupled to the manchette, which in turn is composed of a marginal ring around the nucleus and a fringe-like microtubule structure that extends into the distal cytoplasm. Several mouse models, and microscopic studies, strongly suggest that these structures are integral to the formation of normal sperm head shape.99, 100, 101 Data also suggest that the manchette is involved in the transport of proteins required for sperm tail formation in a process known as intramanchette transport.100, 101, 102
Mouse models have also been critically important in defining the mechanisms of sperm tail formation and have revealed some surprising phenomena and the etiology of several significant human pathologies.52 During the last few years in particular, research has begun to illustrate the importance of axoneme function, not only for sperm motility but also for cilia function in a range of somatic tissues and many aspects of human health. The axoneme is a 9+2 microtubule structure that runs as a core through the sperm tail (modified flagella).103 The same structure is conserved in all motile cilia (and flagella) from all kingdoms ranging from trypanosomes and Chlamydomonas, to mice and humans. While Afzelius et al.104 recognized several decades ago that abnormally formed axonemes lead to both sterility and disorders in other ciliated tissues, including the lung, brain and kidney in a syndrome known as primary ciliary dyskinesia, it is only in the last decade that some of the underlying genes have been revealed.105, 106, 107 In addition, and largely through the use of mouse models, it is now recognized the abnormal cilia (sperm tail) function/formation at least in mice can be induced by a range of other genes involved in sperm tail/cilia development or in transducing environmental signals to the axoneme. Such genes and models include Tektin 2 (Tekt2), which is involved in dynein arm formation in the axoneme;108 Alms1,109, 110 and several other genes involved in basal body function and in initiating the growth of the axoneme; the Catsper genes, which are involved in Ca2+ regulation and onset of hyperactivated motility;111, 112, 113, 114 Slo3, which is involved in K+ flow and in regulating capacitation and hyperpolarization;115 and Gapds and Pgk2, which are involved in the establishment of glycolytic pathways on the sperm tail accessory structures and the generation of ATP for axoneme function.116, 117 For a comprehensive review of mouse models with abonormal sperm tail function, readers are referred to Escalier.52
Spermiation is a hormonally regulated process by which spermatozoa are released into the lumen of the seminiferous tubules. This involves the detachment of elongated spermatids from the Sertoli cells and the removal of most of the cytoplasm within the nascent spermatozoa as residual bodies.118, 119 Several knockout models of genes involving in chromatin packing, nuclear condensation, cytoplasmic exclusion, and signal transduction and protein transport have been shown to contribute male infertility (Table 3).
Table 3. Examples of genes essential for spermiogenesis, epididymal maturation, capacitation and fertilization implicated by mouse model studies.
| Gene | Proposed function | Knockout phenotype | Fertility status | Reference |
|---|---|---|---|---|
| Spem1 (Sperm maturation 1) | Spermatid cytoplasmic removal | Deformed sperm head and tail structure and impaired spermiogenesis | Male infertility | 144 |
| Gopc (Golgi-associated PDZ and coiled-coil motif containing) | Acrosome biogenesis | Fragmented acrosomes; globozoospermia (round-headed spermatozoa) | Male infertility | 145 |
| Prm1 and Prm2 (Protamine 1 and 2) | Sperm chromatin condensation | Haploinsufficiency effect; sperm derived from spermatogenic cells with one copy of the mutated allele are immotile and unable to fertilize eggs | Chimera male/female infertility | 90 |
| Tnp1 (Transition protein 1) | Sperm chromatin condensation | Subtle sperm morphology abnormalities and reduced motility | Male subfertility | 91 |
| Tnp2 (Transition protein 2) | Sperm chromatin condensation | Impaired chromatin condensation, sperm head abnormalities and reduced sperm motility | Male subfertility | 92, 93 |
| Tssk6 (Sstk) (Testis-specific serine kinase 6) | Chromatin remodeling | Abnormal sperm head morphology, reduced DNA compaction in spermatozoa and impaired sperm motility | Male infertility | 146 |
| Cadm1 (Tslc1) (Cell adhesion molecule 1) | Cell adhesion, differentiation and apoptosis | Spermatid maturation arrest, degenerated and sloughed off into the lumen | Male infertility | 147, 148 |
| Pvrl2 (Nectin 2) (Poliovirus receptor-related 2) | Cell adhesion | Abnormal sperm head and sperm tail midpiece structure, impaired zona binding, and lack of oocyte penetration | Male infertility | 149 |
| Csnk2a2 (Casein kinase 2, alpha prime polypeptide) | Sperm head morphogenesis | Oligospermia (low sperm counts) and globozoospermia | Male infertility | 150 |
| Rara (Retinoic acid receptor, alpha) | Ligand-induced transcription factor | Sloughing of immature germ cells in the lumen of seminiferous tubules | Male infertility | 151 |
| Pafah1b1 (Lis1) (Platelet-activating factor acetylhydrolase, isoform 1b) | Acrosome and tail biogenesis | Impaired acrosome and tail formation; the basal tail cuff at the point of insertion but fail to add axonemes to elongate the tail structure | Male infertility | 100a |
| Meig1 (Meiosis-expressed gene 1) | Sperm tail biogenesis, assembly of the axonemes/flagella | Absent sperm flagella, abnormal sperm head, disrupted microtubular organelle essential for sperm head and flagella formation | Male infertility | 101 |
| Tekt2 (Tektin-t) (Tektin 2) | Sperm tail biogenesis, dynein arm formation in the axoneme | Bending of the sperm flagella and marked defects in motility and primary ciliary dyskinesia | Male infertility | 108a |
| Adcy10 (sAC) (Adenylate cyclase 10) | Sperm motility | Normal sperm counts and morphology with severely impaired motility | Male infertility | 152 |
| Gapds (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic) | Generation of energy supply for sperm motility | Normal sperm counts and morphology with impaired motility (loss of forward progression motility) | Male infertility | 117 |
| Pgk2 (Phosphoglycerate kinase 2) | Generation of energy supply for sperm motility | Impaired progressive motility | Male subfertility | 116 |
| Agfg1 (Hrb) (ArfGAP with FG repeats 1) | Acrosome biogenesis | Globozoospermia | Male infertility | 153 |
| Pick1 (Protein interacting with C kinase 1) | Vesicle trafficking from the Golgi apparatus to the acrosome | Fragmented acrosomes Globozoospermia | Male infertility | 154 |
| Vdac3 (Voltage-dependent anion channel 3) | Ion transport for sperm tail formation | Normal sperm counts, abnormal sperm tail structure and markedly reduced motility | Male infertility | 155 |
| Catsper1, Catsper2, Catsper3, Catsper4 (Cation channel, sperm associated 1–4) | Sperm motility and fertilization (ion channels) | Normal sperm counts, normal morphology, markedly reduced motility and unable to fertilize intact eggs | Male infertility | 111, 112, 113, 114 |
| Capza3 (Capping protein (actin filament) muscle Z-line, alpha 3) | Spermatid cytoplasmic extrusion | Excess cytoplasm and impaired motility | Male infertility | 46b |
| Dnahc1 (Dynein, axonemal, heavy chain 1) | Cilia and flagella function; axoneme movement | Normal sperm counts, normal morphology, markedly reduced forward motility | Male infertility and primary ciliary dyskinesia | 156 |
| Izumo1 (Izumo sperm–egg fusion 1) | Membrane fusion | Defect in sperm–egg fusion | Male infertility | 157 |
| Akap4 (A kinase (PRKA) anchor protein 4) | Sperm tail assembly (scaffold protein) | Normal sperm counts, normal morphology, markedly reduced motility | Male infertility | 158 |
| Pcsk4 (Proprotein convertase subtilisin/kexin type 4) | Acrosome reaction and sperm–egg interaction | Impaired fertilization | Male subfertility | 159 |
| Kcnu1 (Slo3) (Potassium channel, subfamily U, member 1) | Capacitation and acrosome reaction (ion transportation) | Impaired motility, abnormal sperm morphology (hairpin shape), impaired sperm capacitation and acrosome reaction | Male infertility | 115 |
| Adam3 (Cyritestin) | Cell adhesions (plasma membrane protein) | Defect in sperm to bind to the zona pellucida | Male infertility | 160 |
| Clgn (Calmegin) | Folding and transport of integral membrane proteins (testis-specific chaperone) | Impaired sperm–zona pellucida adhesion | Male infertility | 161 |
| Adam2 (Fertilin-β) | Cell adhesions (plasma membrane protein) | Impaired sperm–egg membrane adhesion, sperm–egg fusion, migration from the uterus into the oviduct, and binding to the egg zona pellucida | Male subfertility | 162 |
Gene-trapped mouse models.
ENU mutagenized mouse model.
Post-testicular maturation and fertilization defects
Sperm appear morphologically mature following spermiation; however, they do not gain the capacity for fertilization until they have undergone transit through the epididymis in a poorly understood process known as ‘epididymal maturation'. Epididymal maturation is dependent upon factors produced by the epididymis and is characterized by extensive post-translational protein modifications, changes in membrane composition and the progressive attainment of functional competence.120, 121, 122, 123 For a more extensive review of these important processes, readers are referred to Refs 124–126.
Subsequent to epididymal maturation, sperm must undergo the process of capacitation in the female reproductive tract (or in media) to be fully capable of fertilization. Capacitation refers to a series of signal transduction and protein modification processes that occur during a period of time following the ejaculated sperm into the female reproductive tract. Capacitation confers upon sperm the ability to bind the zona pellucida of the oocyte and undergo the acrosome reaction. Sperm capacitation correlates with a massive increase in the tyrosine phosphorylation of many sperm tail and head proteins,127, 128, 129 an increase in membrane fluidity, cholesterol efflux and an increase in intracellular Ca2+ and cyclic AMP concentration.130, 131, 132 Capacitation is also correlated with the ability of sperm to manifest hyperactivated motility and for them to bind to the outer vestments of the oocyte during acrosome reaction.133, 134, 135 Hyperactivated motility, which is characterized by high-amplitude asymmetrical bending of the sperm tail, is proposed to be important for the sperm to detach itself from the mucosal membrane of the oviduct, where they are temporarily stored, and also to penetrate the zona pellucida of the oocyte during the acrosome reaction.135 Ion channels including the CatSper channels play a central role in sperm hyperactivation via the regulation of Ca2+ flow.111, 112, 113, 114
The acrosome reaction is also characterized by a Ca2+ influx that triggers fusion of a number of sites of the initial sperm–zona pellucida interaction.136, 137 This interaction involves complementary, but as yet incompletely defined, sites on the sperm outer acrosomal membrane and ZP3 on the egg plasma membrane.138, 139, 140 The fusion allows development of vesicles, containing hydrolytic enzymes, which are released in a process called acrosomal exocytosis to break down the egg cumulus mass. This exposes the inner acrosomal membrane so that it can bind the zona pellucida and an appropriately reacted sperm enters the perivitelle space. The sperm and the plasma membrane of the egg bind and fuse.141, 142, 143
Several genes have been implicated to play a role in post-testicular maturation, capacitation and fertilization using mouse model studies (Table 3). In contrast to premeiotic and meiotic defects, the majority of genes critical for postmeiotic and post-testicular defects exhibit male-specific infertility defect.
Factors to be considered when evaluating mouse models
Although the use of animal models is ideal for in vivo gene function analysis, phenotypic defects observed in genetically modified animal models can influenced by different genetic backgrounds and environmental factors. It is important that these factors are appropriately addressed to prevent misinterpretation of results.
In the case of knockout/knockin mice, the ES cells and the recipient blastocyst are often obtained from animals carrying genes of different coat colors to facilitate the initial selection of chimeric progeny. The most commonly used ES cells are those derived from the mouse strain 129, which has an agouti coat color. Once targeted, these ES cells are microinjected into blastocysts obtained from a mouse strain with different coat colors (for example, C57BL/6 mice, which have a black coat color). Offspring with a high degree of agouti coat color, as an indication of the transmission of 129 ES cell-derived gene, are mated to produce heterozygous knockout mice. These mice are intercrossed to generate homozygous null progeny, which give rise to a cohort of mice on a mixed genetic background (for example, 129 and C57BL/6). Similarly, mice generated through genome-wide ENU mutagenesis may be on a mixed genetic background as a consequence of the founder mouse strain used for ENU treatment and a mouse strain used for outcrossing for linkage analysis and mutation identification being different. In many cases, the initial phenotypic characterization of the knockout/knockin and point mutant mutagenized mice is performed on a mixed genetic background, which can result in a range of phenotypic variations. Genetic modifiers (for example, single-nucleotide polymorphisms and flanking gene effect) are proposed to be a critical factor that contributes to the phenotypic diversity.163
To minimize the effect of genetic background variations, knockout mice generated by 129 ES, for example, cells can be backcrossed to produce a congenic line, that is, a strain that differs from another in the region of the gene of interest and a short-linked chromosomal segment around the gene. The production of a congenic line involves breeding of the knockout mice with the strain of mice used for the initial chimera breeding (or a new inbred strain of choice) for at least 10 generations. This conventional backcrossing approach can take up to 3 years to generate 99% of the recipient genome at the tenth backcrossing (N10). Recently, a new high-speed congenic approach using round spermatids retrieved from immature males (22–25 days of age) and polymorphic marker analysis was reported.164 This approach has been successfully used to produce congenic line within a 6-month period. An alternative approach to avoid congenic line production is to use C57BL/6 ES cells for the initial gene targeting. Although homologous recombination in the C57BL/6 ES cells is lower than that of the 129 strain, it has been shown that a significant number of targeted ES clones are produced using C57BL/6 ES cells.165
In addition to genetic background, several environmental factors can have a profound influence on the observed phenotype in inbred and congenic strains. These factors include viral and bacterial infections, exposure to loud sounds, and dietary and maternal effects.166
International mouse phenotyping consortiums
Researchers should be aware of the existence of resources that provide access to data and procedures for mouse phenotyping. These include the European Mouse Phenotyping Resource (EuroPhenome: http://www.europhenome.org), the European Mouse Phenotyping Resource of Standardised Screens (EMPReSS: http:empress.har.mrc.ac.uk) and the European Mouse Disease Clinic (EUMODIC: http://www.eumodic.org).167, 168 Within our lab, we broadly categorize the type of male infertility using the strategy outlined in Borg et al.169
EMPReSS provides comprehensive standard operating procedures (SOPs) covering the main physiological systems for mouse phenotypic screens. To date, ∼150 SOPs have been developed. Additionally, SOPs for histology, pathology and gene expression analyses are available. EuroPhenome provides phenotyping data from SOPs contained in the EMPReSS database together with the development of novel mouse phenotyping approaches. EUMODIC aims to perform primary phenotyping of 650 mouse mutant lines derived using the European Conditional Mouse Mutagenesis Program.39 Mouse lines with phenotypic defects of interest will undergo specialized phenotypic assessment. Data from the EUMODIC project are being integrated within the EuroPhenome web interface. These publically available resources will enable researchers to precisely assess the phenotypic defects of mouse models.
Conclusion
The availability of complete genome sequences of human, mouse and a number of organisms has opened up a new era of gene functional analysis. Using mouse models, reproductive biologists have successfully revealed hundreds of genes essential for fertility and many are in the process of being identified and characterized. The next challenge is the translation of the knowledge obtained from studies on the mouse into the human infertility clinic to serve the ultimate goals of: (i) improving the existing infertility treatments; (ii) providing alternative choices of treatment; (iii) developing precise diagnostics; and (iv) formulating contraceptives for males with minimal side effects.
Acknowledgments
The authors thank Ms Anne O'Connor (Department of Anatomy and Developmental Biology, Monash University) for assistance in the review of the manuscript. Grant funding: DJ is a National Health and Medical Research Council (NHMRC) of Australia Peter Doherty Postdoctoral Fellow and MKO'B is an NHMRC Senior Research Fellow.
The authors declare no conflict of interest.
References
- Leifke E, Nieschlag E. Male infertility treatment in the light of evidence-based medicine. Andrologia. 1996;28 Suppl 1:23–30. [PubMed] [Google Scholar]
- de Kretser DM. Male infertility. Lancet. 1997;349:787–90. doi: 10.1016/s0140-6736(96)08341-9. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Baker HW. Infertility in men: recent advances and continuing controversies. J Clin Endocrinol Metab. 1999;84:3443–50. doi: 10.1210/jcem.84.10.6101. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14:1197–213. doi: 10.1038/nm.f.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Flynn O'Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93:1–12. doi: 10.1016/j.fertnstert.2009.10.045. [DOI] [PubMed] [Google Scholar]
- Reijo R, Lee TY, Salo P, Alagappan R, Brown LG, et al. Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet. 1995;10:383–93. doi: 10.1038/ng0895-383. [DOI] [PubMed] [Google Scholar]
- Morris RS, Gleicher N. Genetic abnormalities, male infertility, and ICSI. Lancet. 1996;347:1277. doi: 10.1016/s0140-6736(96)90934-4. [DOI] [PubMed] [Google Scholar]
- Thielemans BF, Spiessens C, D'Hooghe T, Vanderschueren D, Legius E. Genetic abnormalities and male infertility. A comprehensive review. Eur J Obstet Gynecol Reprod Biol. 1998;81:217–25. doi: 10.1016/s0301-2115(98)00194-8. [DOI] [PubMed] [Google Scholar]
- Vicdan A, Vicdan K, Gunalp S, Kence A, Akarsu C, et al. Genetic aspects of human male infertility: the frequency of chromosomal abnormalities and Y chromosome microdeletions in severe male factor infertility. Eur J Obstet Gynecol Reprod Biol. 2004;117:49–54. doi: 10.1016/j.ejogrb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Maduro MR, Lamb DJ. Understanding new genetics of male infertility. J Urol. 2002;168:2197–205. doi: 10.1016/S0022-5347(05)64355-8. [DOI] [PubMed] [Google Scholar]
- McLachlan RI, O'Bryan MK. Clinical review#: state of the art for genetic testing of infertile men. J Clin Endocrinol Metab. 2010;95:1013–24. doi: 10.1210/jc.2009-1925. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Loveland KL, Meinhardt A, Simorangkir D, Wreford N. Spermatogenesis. Hum Reprod. 1998;13 Suppl 1:1–8. doi: 10.1093/humrep/13.suppl_1.1. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Loveland KL, Meehan T, O'Bryan MK, Phillips DJ, et al. Inhibins, activins and follistatin: actions on the testis. Mol Cell Endocrinol. 2001;180:87–92. doi: 10.1016/s0303-7207(01)00502-0. [DOI] [PubMed] [Google Scholar]
- O'Donnell L, Meachem SJ, Stanton PG, McLachlan RI. San Diego, CA: Elsevier; 2005. The Endocrine Regulation of Spermatogenesis; pp. 1177–290. [Google Scholar]
- McLachlan RI, O'Donnell L, Meachem SJ, Stanton PG, de K, et al. Hormonal regulation of spermatogenesis in primates and man: insights for development of the male hormonal contraceptive. J Androl. 2002;23:149–62. [PubMed] [Google Scholar]
- Cooke HJ, Saunders PT. Mouse models of male infertility. Nat Rev Genet. 2002;3:790–801. doi: 10.1038/nrg911. [DOI] [PubMed] [Google Scholar]
- Skinner M, Griswold M. San Diego, CA: Elsevier Academic Press; 2005. Sertoli Cell Biology; p. 494. [Google Scholar]
- Andrews J, Bouffard GG, Cheadle C, Lu J, Becker KG, et al. Gene discovery using computational and microarray analysis of transcription in the Drosophila melanogaster testis. Genome Res. 2000;10:2030–43. doi: 10.1101/gr.10.12.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, van Doren C, et al. A global profile of germline gene expression in C. elegans. Mol Cell. 2000;6:605–16. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Wu SM, Baxendale V, Chen Y, Pang AL, Stitely T, et al. Analysis of mouse germ-cell transcriptome at different stages of spermatogenesis by SAGE: biological significance. Genomics. 2004;84:971–81. doi: 10.1016/j.ygeno.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Page ST, Amory JK, Bremner WJ. Advances in male contraception. Endocr Rev. 2008;29:465–93. doi: 10.1210/er.2007-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church DM, Goodstadt L, Hillier LW, Zody MC, Goldstein S, et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009;7:e1000112. doi: 10.1371/journal.pbio.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel MA, Lessard C, Reinholdt L, Schimenti J, Eppig JJ. Mutagenesis as an unbiased approach to identify novel contraceptive targets. Mol Cell Endocrinol. 2006;250:201–5. doi: 10.1016/j.mce.2005.12.046. [DOI] [PubMed] [Google Scholar]
- Kennedy CL, O'Bryan MK. N-ethyl-N-nitrosourea (ENU) mutagenesis and male fertility research. Hum Reprod Update. 2006;12:293–301. doi: 10.1093/humupd/dmk004. [DOI] [PubMed] [Google Scholar]
- Kennedy CL, O'Connor AE, Sanchez-Partida LG, Holland MK, Goodnow CC, et al. A repository of ENU mutant mouse lines and their potential for male fertility research. Mol Hum Reprod. 2005;11:871–80. doi: 10.1093/molehr/gah251. [DOI] [PubMed] [Google Scholar]
- Lessard C, Lothrop H, Schimenti JC, Handel MA. Mutagenesis-generated mouse models of human infertility with abnormal sperm. Hum Reprod. 2007;22:159–66. doi: 10.1093/humrep/del322. [DOI] [PubMed] [Google Scholar]
- Lessard C, Pendola JK, Hartford SA, Schimenti JC, Handel MA, et al. New mouse genetic models for human contraceptive development. Cytogenet Genome Res. 2004;105:222–7. doi: 10.1159/000078192. [DOI] [PubMed] [Google Scholar]
- Jamsai D, O'Bryan MK. Genome-wide ENU mutagenesis for the discovery of novel male fertility regulators. Syst Biol Reprod Med. 2010;56:246–59. doi: 10.3109/19396361003706424. [DOI] [PubMed] [Google Scholar]
- Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature. 1985;317:230–4. doi: 10.1038/317230a0. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–12. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–92. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. The new mouse genetics: altering the genome by gene targeting. Trends Genet. 1989;5:70–6. doi: 10.1016/0168-9525(89)90029-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–40. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Brennan J, Skarnes WC. Gene trapping in mouse embryonic stem cells. Methods Mol Biol. 2008;461:133–48. doi: 10.1007/978-1-60327-483-8_9. [DOI] [PubMed] [Google Scholar]
- Nord AS, Chang PJ, Conklin BR, Cox AV, Harper CA, et al. The International Gene Trap Consortium Website: a portal to all publicly available gene trap cell lines in mouse. Nucleic Acids Res. 2006;34:D642–8. doi: 10.1093/nar/gkj097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet. 2001;2:756–68. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–81. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondo Y. Trends in large-scale mouse mutagenesis: from genetics to functional genomics. Nat Rev Genet. 2008;9:803–10. doi: 10.1038/nrg2431. [DOI] [PubMed] [Google Scholar]
- Friedel RH, Seisenberger C, Kaloff C, Wurst W. EUCOMM—the European Conditional Mouse Mutagenesis Program. Brief Funct Genomic Proteomic. 2007;6:180–5. doi: 10.1093/bfgp/elm022. [DOI] [PubMed] [Google Scholar]
- Acevedo-Arozena A, Wells S, Potter P, Kelly M, Cox RD, et al. ENU mutagenesis, a way forward to understand gene function. Annu Rev Genomics Hum Genet. 2008;9:49–69. doi: 10.1146/annurev.genom.9.081307.164224. [DOI] [PubMed] [Google Scholar]
- Georgel P, Du X, Hoebe K, Beutler B. ENU mutagenesis in mice. Methods Mol Biol. 2008;415:1–16. doi: 10.1007/978-1-59745-570-1_1. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8:1955–63. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis M, Balling R. Large scale ENU screens in the mouse: genetics meets genomics. Mutat Res. 1998;400:25–32. doi: 10.1016/s0027-5107(98)00061-x. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–7. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- Gondo Y, Fukumura R, Murata T, Makino S. Next-generation gene targeting in the mouse for functional genomics. BMB Rep. 2009;42:315–23. doi: 10.5483/bmbrep.2009.42.6.315. [DOI] [PubMed] [Google Scholar]
- Geyer CB, Inselman AL, Sunman JA, Bornstein S, Handel MA, et al. A missense mutation in the Capza3 gene and disruption of F-actin organization in spermatids of repro32 infertile male mice. Dev Biol. 2009;330:142–52. doi: 10.1016/j.ydbio.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T, Marquez B, Suarez S, Schimenti J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol Reprod. 2007;77:376–82. doi: 10.1095/biolreprod.106.058669. [DOI] [PubMed] [Google Scholar]
- Schneider M, Forster H, Boersma A, Seiler A, Wehnes H, et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. Faseb J. 2009;23:3233–42. doi: 10.1096/fj.09-132795. [DOI] [PubMed] [Google Scholar]
- Sun F, Palmer K, Handel MA. Mutation of Eif4g3, encoding a eukaryotic translation initiation factor, causes male infertility and meiotic arrest of mouse spermatocytes. Development. 2010;137:1699–707. doi: 10.1242/dev.043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility pathways. Nat Cell Biol. 2002;4 Suppl:s41–9. doi: 10.1038/ncb-nm-fertilityS41. [DOI] [PubMed] [Google Scholar]
- O'Bryan MK, de Kretser D. Mouse models for genes involved in impaired spermatogenesis. Int J Androl. 2006;29:76–89; discussion 105–8. doi: 10.1111/j.1365-2605.2005.00614.x. [DOI] [PubMed] [Google Scholar]
- Escalier D. Knockout mouse models of sperm flagellum anomalies. Hum Reprod Update. 2006;12:449–61. doi: 10.1093/humupd/dml013. [DOI] [PubMed] [Google Scholar]
- McLachlan RI, Rajpert-De Meyts E, Hoei-Hansen CE, de Kretser DM, Skakkebaek NE. Histological evaluation of the human testis—approaches to optimizing the clinical value of the assessment: mini review. Hum Reprod. 2007;22:2–16. doi: 10.1093/humrep/del279. [DOI] [PubMed] [Google Scholar]
- Chen C, Ouyang W, Grigura V, Zhou Q, Carnes K, et al. ERM is required for transcriptional control of the spermatogonial stem cell niche. Nature. 2005;436:1030–4. doi: 10.1038/nature03894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–9. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Jiang G, Hyman R, Lee KF, O'Gorman S, et al. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3′-kinase is essential for male fertility. Nat Genet. 2000;24:157–62. doi: 10.1038/72814. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, et al. Conserved role of nanos proteins in germ cell development. Science. 2003;301:1239–41. doi: 10.1126/science.1085222. [DOI] [PubMed] [Google Scholar]
- Sada A, Suzuki A, Suzuki H, Saga Y. The RNA-binding protein NANOS2 is required to maintain murine spermatogonial stem cells. Science. 2009;325:1394–8. doi: 10.1126/science.1172645. [DOI] [PubMed] [Google Scholar]
- Tanaka SS, Toyooka Y, Akasu R, Katoh-Fukui Y, Nakahara Y, et al. The mouse homolog of Drosophila Vasa is required for the development of male germ cells. Genes Dev. 2000;14:841–53. [PMC free article] [PubMed] [Google Scholar]
- Ruggiu M, Speed R, Taggart M, McKay SJ, Kilanowski F, et al. The mouse Dazla gene encodes a cytoplasmic protein essential for gametogenesis. Nature. 1997;389:73–7. doi: 10.1038/37987. [DOI] [PubMed] [Google Scholar]
- Roeder GS. Meiotic chromosomes: it takes two to tango. Genes Dev. 1997;11:2600–21. doi: 10.1101/gad.11.20.2600. [DOI] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet. 2010;11:124–36. doi: 10.1038/nrg2723. [DOI] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell. 2000;6:989–98. doi: 10.1016/s1097-2765(00)00098-8. [DOI] [PubMed] [Google Scholar]
- Libby BJ, Reinholdt LG, Schimenti JC. Positional cloning and characterization of Mei1, a vertebrate-specific gene required for normal meiotic chromosome synapsis in mice. Proc Natl Acad Sci USA. 2003;100:15706–11. doi: 10.1073/pnas.2432067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, et al. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–22. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Pittman DL, Cobb J, Schimenti KJ, Wilson LA, Cooper DM, et al. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell. 1998;1:697–705. doi: 10.1016/s1097-2765(00)80069-6. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XC, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. 2007;3:e130. doi: 10.1371/journal.pgen.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–42. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Lipkin SM, Moens PB, Wang V, Lenzi M, Shanmugarajah D, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31:385–90. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–19. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Kneitz B, Cohen PE, Avdievich E, Zhu L, Kane MF, et al. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000;14:1085–97. [PMC free article] [PubMed] [Google Scholar]
- de Vries SS, Baart EB, Dekker M, Siezen A, de Rooij DG, et al. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 1999;13:523–31. doi: 10.1101/gad.13.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, et al. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–14. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- Liu D, Matzuk MM, Sung WK, Guo Q, Wang P, et al. Cyclin A1 is required for meiosis in the male mouse. Nat Genet. 1998;20:377–80. doi: 10.1038/3855. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Kolas NK, Noguchi J, Sarao R, Kikuchi K, et al. Essential role of Fkbp6 in male fertility and homologous chromosome pairing in meiosis. Science. 2003;300:1291–5. doi: 10.1126/science.1083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova GV, Romanienko PJ, Camerini-Otero RD. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell. 2003;5:927–36. doi: 10.1016/s1534-5807(03)00369-1. [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- Webster KE, O'Bryan MK, Fletcher S, Crewther PE, Aapola U, et al. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc Natl Acad Sci USA. 2005;102:4068–73. doi: 10.1073/pnas.0500702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Frew IJ, House CM, O'Bryan MK, Holloway AJ, et al. The ubiquitin ligase component Siah1a is required for completion of meiosis I in male mice. Mol Cell Biol. 2002;22:2294–303. doi: 10.1128/MCB.22.7.2294-2303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–8. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- Xu H, Beasley MD, Warren WD, van der Horst GT, McKay MJ. Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell. 2005;8:949–61. doi: 10.1016/j.devcel.2005.03.018. [DOI] [PubMed] [Google Scholar]
- de Vries FA, de Boer E, van den Bosch M, Baarends WM, Ooms M, et al. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005;19:1376–89. doi: 10.1101/gad.329705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, de la Fuente R, Leu NA, Baumann C, McLaughlin KJ, et al. Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J Cell Biol. 2006;173:497–507. doi: 10.1083/jcb.200603063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Liu JG, Zhao J, Brundell E, Daneholt B, et al. The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol Cell. 2000;5:73–83. doi: 10.1016/s1097-2765(00)80404-9. [DOI] [PubMed] [Google Scholar]
- Bolcun-Filas E, Hall E, Speed R, Taggart M, Grey C, et al. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009;5:e1000393. doi: 10.1371/journal.pgen.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcun-Filas E, Costa Y, Speed R, Taggart M, Benavente R, et al. SYCE2 is required for synaptonemal complex assembly, double strand break repair, and homologous recombination. J Cell Biol. 2007;176:741–7. doi: 10.1083/jcb.200610027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenkova E, Eijpe M, Heyting C, Hodges CA, Hunt PA, et al. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol. 2004;6:555–62. doi: 10.1038/ncb1135. [DOI] [PubMed] [Google Scholar]
- Cho C, Willis WD, Goulding EH, Jung-Ha H, Choi YC, et al. Haploinsufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet. 2001;28:82–6. doi: 10.1038/ng0501-82. [DOI] [PubMed] [Google Scholar]
- Yu YE, Zhang Y, Unni E, Shirley CR, Deng JM, et al. Abnormal spermatogenesis and reduced fertility in transition nuclear protein 1-deficient mice. Proc Natl Acad Sci USA. 2000;97:4683–8. doi: 10.1073/pnas.97.9.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Shirley CR, Yu YE, Mohapatra B, Zhang Y, et al. Targeted disruption of the transition protein 2 gene affects sperm chromatin structure and reduces fertility in mice. Mol Cell Biol. 2001;21:7243–55. doi: 10.1128/MCB.21.21.7243-7255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adham IM, Nayernia K, Burkhardt-Gottges E, Topaloglu O, Dixkens C, et al. Teratozoospermia in mice lacking the transition protein 2 (Tnp2) Mol Hum Reprod. 2001;7:513–20. doi: 10.1093/molehr/7.6.513. [DOI] [PubMed] [Google Scholar]
- Carrell DT, Emery BR, Hammoud S. Altered protamine expression and diminished spermatogenesis: what is the link. Hum Reprod Update. 2007;13:313–27. doi: 10.1093/humupd/dml057. [DOI] [PubMed] [Google Scholar]
- Oliva R. Protamines and male infertility. Hum Reprod Update. 2006;12:417–35. doi: 10.1093/humupd/dml009. [DOI] [PubMed] [Google Scholar]
- Kierszenbaum AL, Tres LL. The acrosome–acroplaxome–manchette complex and the shaping of the spermatid head. Arch Histol Cytol. 2004;67:271–84. doi: 10.1679/aohc.67.271. [DOI] [PubMed] [Google Scholar]
- Kierszenbaum AL, Rivkin E, Tres LL. Acroplaxome, an F-actin-keratin-containing plate, anchors the acrosome to the nucleus during shaping of the spermatid head. Mol Biol Cell. 2003;14:4628–40. doi: 10.1091/mbc.E03-04-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liska F, Gosele C, Rivkin E, Tres L, Cardoso MC, et al. Rat hd mutation reveals an essential role of centrobin in spermatid head shaping and assembly of the head-tail coupling apparatus. Biol Reprod. 2009;81:1196–205. doi: 10.1095/biolreprod.109.078980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC. Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol. 2005;17:47–54. doi: 10.1016/j.ceb.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Nayernia K, Vauti F, Meinhardt A, Cadenas C, Schweyer S, et al. Inactivation of a testis-specific Lis1 transcript in mice prevents spermatid differentiation and causes male infertility. J Biol Chem. 2003;278:48377–85. doi: 10.1074/jbc.M309583200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Shen X, Gude DR, Wilkinson BM, Justice MJ, et al. MEIG1 is essential for spermiogenesis in mice. Proc Natl Acad Sci USA. 2009;106:17055–60. doi: 10.1073/pnas.0906414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierszenbaum AL. Intramanchette transport (IMT): managing the making of the spermatid head, centrosome, and tail. Mol Reprod Dev. 2002;63:1–4. doi: 10.1002/mrd.10179. [DOI] [PubMed] [Google Scholar]
- Mitchell DR. Speculations on the evolution of 9+2 organelles and the role of central pair microtubules. Biol Cell. 2004;96:691–6. doi: 10.1016/j.biolcel.2004.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzelius BA, Eliasson R, Johnsen O, Lindholmer C. Lack of dynein arms in immotile human spermatozoa. J Cell Biol. 1975;66:225–32. doi: 10.1083/jcb.66.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120–6. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loges NT, Olbrich H, Becker-Heck A, Haffner K, Heer A, et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am J Hum Genet. 2009;85:883–9. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noone PG, et al. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am J Respir Crit Care Med. 2006;174:858–66. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Iguchi N, Toyama Y, Kitamura K, Takahashi T, et al. Mice deficient in the axonemal protein Tektin-t exhibit male infertility and immotile-cilium syndrome due to impaired inner arm dynein function. Mol Cell Biol. 2004;24:7958–64. doi: 10.1128/MCB.24.18.7958-7964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsov T, Silva DG, O'Bryan MK, Sainsbury A, Lee NJ, et al. Fat aussie—a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20:1610–22. doi: 10.1210/me.2005-0494. [DOI] [PubMed] [Google Scholar]
- Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323–33. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Navarro B, Perez G, Jackson AC, Hsu S, et al. A sperm ion channel required for sperm motility and male fertility. Nature. 2001;413:603–9. doi: 10.1038/35098027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quill TA, Sugden SA, Rossi KL, Doolittle LK, Hammer RE, et al. Hyperactivated sperm motility driven by CatSper2 is required for fertilization. Proc Natl Acad Sci USA. 2003;100:14869–74. doi: 10.1073/pnas.2136654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Moran MM, Navarro B, Chong JA, Krapivinsky G, et al. All four CatSper ion channel proteins are required for male fertility and sperm cell hyperactivated motility. Proc Natl Acad Sci USA. 2007;104:1219–23. doi: 10.1073/pnas.0610286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Jin N, Zheng H, Ro S, Tafolla D, et al. Catsper3 and Catsper4 are essential for sperm hyperactivated motility and male fertility in the mouse. Biol Reprod. 2007;77:37–44. doi: 10.1095/biolreprod.107.060186. [DOI] [PubMed] [Google Scholar]
- Santi CM, Martinez-Lopez P, de la Vega-Beltran JL, Butler A, Alisio A, et al. The SLO3 sperm-specific potassium channel plays a vital role in male fertility. FEBS Lett. 2010;584:1041–6. doi: 10.1016/j.febslet.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danshina PV, Geyer CB, Dai Q, Goulding EH, Willis WD, et al. Phosphoglycerate kinase 2 (PGK2) is essential for sperm function and male fertility in mice. Biol Reprod. 2010;82:136–45. doi: 10.1095/biolreprod.109.079699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki K, Qu W, Goulding EH, Willis WD, Bunch DO, et al. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci USA. 2004;101:16501–6. doi: 10.1073/pnas.0407708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley A, O'Donnell L. Characterization of normal spermiation and spermiation failure induced by hormone suppression in adult rats. Biol Reprod. 2003;68:1299–307. doi: 10.1095/biolreprod.102.009811. [DOI] [PubMed] [Google Scholar]
- de Kretser DM. Clinical male infertility. I. Prevalence of and progress in understanding male infertility. Reprod Fertil Dev. 1994;6:3–8. doi: 10.1071/rd9940003. [DOI] [PubMed] [Google Scholar]
- Girouard J, Frenette G, Sullivan R. Compartmentalization of proteins in epididymosomes coordinates the association of epididymal proteins with the different functional structures of bovine spermatozoa. Biol Reprod. 2009;80:965–72. doi: 10.1095/biolreprod.108.073551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins DD, Brandt H, Acott TS. Initiation of sperm motility in the mammalian epididymis. Fed Proc. 1978;37:2534–42. [PubMed] [Google Scholar]
- Jones R. Plasma membrane structure and remodelling during sperm maturation in the epididymis. J Reprod Fertil Suppl. 1998;53:73–84. [PubMed] [Google Scholar]
- Lin M, Lee YH, Xu W, Baker MA, Aitken RJ. Ontogeny of tyrosine phosphorylation-signaling pathways during spermatogenesis and epididymal maturation in the mouse. Biol Reprod. 2006;75:588–97. doi: 10.1095/biolreprod.106.052712. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Nixon B, Lin M, Koppers AJ, Lee YH, et al. Proteomic changes in mammalian spermatozoa during epididymal maturation. Asian J Androl. 2007;9:554–64. doi: 10.1111/j.1745-7262.2007.00280.x. [DOI] [PubMed] [Google Scholar]
- Cornwall GA. New insights into epididymal biology and function. Hum Reprod Update. 2009;15:213–27. doi: 10.1093/humupd/dmn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacheux JL, Belleannee C, Jones R, Labas V, Belghazi M, et al. Mammalian epididymal proteome. Mol Cell Endocrinol. 2009;306:45–50. doi: 10.1016/j.mce.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Baker MA, Reeves G, Hetherington L, Aitken RJ. Analysis of proteomic changes associated with sperm capacitation through the combined use of IPG-strip pre-fractionation followed by RP chromatography LC-MS/MS analysis. Proteomics. 2010;10:482–95. doi: 10.1002/pmic.200900574. [DOI] [PubMed] [Google Scholar]
- Ficarro S, Chertihin O, Westbrook VA, White F, Jayes F, et al. Phosphoproteome analysis of capacitated human sperm. Evidence of tyrosine phosphorylation of a kinase-anchoring protein 3 and valosin-containing protein/p97 during capacitation. J Biol Chem. 2003;278:11579–89. doi: 10.1074/jbc.M202325200. [DOI] [PubMed] [Google Scholar]
- Platt MD, Salicioni AM, Hunt DF, Visconti PE. Use of differential isotopic labeling and mass spectrometry to analyze capacitation-associated changes in the phosphorylation status of mouse sperm proteins. J Proteome Res. 2009;8:1431–40. doi: 10.1021/pr800796j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK. Timing of fertilization in mammals: sperm cholesterol/phospholipid ratio as a determinant of the capacitation interval. Proc Natl Acad Sci USA. 1981;78:7560–4. doi: 10.1073/pnas.78.12.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis AJ, Kopf GS. The role of cholesterol efflux in regulating the fertilization potential of mammalian spermatozoa. J Clin Invest. 2002;110:731–6. doi: 10.1172/JCI16392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconti PE, Ning X, Fornes MW, Alvarez JG, Stein P, et al. Cholesterol efflux-mediated signal transduction in mammalian sperm: cholesterol release signals an increase in protein tyrosine phosphorylation during mouse sperm capacitation. Dev Biol. 1999;214:429–43. doi: 10.1006/dbio.1999.9428. [DOI] [PubMed] [Google Scholar]
- de Jonge C. Biological basis for human capacitation. Hum Reprod Update. 2005;11:205–14. doi: 10.1093/humupd/dmi010. [DOI] [PubMed] [Google Scholar]
- Gadella BM. The assembly of a zona pellucida binding protein complex in sperm. Reprod Domest Anim. 2008;43 Suppl 5:12–9. doi: 10.1111/j.1439-0531.2008.01255.x. [DOI] [PubMed] [Google Scholar]
- Suarez SS. Control of hyperactivation in sperm. Hum Reprod Update. 2008;14:647–57. doi: 10.1093/humupd/dmn029. [DOI] [PubMed] [Google Scholar]
- Florman HM, Jungnickel MK, Sutton KA. Regulating the acrosome reaction. Int J Dev Biol. 2008;52:503–10. doi: 10.1387/ijdb.082696hf. [DOI] [PubMed] [Google Scholar]
- Jungnickel MK, Marrero H, Birnbaumer L, Lemos JR, Florman HM. Trp2 regulates entry of Ca2+ into mouse sperm triggered by egg ZP3. Nat Cell Biol. 2001;3:499–502. doi: 10.1038/35074570. [DOI] [PubMed] [Google Scholar]
- Gibbs GM, Lo JC, Nixon B, Jamsai D, O'Connor AE, et al. Glioma pathogenesis-related 1-like 1 is testis enriched, dynamically modified, and redistributed during male germ cell maturation and has a potential role in sperm–oocyte binding. Endocrinology. 2010;151:2331–42. doi: 10.1210/en.2009-1255. [DOI] [PubMed] [Google Scholar]
- Raymond AS, Shur BD. A novel role for SED1 (MFG-E8) in maintaining the integrity of the epididymal epithelium. J Cell Sci. 2009;122:849–58. doi: 10.1242/jcs.041731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler CD, Cardullo RA. Distinct membrane fractions from mouse sperm bind different zona pellucida glycoproteins. Biol Reprod. 2002;66:65–9. doi: 10.1095/biolreprod66.1.65. [DOI] [PubMed] [Google Scholar]
- Ikawa M, Inoue N, Benham AM, Okabe M. Fertilization: a sperm's journey to and interaction with the oocyte. J Clin Invest. 2010;120:984–94. doi: 10.1172/JCI41585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon B, Aitken RJ, McLaughlin EA. New insights into the molecular mechanisms of sperm–egg interaction. Cell Mol Life Sci. 2007;64:1805–23. doi: 10.1007/s00018-007-6552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vjugina U, Evans JP. New insights into the molecular basis of mammalian sperm–egg membrane interactions. Front Biosci. 2008;13:462–76. doi: 10.2741/2693. [DOI] [PubMed] [Google Scholar]
- Zheng H, Stratton CJ, Morozumi K, Jin J, Yanagimachi R, et al. Lack of Spem1 causes aberrant cytoplasm removal, sperm deformation, and male infertility. Proc Natl Acad Sci USA. 2007;104:6852–7. doi: 10.1073/pnas.0701669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Ito C, Natsume Y, Sugitani Y, Yamanaka H, et al. Lack of acrosome formation in mice lacking a Golgi protein, GOPC. Proc Natl Acad Sci USA. 2002;99:11211–6. doi: 10.1073/pnas.162027899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiridonov NA, Wong L, Zerfas PM, Starost MF, Pack SD, et al. Identification and characterization of SSTK, a serine/threonine protein kinase essential for male fertility. Mol Cell Biol. 2005;25:4250–61. doi: 10.1128/MCB.25.10.4250-4261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden L, Arends MJ, Chausiaux OE, Ellis PJ, Lange UC, et al. Loss of TSLC1 causes male infertility due to a defect at the spermatid stage of spermatogenesis. Mol Cell Biol. 2006;26:3595–609. doi: 10.1128/MCB.26.9.3595-3609.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada D, Yoshida M, Williams YN, Fukami T, Kikuchi S, et al. Disruption of spermatogenic cell adhesion and male infertility in mice lacking TSLC1/IGSF4, an immunoglobulin superfamily cell adhesion molecule. Mol Cell Biol. 2006;26:3610–24. doi: 10.1128/MCB.26.9.3610-3624.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MJ, Dong Y, McDermott BM, Jr, Lam DH, Brown KR, et al. Defects in nuclear and cytoskeletal morphology and mitochondrial localization in spermatozoa of mice lacking nectin-2, a component of cell–cell adherens junctions. Mol Cell Biol. 2000;20:2865–73. doi: 10.1128/mcb.20.8.2865-2873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Toselli PA, Russell LD, Seldin DC. Globozoospermia in mice lacking the casein kinase II alpha' catalytic subunit. Nat Genet. 1999;23:118–21. doi: 10.1038/12729. [DOI] [PubMed] [Google Scholar]
- Chapellier B, Mark M, Garnier JM, LeMeur M, Chambon P, et al. A conditional floxed (loxP-flanked) allele for the retinoic acid receptor alpha (RARalpha) gene. Genesis. 2002;32:87–90. doi: 10.1002/gene.10071. [DOI] [PubMed] [Google Scholar]
- Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, et al. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA. 2004;101:2993–8. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Decker N, Mantchev GT, Juneja SC, McNiven MA, van Deursen JM. Lack of acrosome formation in Hrb-deficient mice. Science. 2001;294:1531–3. doi: 10.1126/science.1063665. [DOI] [PubMed] [Google Scholar]
- Xiao N, Kam C, Shen C, Jin W, Wang J, et al. PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. J Clin Invest. 2009;119:802–12. doi: 10.1172/JCI36230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson MJ, Decker WK, Beaudet AL, Ruitenbeek W, Armstrong D, et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem. 2001;276:39206–12. doi: 10.1074/jbc.M104724200. [DOI] [PubMed] [Google Scholar]
- Neesen J, Kirschner R, Ochs M, Schmiedl A, Habermann B, et al. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet. 2001;10:1117–28. doi: 10.1093/hmg/10.11.1117. [DOI] [PubMed] [Google Scholar]
- Inoue N, Ikawa M, Isotani A, Okabe M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature. 2005;434:234–8. doi: 10.1038/nature03362. [DOI] [PubMed] [Google Scholar]
- Miki K, Willis WD, Brown PR, Goulding EH, Fulcher KD, et al. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev Biol. 2002;248:331–42. doi: 10.1006/dbio.2002.0728. [DOI] [PubMed] [Google Scholar]
- Mbikay M, Tadros H, Ishida N, Lerner CP, de Lamirande E, et al. Impaired fertility in mice deficient for the testicular germ-cell protease PC4. Proc Natl Acad Sci USA. 1997;94:6842–6. doi: 10.1073/pnas.94.13.6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsadin R, Adham IM, Nayernia K, Heinlein UA, Oberwinkler H, et al. Male mice deficient for germ-cell cyritestin are infertile. Biol Reprod. 1999;61:1445–51. doi: 10.1095/biolreprod61.6.1445. [DOI] [PubMed] [Google Scholar]
- Ikawa M, Wada I, Kominami K, Watanabe D, Toshimori K, et al. The putative chaperone calmegin is required for sperm fertility. Nature. 1997;387:607–11. doi: 10.1038/42484. [DOI] [PubMed] [Google Scholar]
- Cho C, Bunch DO, Faure JE, Goulding EH, Eddy EM, et al. Fertilization defects in sperm from mice lacking fertilin beta. Science. 1998;281:1857–9. doi: 10.1126/science.281.5384.1857. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–40. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- Ogonuki N, Inoue K, Hirose M, Miura I, Mochida K, et al. A high-speed congenic strategy using first-wave male germ cells. PLoS One. 2009;4:e4943. doi: 10.1371/journal.pone.0004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CP, Battey JF, Bradley A, Bucan M, Capecchi M, et al. The knockout mouse project. Nat Genet. 2004;36:921–4. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers J, Wurst W, de Angelis MH. Towards better mouse models: enhanced genotypes, systemic phenotyping and envirotype modelling. Nat Rev Genet. 2009;10:371–80. doi: 10.1038/nrg2578. [DOI] [PubMed] [Google Scholar]
- Hancock JM, Adams NC, Aidinis V, Blake A, Bogue M, et al. Mouse Phenotype Database Integration Consortium: integration [corrected] of mouse phenome data resources. Mamm Genome. 2007;18:157–63. doi: 10.1007/s00335-007-9004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon AM, Blake A, Hancock JM. EuroPhenome and EMPReSS: online mouse phenotyping resource. Nucleic Acids Res. 2008;36:D715–8. doi: 10.1093/nar/gkm728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg CL, Wolski KM, Gibbs GM, O'Bryan MK. Phenotyping male infertility in the mouse: how to get the most out of a ‘non-performer'. Hum Reprod Update. 2010;16:205–24. doi: 10.1093/humupd/dmp032. [DOI] [PMC free article] [PubMed] [Google Scholar]