

Abstract
DNA damage is frequently encountered in spermatozoa of subfertile males and is correlated with a range of adverse clinical outcomes including impaired fertilization, disrupted preimplantation embryonic development, increased rates of miscarriage and an enhanced risk of disease in the progeny. The etiology of DNA fragmentation in human spermatozoa is closely correlated with the appearance of oxidative base adducts and evidence of impaired spermiogenesis. We hypothesize that oxidative stress impedes spermiogenesis, resulting in the generation of spermatozoa with poorly remodelled chromatin. These defective cells have a tendency to default to an apoptotic pathway associated with motility loss, caspase activation, phosphatidylserine exteriorization and the activation of free radical generation by the mitochondria. The latter induces lipid peroxidation and oxidative DNA damage, which then leads to DNA fragmentation and cell death. The physical architecture of spermatozoa prevents any nucleases activated as a result of this apoptotic process from gaining access to the nuclear DNA and inducing its fragmentation. It is for this reason that a majority of the DNA damage encountered in human spermatozoa seems to be oxidative. Given the important role that oxidative stress seems to have in the etiology of DNA damage, there should be an important role for antioxidants in the treatment of this condition. If oxidative DNA damage in spermatozoa is providing a sensitive readout of systemic oxidative stress, the implications of these findings could stretch beyond our immediate goal of trying to minimize DNA damage in spermatozoa as a prelude to assisted conception therapy.
Keywords: apoptosis, DNA damage, oxidative stress, spermatozoa
Introduction
The use of assisted reproductive technologies (ARTs) to treat human subfertility has seen unprecedented growth in recent years. At the present time, 1 in 80 children born in the United States, 1 in 50 born in Sweden, 1 in 35 born in Australia and 1 in 24 born in Denmark are the products of this form of treatment. In 2003, more than 100 000 in vitro fertilization cycles were reported from 399 clinics in the United States, resulting in the birth of more than 48 000 babies.1, 2, 3, 4 Worldwide, this figure has now exceeded 200 000 births per annum3 and is continuing to increase with every year that passes.
There are two major reasons why patients are referred for this kind of treatment. One of the most common is advanced maternal age. The age at which the first child is born in developed countries is typically around 30 years and yet, from the age of 35 years onward, we know that female fecundity declines. This provides a very narrow window within which couples are attempting to achieve their desired family size. Unfortunately, there is very little that ARTs can do to help such patients given that the decline in live birth with maternal age follows exactly the same trajectory in ART cycles as it does in the general population.5 The use of ART to treat age-related female infecundity is not rational, because for the ageing oocyte, failed fertilization is not the issue at hand. It is the ability of the oocyte to support normal embryonic development after fertilization that is defective and, by definition, assisted conception technologies cannot address this issue. On the other hand, the second major reason why patients are referred for ART is male factor infertility, and for this cohort, assisted conception does represent a rational form of treatment. Indeed, it has been known for some time that the largest, single, defined cause of human infertility is defective sperm function, resulting in failed fertilization.6 In such instances, assisting fertilization by either concentrating the spermatozoa and placing them in close proximity to the oocyte (in vitro fertilization) or, in severe cases, physically injecting a single spermatozoon into the oocyte (intracytoplasmic sperm injection (ICSI)), can readily rescue the male infertility phenotype. However, there is a price to pay for the effectiveness of ART in treating subfertile males.
When ART is used to address defective sperm function, many, if not all, of the sperm selection mechanisms that nature has put in place to ensure fertilization of the oocyte with healthy spermatozoa are circumvented. As a result, fertilization is being achieved in vitro with spermatozoa that would have been excluded from this process in vivo. There are at least two possible consequences of such action. The first is that, in as much as male infertility has a genetic familial component,7 we may be encouraging the vertical transmission of male subfertility to the progeny, as is patently the case when Y-chromosome deletions are involved. There is, therefore, a risk that the more we use ART to address male infertility in one generation, the more we shall need it in the next. A second potential issue raised by the extensive use of such treatment also relates to the health and wellbeing of the offspring. Male subfertility is commonly associated with high rates of DNA damage in the spermatozoa, and such damage has, in turn, been correlated with a wide range of adverse clinical outcomes including impaired fertility, disordered embryonic development, high rates of miscarriage and an increased risk of defects in the offspring.8, 9, 10 As a result of these linkages, there is an inevitable risk that the use of DNA-damaged spermatozoa in ART will compromise the health of the progeny. We already know that the incidence of birth defects following assisted conception is double that seen in the naturally conceived population11 and that imprinting disorders, notably the Beckwith–Wiedemann and Angelman syndromes, seem to be increased in such children.12 Infants conceived by ART are also significantly more likely to be admitted to a neonatal intensive care unit, to be hospitalized and to stay in hospital longer than their naturally conceived counterparts.13 Recent studies have also shown an increase in the hospitalization of ART offspring in infancy and early childhood compared with spontaneously conceived children,14, 15, 16 whereas other investigations have revealed abnormal retinal vascularization and an eightfold increase in the incidence of undescended testicles in boys conceived by ICSI.17, 18
We have proposed that the etiology of such defects in the F1 generation involves aberrant repair of DNA lesions brought into the oocyte by the fertilizing spermatozoon (Figure 1). Thus, immediately after fertilization, the oocyte surveys the integrity of the DNA introduced into the zygote by the spermatozoon and, if damaged, puts DNA replication on hold in both male and female pronuclei, until DNA repair has been completed.19 If the oocyte makes a mistake at this point, there is the potential to create a mutation which, because it precedes S phase of the first mitotic division, will be in every cell in the body. Through such a mechanism, DNA damage induced in spermatozoa by such factors as smoking,20 exposure to radiofrequency electromagnetic radiation21 or age22 could increase the mutational load carried by the embryo, resulting in miscarriage or serious disease in the offspring, including cancer.9, 10 A corollary of this hypothesis is that the mutational load carried by the embryo will also be influenced by the DNA repair capacity of the oocyte. At present, very little is known about the nature and fidelity of these repair mechanisms other than the fact that they are negatively impacted by age.23 As a consequence of such action, age creates a ‘perfect storm', increasing levels of DNA damage in the male gamete on the one hand, while compromising the competence of the oocyte for DNA repair on the other (Figure 1). In light of this chain of cause and effect from DNA damage in spermatozoa, to increased mutational loads in the embryo and birth defects in the offspring, it is imperative that we understand the origins of this damage and take steps to prevent its occurrence.
Figure 1.
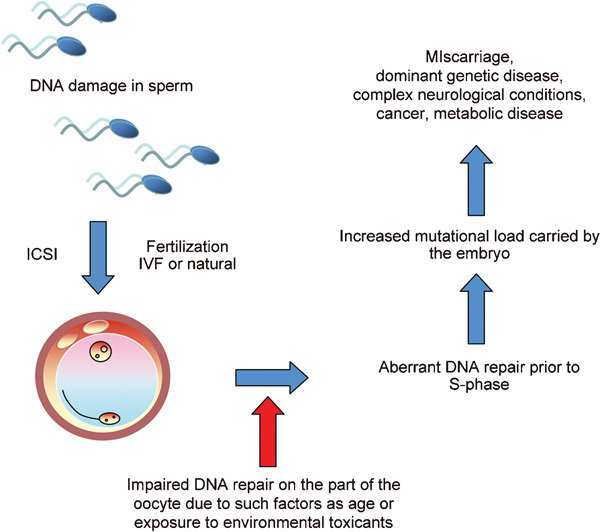
A possible mechanism by which DNA damage in human spermatozoa can impact on the health and wellbeing of the offspring. As soon as fertilization occurs, the oocyte scans the DNA introduced into the ooplasm by the fertilizing spermatozoon to determine the level of DNA damage. If excessive, the oocyte embarks on a round of DNA repair and will put DNA replication on hold until this process has been completed. If the oocyte should make a mistake at this point, a mutation may be created that will subsequently impact on the normality of embryonic development and the incidence of disease in the progeny. Such an effect will be compounded if the capacity of the oocyte for DNA repair is diminished by, for example, age or exposure to environmental toxicants.
Possible causes of DNA damage in the germ line
Physiological strand breaks
DNA fragmentation in spermatozoa may be the result of unresolved strand breaks created during the normal process of spermiogenesis in order to relieve the torsional stresses involved in packaging a very large amount of DNA into a very small sperm head. Normally, these ‘physiological' strand breaks are corrected by a complex process involving H2Ax phosphorylation and the subsequent activation of nuclear poly(ADP-ribose) polymerase and topoisomerase.24 However, if spermiogenesis should be disrupted for some reason, then the restoration of these cleavage sites might be impaired and the spermatozoa, lacking any capacity for DNA repair in their own right, would be released from the germinal epithelium still carrying their unresolved strand breaks.
Lack of antioxidant protection
While such a mechanism is clearly plausible, it does not account for the fact that a majority of the DNA damage found in human spermatozoa seems to be oxidatively induced. This conclusion is based on the known capacity of human spermatozoa to generate reactive oxygen species (ROS) such as superoxide anion,25, 26 the proven ability of ROS to induce DNA damage in human spermatozoa27 and the fact that a strong correlation exists between DNA strand breaks in these cells on the one hand and the presence of 8-hydroxy-2′-deoxyguanosine (8OHdG) adducts on the other. The latter is a sensitive marker of oxidative attacks on DNA and is a clear indicator that oxidative stress is central to the etiology of DNA damage in the male germ line.
Oxidative stress could arise as a consequence of excess exposure to ROS and/or might be a consequence of deficiencies in the antioxidant strategies that the male tract puts in place to protect the spermatozoa from free radical attack. The spermatozoon is particularly susceptible to oxidative stress because it possesses abundant targets for oxidation in the form of unsaturated fatty acids (∼50% of the fatty acid in human spermatozoa is docosahexaenoic acid with six double bonds per molecule) and the DNA present in the sperm nucleus and mitochondria. These cells also possess little in the way of intrinsic antioxidant defenses, because they have shed a majority of their cytoplasm, and hence possess only low levels of cytoplasmic antioxidant enzymes such as glutathione peroxidase (GPx), catalase and superoxide dismutase. Moreover, whatever cytoplasm they do possess is largely confined to one compartment of this cell, the midpiece. There is very little the spermatozoon can do to protect the large area of plasma membrane overlying the sperm head and tail. There is also very little that the cell can do to actively protect its DNA other than to compact it with protamines.27 However, the latter brings with it its own problems because the tightly compacted, almost crystalline DNA present in the sperm head is not amenable to DNA repair. Rather, DNA damage just accumulates in these cells over their life span for eventual repair within the oocyte after fertilization. The only real protection that these cells possess is to be found in the extracellular antioxidants provided by the secretions of the male reproductive tract. The epididymis, for example, produces highly specialized enzymes in the form of extracellular superoxide dismutase and a specific form of selenocysteine-independent GPx, GPx5.28 When the latter was knocked out, the animals developed an age-related phenotype associated with the induction of oxidative DNA damage in the spermatozoa and the appearance of birth defects in the offspring.29
In addition to such antioxidant enzymes, small molecular mass free radical scavengers are also generated in the male tract, and are major constituents of the antioxidant protection afforded to the spermatozoa by seminal plasma. The most important of these scavengers are vitamin C, uric acid, tryptophan, spermine and taurine.30, 31 Just as we have seen with GPx5, if antioxidants such as ascorbate are depleted, for example, by heavy smoking, the result is oxidative stress within the male reproductive tract and DNA damage to the spermatozoa.20
The importance of adequate antioxidant protection is also indicated by the apparent beneficial effects of antioxidant therapy in men exhibiting high levels of DNA damage in their spermatozoa. Thus Greco et al.32 found that 2 months' treatment with vitamins E and C was sufficient to significantly reduce the percentage of DNA-fragmented spermatozoa in the ejaculates of male infertility patients compared with a cohort of placebo controls.
Excess exposure to ROS
In addition to possible deficiencies in the antioxidant protection afforded to human spermatozoa in the male tract, it is also possible that DNA damage in these cells is a consequence of excess exposure to ROS emanating from either the spermatozoa themselves or infiltrating leukocytes. With respect to the latter, every human sperm sample is contaminated with leukocytes, particularly neutrophils and macrophages. Unfortunately, these phagocytes are much more powerful generators of ROS than spermatozoa, so that even a low level of leukocyte contamination can overwhelm the signal generated by the spermatozoa. Although seminal leukocytes are clearly capable of generating ROS,33 the presence of these cells in subclinical concentrations (<1×106 cells ml−1) does not seem to have any impact on sperm quality.34 A possible reason for this is that a majority of seminal phagocytes originate from the secondary sexual glands and only make contact with the spermatozoa at the moment of ejaculation. At this juncture, spermatozoa are protected from leukocyte-derived ROS by the powerful antioxidants present in seminal plasma. Once the seminal plasma has been removed; however, as occurs when spermatozoa are being prepared for ART, then the free radicals generated by the leukocyte population have unfettered access to the spermatozoa and are capable of inducing significant damage to these cells.35 Thus, the use of a formyl peptide provocation test to examine the presence of leukocytes in sperm suspensions used for ART has confirmed that not only are such cells present in these preparations but also that their presence significantly disrupts fertilization.36 The existence of a cause and effect relationship between leukocytic infiltration and impaired fertilization is suggested by the suppression of sperm function observed after the addition of activated leukocytes to human sperm suspensions.37 Furthermore, the physical removal of these cellular contaminants using magnetic beads coated with a monoclonal antibody against the common leukocyte antigen, CD45, has been found to significantly enhance fertilization rates in vitro.38 It has also been shown that the disruptive effect of leukocytes in vitro can be reversed by the addition of antioxidants to the medium including reduced glutathione, N-acetylcysteine, hypotaurine and catalase.37 In the context of DNA damage, the protective action of seminal plasma antioxidants ensures that leukocytic infiltration has minimal effect on DNA damage in vivo.39, 40 However, DNA damage can be induced by leukocytes in vitro during the preparation of the spermatozoa.41 The addition of antioxidants to the sperm preparation media used in ARTs would be a rational approach to circumventing this problem.
If leukocytes are not generating the ROS that account for the high levels of oxidative DNA damage seen in human sperm suspensions, then these pernicious oxygen metabolites must be coming from the spermatozoa themselves. The ability of spermatozoa to produce ROS has been recognized since the 1940s when Tosic and Walton42 published their pioneering paper on hydrogen peroxide production by bovine spermatozoa. In this instance, the ROS seemed to arise from an amino acid oxidase that used aromatic amino acids as substrate. This pathway has not been reported for other species and, at present, the consensus is that the ROS originate from either the sperm mitochondria or an, as yet uncharacterized, plasma membrane nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.43, 44 With respect to the latter evidence for the NADPH oxidase, NOX5, has been obtained in both human (unpublished observations) and equine spermatozoa45 and elements of the NOX2 complex have been reported for mouse spermatozoa.46 However, there is no evidence that the activity of such oxidases is involved in the etiology of defective sperm function or DNA damage. On the contrary, there are very good data to indicate that mitochondrial ROS generation is enhanced in defective spermatozoa and that such activity is capable of inducing DNA damage, possibly as a component of an apoptotic cascade.47
Mitochondrial ROS and apoptosis
The ability of mitochondria to generate ROS is inversely related to sperm motility.47 This link is causative because if mitochondrial ROS generation is artificially triggered using rotenone to stimulate free radical generation at complex I of the electron transport chain, then the spermatozoa exhibit both lipid peroxidation and a loss of motility that can be reversed by the concomitant presence of an antioxidant such as α-tocopherol.47 The stimulation of mitochondrial ROS generation can also result in oxidative DNA damage to spermatozoa, for example, when spermatozoa are irradiated with radiofrequency electromagnetic radiation21 or when mitochondrial ROS is triggered after exposure to unsaturated fatty acids.48 Thus, there is a plausible role for mitochondrial ROS in the etiology of DNA damage in human spermatozoa, although the factors responsible for stimulating free radical leakage from the mitochondria are still unresolved. One important contributor to such aberrant activity might be the induction of apoptosis.
Mature spermatozoa have the potential to exhibit many of the features of apoptosis including activation of caspases 1, 3, 8 and 9, annexin-V binding and the mitochondrial generation of ROS.49, 50, 51 Although many of the reagents that have been shown to induce apoptosis in somatic cells (staurosporine, lipopolysaccharide, 3-deoxy-𝒹-manno-octulosonic acid (Kdo) and genistein) are ineffective with human spermatozoa, these cells will default to the intrinsic apoptotic pathway in response to oxidative stress. Thus, exposure of human spermatozoa to H2O2 can readily trigger an apoptotic cascade characterized by the activation of caspase 3 and the appearance of annexin-V binding positivity.52 Moreover, pre-exposure of human spermatozoa to antioxidants, such as melatonin or catalase, will prevent this apoptotic response.53, 54 Such an apoptotic cascade can also be precipitated by a variety of factors that induce oxidative stress in spermatozoa by triggering free radical generation by the mitochondria, including exposure to radiofrequency electromagnetic radiation,21 treatment with unsaturated fatty acids48 and dosing with the phosphatidylinositol 3-kinase inhibitor, wortmannin (Koppers AJ and Aitken RJ, unpubl. obs.).
It might be anticipated that the culmination of this apoptotic cascade in spermatozoa would be the activation of endonucleases such as caspase-activated DNAse that, in combination with other nucleases released from the mitochondria (endonuclease G and apoptosis-inducing factor), would move to the nucleus to induce DNA cleavage and complete the pathway to cell death. However, in spermatozoa, this process is prevented by the physical architecture of the cell (the mitochondria and nucleus are in different cellular compartments) and the condensed nature of the chromatin. As a result of these physical constraints, apoptosis in spermatozoa is quite different from somatic cells in that endonuclease activation cannot be immediately followed by DNA fragmentation. Nevertheless, apoptosis does induce significant motility loss which deletes the spermatozoa in an immediate functional sense, but cannot precipitate the immediate DNA cleavage typical of apoptosis in somatic cells. As a consequence of this impediment to complete apoptosis, the latter may not completely eliminate the cells' competence for fertilization.26 It is therefore still perfectly possible for apoptotic spermatozoa, possessing high levels of DNA damage (particularly oxidative damage), to fertilize the oocyte. The inefficiency of apoptosis as a cell depletion strategy is clearly emphasized by the associations that have been repeatedly observed between DNA damage in the germ line and miscarriage or morbidity in the offspring. In such cases, conception must have occurred despite the DNA damage in the sperm nucleus.8, 9 Thus, while spermatozoa have evolved to be extremely resilient cells that are competent to undertake the perilous journey from the germinal epithelium of the testes to the oocyte in the fallopian tubes, the downside of this strategy is that spermatozoa are extremely difficult to destroy using the intrinsic apoptotic cascade.
However, apoptosis does result in DNA damage, so how does it occur? The only element of the apoptotic cascade that can induce such damage is the ROS released from the mitochondria; it is for this reason that most of the DNA damage seen in human spermatozoa is oxidative in nature.55 More extensive DNA fragmentation may occur post-mortem as a result of the activation of endonucleases that are integrated into the sperm chromatin, thus finalizing the destruction of the cell.56 However, the DNA damage induced in live cells, which is the clinically relevant damage, is entirely oxidative in nature.57
Triggers of apoptosis
There are a large number of different biological situations in which DNA damage is observed in human spermatozoa that could be due to the induction of an apoptotic cascade. Thus, cryostorage is well-known to be associated with the induction of DNA damage and, again, a majority of this damage seems to be oxidative.58, 59 Similarly, heat exposure will induce free radical generation by the spermatozoa as a consequence of the initiation of apoptotic cascade, and so will exposure to radiofrequency electromagnetic radiation21 or hydrogen peroxide.52 All of these conditions are associated with the creation of oxidative stress. To our knowledge, the only other way to induce apoptosis in human spermatozoa is through the withdrawal of prosurvival factors.
In vivo spermatozoa have to survive for around a week, as they migrate through the lumen of the epididymis, and for another week or so in the female reproductive tract, as they wait for an egg to arrive. The need for such prolonged survival time periods is an unusual human attribute, because uniquely in our species (and certain species of bats) insemination and ovulation are not synchronized. In contrast to this prolonged survival in vivo, in vitro human spermatozoa frequently suffer from a major loss of motility when incubated for 12 h or more at 37 °C.60 Moreover, this time-dependent motility loss is associated with signs of spontaneous apoptosis in the spermatozoa, including phosphatidylserine exteriorization, caspase activation and DNA damage.61, 62 The reason for this time-dependent deterioration of semen quality can be found in the simple nature of the incubation media used to incubate these cells. Thus, most ART culture media are nothing more than balanced salt solutions supplemented with energy substrates and, possibly, serum albumin. There is nothing in these preparations to support the spermatozoa and prevent them from defaulting to an apoptotic state. In contrast, the relative longevity of human spermatozoa in vivo may be due to the presence of prosurvival factors in the epididymal plasma and uterotubal fluids that prevent apoptosis from taking place.
A clue to the possible nature of these putative prosurvival factors was provided with publication of the first draft of the human sperm proteome.63 Up to that point, no receptors had been definitively identified on the sperm surface. Completion of the sperm proteome resulted in the identification of around 20 different receptors on the surface of these cells. From a prosurvival perspective, the first candidate prosurvival receptor to be investigated was the prolactin receptor. Detailed analysis of this receptor showed that the human spermatozoon possesses four different isoforms of the prolactin receptor on its surface including the long form and three variants of the short form, one of which is unique to the male gamete. Critically, prolactin stimulates phosphatidylinositol 3-kinase activity, maintains Akt phosphorylation and has a powerful prosurvival effect on human spermatozoa.60 There is no reason to believe that prolactin is the only or even the most powerful prosurvival factor for human spermatozoa; however, it is the first such factor to be identified. Its discovery heralds a new era in our understanding of sperm cell biology by emphasizing the dependence of these cells on survival factors that, through activation of phosphatidylinositol 3-kinase–Akt phosphorylation, prevent spermatozoa from entering an apoptotic pathway that culminates in cell death. When spermatozoa are incubated in simple-defined culture medium in vitro in the absence of such prosurvival factors, they ultimately revert to an apoptotic state characterized by mitochondrial ROS generation. As a consequence of the latter, the sperm DNA becomes oxidatively damaged, generating 8OHdG adducts and, ultimately, DNA strand breaks. Further research is clearly needed in this area to identify the optimal combination of prosurvival factors that would allow spermatozoa to survive for prolonged periods of time in vitro, without exhibiting signs of DNA damage. Such reagents might also be useful in the design and development of optimized cryostorage media.
Why do spermatozoa undergo apoptosis
One of the major reasons why spermatozoa have to undergo a regulated, programmed senescence relates to their ultimate disposal post-coitum. Thus, after insemination, the female tract responds to the presence of millions of dead and moribund spermatozoa by triggering a massive leukocyte infiltration into the cervix and uterine cavity. The phagocytic activity exhibited by these cells must be silent; in other words, the spermatozoa must be efficiently phagocytosed and removed from the tract in the absence of an oxidative burst or the production of proinflammatory cytokines. Furthermore, this phagocytosis must be silent even though spermatozoa are patently foreign cells containing potent antigens. There are many examples of silent phagocytosis in biology and a common feature of this phenomenon is the expression of apoptotic markers, such as phosphatidylserine, on the surface of the target cell. This apoptotic marker is thought to instruct the phagocyte that the target cell should be engulfed in a non-phlogistic manner.64 The activation of this apoptotic cascade in oxidatively stressed spermatozoa could, therefore, be viewed as an adaptation that permits the efficient removal of these cells from the female tract by phagocytic leukocytes, without provoking a damaging inflammatory response.
The second important purpose of apoptosis in the germ line is to prevent DNA-damaged cells from participating in the process of fertilization. Thus, as soon as these cells sense that they are oxidatively damaged, they initiate an apoptotic cascade, one of the early features of which is a rapid loss of motility that prevents these cells from participating in fertilization. This preventative mechanism is not perfect; otherwise we would not see paternally mediated defects in embryonic development or morbidity in the offspring.8, 9, 10 Furthermore, this natural mechanism for preventing fertilization by defective human spermatozoa can be, and is, circumvented by assisted conception therapy, particularly ICSI. In this light, selecting spermatozoa for ICSI on the basis that they do not express markers of apoptosis such as annexin-V binding, would seem a rational approach towards reducing the risk that DNA-damaged spermatozoa are used in assisted conception.65
Conclusions and future directions
The purpose of this review has been to emphasize the importance of DNA damage in human spermatozoa as a determinant of both fertility and the subsequent normality of embryonic development. Given the associations that have been highlighted between DNA damage in spermatozoa and adverse clinical outcomes for both the patient and his offspring, it is clearly important that we understand the etiology of these lesions. A significant insight into this problem was generated when it was found that a majority of the DNA damage is oxidative and associated with the formation of 8OHdG adducts. A second factor in the DNA damage equation is the close correlation that has been observed by several independent groups between DNA damage in the spermatozoa and evidence of poor chromatin remodelling during spermiogenesis, as revealed by the CMA3 fluorescence.9 To accommodate these observations into a single hypothesis, we have proposed that the spermiogenetic process is extremely vulnerable to oxidative stress (Figure 2). The vulnerability of spermiogenesis to oxidative stress has been ascribed to the importance of regulated protein translation in this process and the unique sensitivity of the protein translation machinery to such stress.66 The result of this ROS-mediated disruption of spermiogenesis is not to halt the morphogenesis of spermatozoa but rather to impair this process so that the spermatozoa that are produced are imperfect. Specifically, we propose that these defective spermatozoa with their poorly compacted chromatin are particularly vulnerable to stress and respond to such adversity by defaulting to apoptosis. The latter then results in mitochondrial ROS generation that attacks the vulnerable chromatin resulting in the 8OHdG formation55, 67 and ultimately DNA fragmentation.68
Figure 2.
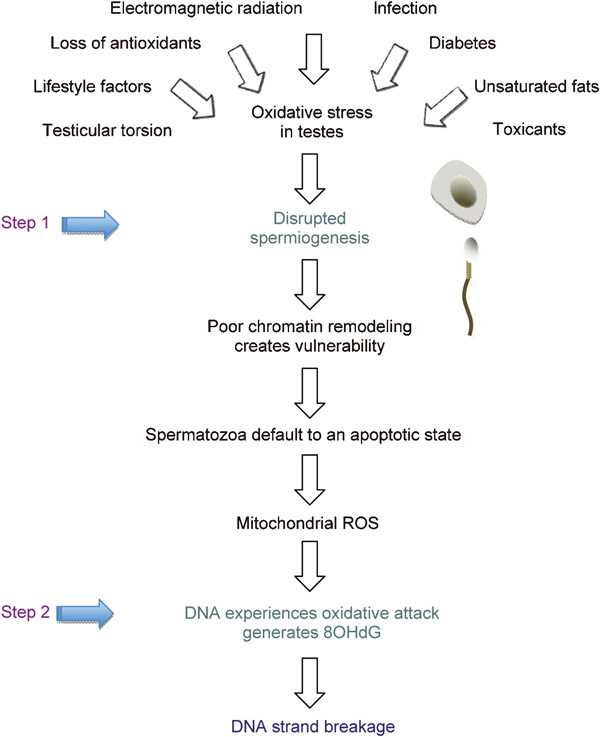
Two-step hypothesis for the origins of DNA damage in the male germ line. This hypothesis posits that a wide variety of clinical and environmental factors are capable, alone or in combination, of creating oxidative stress in the testes. In step 1, this stress impairs spermiogenesis resulting in the production of defective spermatozoa possessing poorly protaminated chromatin. These defective spermatozoa have a tendency to default to an apoptotic cascade that involves the generation of ROS by the sperm mitochondria. In step 2, these ROS attack the poorly remodelled chromatin generating oxidized DNA base adducts (8OHdG) that ultimately result in DNA strand breakage and cell death.
If oxidative stress is at the heart of this process, then antioxidants should surely be part of the cure. Surprisingly, despite an awareness of the importance of antioxidant vitamins to male infertility that stretches back to the 1930s,69 there have been very few rigorous attempts to evaluate the benefits, and potential risks,70 of antioxidant therapy in the treatment of male infertility. Studies that have been conducted in this regard have returned generally positive results,32, 71, 72, 73 but there is a desperate need for randomized, double-blind, crossover trials in this area using patients who have been selected with a robust marker of oxidative stress such as 8OHdG. A key question that should be addressed in the context of such trials is whether the oxidative stress associated with disrupted spermiogenesis and DNA damage to spermatozoa is systemic or localized to the male reproductive tract. A number of articles have appeared recently, suggesting that there is a chain of cause and effect relationships between body mass index, systemic oxidative stress, semen quality, DNA damage in the spermatozoa and subfertility.74, 75, 76, 77, 78 If these associations are confirmed and DNA damage in the male germ line is indeed providing a readout of the systemic level of oxidative stress, then there are important implications in these data that stretch beyond the diagnosis, treatment and prevention of male infertility.
Acknowledgments
We gratefully acknowledge the support of the ARC Centre of Excellence in Biotechnology and Development and the National Health and Medical Research Council (NHMRC).
The authors declare no competing financial interests.
References
- van Voorhis BJ. Clinical practice. In vitro fertilization. N Engl J Med. 2007;356:379–86. doi: 10.1056/NEJMcp065743. [DOI] [PubMed] [Google Scholar]
- Australian Bureau of Statistics Australian Babies. 4102.0. Australian Social Trends Canberra: Australian Bureau of Statistics; 2007 . http://www.abs.gov.au/AUSSTATS [Google Scholar]
- International Committee for Monitoring Assisted Reproductive Technology Adamson GD, de Mouzon J, Lancaster P, Nygren KG, et al. World collaborative report on in vitro fertilization, 2000. Fertil Steril. 2006;85:1586–622. doi: 10.1016/j.fertnstert.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Aitken RJ. Just how safe is assisted conception therapy. Exp Rev Obstet Gynecol. 2008;3:267–71. [Google Scholar]
- Jansen RP. The effect of female age on the likelihood of a live birth from one in-vitro fertilisation treatment. Med J Aust. 2003;178:258–61. [PubMed] [Google Scholar]
- Hull MGR, Glazener CMA, Kelly NJ, Conway DI, Foster PA, et al. Population study of causes, treatment and outcome of infertility. BMJ. 1985;291:1693–7. doi: 10.1136/bmj.291.6510.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poongothai J, Gopenath TS, Manonayaki S. Genetics of human male infertility. Singapore Med J. 2009;50:336–47. [PubMed] [Google Scholar]
- Aitken RJ, de Iuliis GN. On the possible origins of DNA damage in human spermatozoa. Mol Hum Reprod. 2010;16:3–13. doi: 10.1093/molehr/gap059. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, de Iuliis GN, McLachlan RI. Biological and clinical significance of DNA damage in the male germ line. Int J Androl. 2009;32:46–56. doi: 10.1111/j.1365-2605.2008.00943.x. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Koopman P, Lewis SE. Seeds of concern. Nature. 2004;432:48–52. doi: 10.1038/432048a. [DOI] [PubMed] [Google Scholar]
- Hansen M, Kurinczuk JJ, Bower C, Webb S. The risk of major birth defects after intracytoplasmic sperm injection and in vitro fertilization. N Engl J Med. 2002;346:725–30. doi: 10.1056/NEJMoa010035. [DOI] [PubMed] [Google Scholar]
- Shiota K, Yamada S. Intrauterine environment-genome interaction and children's development (3): assisted reproductive technologies and developmental disorders. J Toxicol Sci. 2009;34 Suppl 2:SP287–91. doi: 10.2131/jts.34.sp287. [DOI] [PubMed] [Google Scholar]
- Hansen M, Colvin L, Petterson B, Kurinczuk JJ, de Klerk N, et al. Admission to hospital of singleton children born following assisted reproductive technology (ART) Hum Reprod. 2008;23:297–305. doi: 10.1093/humrep/den093. [DOI] [PubMed] [Google Scholar]
- Ericson A, Nygren KG, Olausson PO, Kallen B. Hospital care utilization of infants born after IVF. Hum Reprod. 2002;7:929–32. doi: 10.1093/humrep/17.4.929. [DOI] [PubMed] [Google Scholar]
- Kallen B, Finnstrom O, Nygren KG, Olausson PO. In vitro fertilization in Sweden: child morbidity including cancer risk. Fertil Steril. 2005;84:605–10. doi: 10.1016/j.fertnstert.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Klemetti R, Sevon T, Gissler M, Hemminki E. Health of children born as a result of in vitro fertilization. Pediatrics. 2006;118:1819–27. doi: 10.1542/peds.2006-0735. [DOI] [PubMed] [Google Scholar]
- Ludwig AK, Katalinic A, Thyen U, Sutcliffe AG, Diedrich K, et al. Physical health at 5.5 years of age of term-born singletons after intracytoplasmic sperm injection: results of a prospective, controlled, single-blinded study. Fertil Steril. 2009;91:115–24. doi: 10.1016/j.fertnstert.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Wikstrand MH, Niklasson A, Strömland K, Hellström A. Abnormal vessel morphology in boys born after intracytoplasmic sperm injection. Acta Paediatr. 2008;97:1512–7. doi: 10.1111/j.1651-2227.2008.00959.x. [DOI] [PubMed] [Google Scholar]
- Shimura T, Inoue M, Taga M, Shiraishi K, Uematsu N, et al. p53-dependent S-phase damage checkpoint and pronuclear cross talk in mouse zygotes with X-irradiated sperm. Mol Cell Biol. 2002;22:2220–8. doi: 10.1128/MCB.22.7.2220-2228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga CG, Motchnik PA, Wyrobek AJ, Rempel DM, Ames BN. Smoking and low antioxidant levels increase oxidative damage to DNA. Mut Res. 1996;351:199–203. doi: 10.1016/0027-5107(95)00251-0. [DOI] [PubMed] [Google Scholar]
- de Iuliis GN, Newey RJ, King BV, Aitken RJ. Mobile phone radiation induces reactive oxygen species production and DNA damage in human spermatozoa in vitro. PLoS One. 2009;4:e6446. doi: 10.1371/journal.pone.0006446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, Muller CH, Berger RE. Effects of age on DNA double-strand breaks and apoptosis in human sperm. Fertil Steril. 2003;80:1420–30. doi: 10.1016/j.fertnstert.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Tease C, Fisher G. The influence of maternal age on radiation-induced chromosome aberrations in mouse oocytes. Mutat Res. 1991;262:57–62. doi: 10.1016/0165-7992(91)90107-f. [DOI] [PubMed] [Google Scholar]
- Meyer-Ficca ML, Lonchar J, Credidio C, Ihara M, Li Y, et al. Disruption of poly(ADP-ribose) homeostasis affects spermiogenesis and sperm chromatin integrity in mice. Biol Reprod. 2009;81:46–55. doi: 10.1095/biolreprod.108.075390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken RJ, Clarkson JS. Cellular basis of defective sperm function and its association with the genesis of reactive oxygen species by human spermatozoa. J Reprod Fertil. 1987;81:459–69. doi: 10.1530/jrf.0.0810459. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Gordon E, Harkiss D, Twigg JP, Milne P, et al. Relative impact of oxidative stress on the functional competence and genomic integrity of human spermatozoa. Biol Reprod. 1998;59:1037–46. doi: 10.1095/biolreprod59.5.1037. [DOI] [PubMed] [Google Scholar]
- Bennetts LE, Aitken RJ. A comparative study of oxidative DNA damage in mammalian spermatozoa. Mol Reprod Dev. 2005;71:77–87. doi: 10.1002/mrd.20285. [DOI] [PubMed] [Google Scholar]
- Vernet P, Rigaudiére N, Ghyselinck N, Dufaure JP, Drevet JR. In vitro expression of a mouse tissue specific glutathione-peroxidase-like protein lacking the selenocysteine can protect stably transfected mammalian cells against oxidative damage. Biochem Cell Biol. 1996;74:125–31. doi: 10.1139/o96-014. [DOI] [PubMed] [Google Scholar]
- Chabory E, Damon C, Lenoir A, Kauselmann G, Kern H, et al. Epididymis seleno-independent glutathione peroxidase 5 maintains sperm DNA integrity in mice. J Clin Invest. 2009;119:2074–85. doi: 10.1172/JCI38940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel RR, Schill WB. Sperm preparation for ART. Reprod Biol Endocrinol. 2003;1:108. doi: 10.1186/1477-7827-1-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhemrev JP, van Overveld FW, Haenen GR, Teerlink T, Bast A, et al. Quantification of the nonenzymatic fast and slow TRAP in a postaddition assay in human seminal plasma and the antioxidant contributions of various seminal compounds. J Androl. 2000;21:913–20. [PubMed] [Google Scholar]
- Greco E, Iacobelli M, Rienzi L, Ubaldi F, Ferrero S, et al. Reduction of the incidence of sperm DNA fragmentation by oral antioxidant treatment. J Androl. 2005;26:349–53. doi: 10.2164/jandrol.04146. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, West KM. Analysis of the relationship between reactive oxygen species production and leucocyte infiltration in fractions of human semen separated on Percoll gradients. Int J Androl. 1990;13:433–51. doi: 10.1111/j.1365-2605.1990.tb01051.x. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Buckingham DW, Brindle J, Gomez E, Baker HW, et al. Analysis of sperm movement in relation to the oxidative stress created by leukocytes in washed sperm preparations and seminal plasma. Hum Reprod. 1995;10:2061–71. doi: 10.1093/oxfordjournals.humrep.a136237. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Clarkson JS. Significance of reactive oxygen species and antioxidants in defining the efficacy of sperm preparation techniques. J Androl. 1988;9:367–76. doi: 10.1002/j.1939-4640.1988.tb01067.x. [DOI] [PubMed] [Google Scholar]
- Krausz C, Mills C, Rogers S, Tan SL, Aitken RJ. Stimulation of oxidant generation by human sperm suspensions using phorbol esters and formyl peptides: relationships with motility and fertilization in vitro. Fertil Steril. 1994;62:599–605. doi: 10.1016/s0015-0282(16)56952-0. [DOI] [PubMed] [Google Scholar]
- Baker HW, Brindle J, Irvine DS, Aitken RJ. Protective effect of antioxidants on the impairment of sperm motility by activated polymorphonuclear leukocytes. Fertil Steril. 1996;65:411–9. doi: 10.1016/s0015-0282(16)58109-6. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Buckingham DW, West K, Brindle J. On the use of paramagnetic beads and ferrofluids to assess and eliminate the leukocytic contribution to oxygen radical generation by human sperm suspensions. Am J Reprod Immunol. 1996;35:541–51. doi: 10.1111/j.1600-0897.1996.tb00055.x. [DOI] [PubMed] [Google Scholar]
- Moskovtsev SI, Willis J, White J, Mullen JB. Leukocytospermia: relationship to sperm deoxyribonucleic acid integrity in patients evaluated for male factor infertility. Fertil Steril. 2007;88:737–40. doi: 10.1016/j.fertnstert.2006.11.132. [DOI] [PubMed] [Google Scholar]
- Henkel R, Kierspel E, Stalf T, Mehnert C, Menkveld R, et al. Effect of reactive oxygen species produced by spermatozoa and leukocytes on sperm functions in non-leukocytospermic patients. Fertil Steril. 2005;83:635–42. doi: 10.1016/j.fertnstert.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Twigg J, Irvine DS, Houston P, Fulton N, Michael L, et al. Iatrogenic DNA damage induced in human spermatozoa during sperm preparation: protective significance of seminal plasma. Mol Hum Reprod. 1998;4:439–45. doi: 10.1093/molehr/4.5.439. [DOI] [PubMed] [Google Scholar]
- Tosic J, Walton A. Formation of hydrogen peroxide by spermatozoa and its inhibitory effect on respiration. Nature. 1946;158:485. doi: 10.1038/158485a0. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Ryan AL, Curry BJ, Baker MA. Multiple forms of redox activity in populations of human spermatozoa. Mol Hum Reprod. 2003;9:645–61. doi: 10.1093/molehr/gag086. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Fisher HM, Fulton N, Gomez E, Knox W, et al. Reactive oxygen species generation by human spermatozoa is induced by exogenous NADPH and inhibited by the flavoprotein inhibitors diphenylene iodonium and quinacrine. Mol Reprod Dev. 1997;47:468–82. doi: 10.1002/(SICI)1098-2795(199708)47:4<468::AID-MRD14>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Sabeur K, Ball BA. Characterization of NADPH oxidase 5 in equine testis and spermatozoa. Reproduction. 2007;34:263–7. doi: 10.1530/REP-06-0120. [DOI] [PubMed] [Google Scholar]
- Shukla S, Jha RK, Laloraya M, Kumar PG. Identification of non-mitochondrial NADPH oxidase and the spatio-temporal organization of its components in mouse spermatozoa. Biochem Biophys Res Commun. 2005;331:476–83. doi: 10.1016/j.bbrc.2005.03.198. [DOI] [PubMed] [Google Scholar]
- Koppers AJ, de Iuliis GN, Finnie JM, McLaughlin EA, Aitken RJ. Significance of mitochondrial reactive oxygen species in the generation of oxidative stress in spermatozoa. J Clin Endocrinol Metab. 2008;93:3199–207. doi: 10.1210/jc.2007-2616. [DOI] [PubMed] [Google Scholar]
- Koppers AJ, Garg ML, Aitken RJ. Stimulation of mitochondrial reactive oxygen species production by unesterified, unsaturated fatty acids in defective human spermatozoa. Free Radic Biol Med. 2010;48:112–9. doi: 10.1016/j.freeradbiomed.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Barroso G, Morshedi M, Oehninger S. Analysis of DNA fragmentation, plasma membrane translocation of phosphatidylserine and oxidative stress in human spermatozoa. Hum Reprod. 2000;15:1338–44. doi: 10.1093/humrep/15.6.1338. [DOI] [PubMed] [Google Scholar]
- Paasch U, Grunewald S, Agarwal A, Glandera HJ. Activation pattern of caspases in human spermatozoa. Fertil Steril. 2004;81 Suppl 1:802–9. doi: 10.1016/j.fertnstert.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Grunewald S, Sharma R, Paasch U, Glander HJ, Agarwal A. Impact of caspase activation in human spermatozoa. Microsc Res Tech. 2009;72:878–88. doi: 10.1002/jemt.20732. [DOI] [PubMed] [Google Scholar]
- Lozano GM, Bejarano I, Espino J, González D, Ortiz A, et al. Relationship between caspase activity and apoptotic markers in human sperm in response to hydrogen peroxide and progesterone. J Reprod Dev. 2009;55:615–21. doi: 10.1262/jrd.20250. [DOI] [PubMed] [Google Scholar]
- Libman J, Gabriel MS, Sairam MR, Zini A.Catalase can protect spermatozoa of FSH receptor knock-out mice against oxidant-induced DNA damage in vitro Int J Androle-pub ahead of print 4 January 2010; doi: 10.1111/j.1365-2605.2009.01032.x. [DOI] [PubMed]
- Espino J, Bejarano I, Ortiz A, Lozano GM, García JF, et al. Melatonin as a potential tool against oxidative damage and apoptosis in ejaculated human spermatozoa Fertil Sterile-pub ahead of print 10 February 2010; doi: 10.1016/j.fertnstert.2009.12.082. [DOI] [PubMed]
- de Iuliis GN, Thomson LK, Mitchell LA, Finnie JM, Koppers AJ, et al. DNA damage in human spermatozoa is highly correlated with the efficiency of chromatin remodeling and the formation of 8-hydroxy-2′-deoxyguanosine, a marker of oxidative stress. Biol Reprod. 2009;81:517–24. doi: 10.1095/biolreprod.109.076836. [DOI] [PubMed] [Google Scholar]
- Sotolongo B, Huang TT, Isenberger E, Ward WS. An endogenous nuclease in hamster, mouse, and human spermatozoa cleaves DNA into loop-sized fragments. J Androl. 2005;26:272–80. doi: 10.1002/j.1939-4640.2005.tb01095.x. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, de Iuliis GN, Finnie JM, Hedges A, McLachlan RI.Analysis of the relationships between oxidative stress, DNA damage and sperm vitality in a patient population: development of diagnostic criteria Hum Reprode-pub ahead of print 17 August 2010; doi: 10.1093/humrep/deq214. [DOI] [PubMed]
- Zribi N, Feki Chakroun N, El Euch H, Gargouri J, Bahloul A, et al. Effects of cryopreservation on human sperm deoxyribonucleic acid integrity. Fertil Steril. 2010;93:159–66. doi: 10.1016/j.fertnstert.2008.09.038. [DOI] [PubMed] [Google Scholar]
- Donnelly ET, Steele EK, McClure N, Lewis SE. Assessment of DNA integrity and morphology of ejaculated spermatozoa from fertile and infertile men before and after cryopreservation. Hum Reprod. 2001;16:1191–9. doi: 10.1093/humrep/16.6.1191. [DOI] [PubMed] [Google Scholar]
- Pujianto DA, Curry BJ, Aitken RJ. Prolactin exerts a prosurvival effect on human spermatozoa via mechanisms that involve the stimulation of Akt phosphorylation and suppression of caspase activation and capacitation. Endocrinology. 2010;151:1269–79. doi: 10.1210/en.2009-0964. [DOI] [PubMed] [Google Scholar]
- Schuffner A, Morshedi M, Vaamonde D, Duran EH, Oehninger S. Effect of different incubation conditions on phosphatidylserine externalization and motion parameters of purified fractions of highly motile human spermatozoa. J Androl. 2002;23:194–201. [PubMed] [Google Scholar]
- Muratori M, Maggi M, Spinelli S, Filimberti E, Forti G, et al. Spontaneous DNA fragmentation in swim-up selected human spermatozoa during long term incubation. J Androl. 2003;24:253–62. doi: 10.1002/j.1939-4640.2003.tb02670.x. [DOI] [PubMed] [Google Scholar]
- Baker MA, Reeves G, Hetherington L, Müller J, Baur I, et al. Identification of gene products present in Triton X-100 soluble and insoluble fractions of human spermatozoa lysates using LC-MS/MS analysis. Proteomics Clin Appl. 2007;1:524–32. doi: 10.1002/prca.200601013. [DOI] [PubMed] [Google Scholar]
- Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y. Silent cleanup of very early apoptotic cells by macrophages. J Immunol. 2003;171:4672–9. doi: 10.4049/jimmunol.171.9.4672. [DOI] [PubMed] [Google Scholar]
- Said TM, Agarwal A, Zborowski M, Grunewald S, Glander HJ, et al. Utility of magnetic cell separation as a molecular sperm preparation technique. J Androl. 2008;29:134–42. doi: 10.2164/jandrol.107.003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Söll D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc Natl Acad Sci USA. 2010;107:4028–33. doi: 10.1073/pnas.1000315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama H, Yamaguchi R, Fukuda J, Kasi H, Tanak T. Increased deoxyribonucleic acid damage in the spermatozoa of infertile male patients. Fertil Steril. 1997;65:519–24. doi: 10.1016/s0015-0282(97)00236-7. [DOI] [PubMed] [Google Scholar]
- Irvine DS, Twigg JP, Gordon EL, Fulton N, Milne PA, et al. 2000 DNA integrity in human spermatozoa: relationships with semen quality. J Androl. 2000;21:33–44. [PubMed] [Google Scholar]
- Mason KE. The specifcity of vitamin E for the testis. I. Relation between vitamins A and E. J Exp Zool. 1930;55:101–22. [Google Scholar]
- Ménézo YJ, Hazout A, Panteix G, Robert F, Rollet J, et al. Antioxidants to reduce sperm DNA fragmentation: an unexpected adverse effect. Reprod Biomed Online. 2007;14:418–21. doi: 10.1016/s1472-6483(10)60887-5. [DOI] [PubMed] [Google Scholar]
- Suleiman SA, Elamin Ali M, Zaki ZM, el-Malik EM, Nasr MA. Lipid peroxidation and human sperm motility: protective role of vitamin E. J Androl. 1996;17:530–7. [PubMed] [Google Scholar]
- Tremellen K. Oxidative stress and male infertility-a clinical perspective. Hum Reprod Update. 2008;14:243–58. doi: 10.1093/humupd/dmn004. [DOI] [PubMed] [Google Scholar]
- Ross C, Morriss A, Khairy M, Khalaf Y, Braude P, et al. A systematic review of the effect of oral antioxidants on male infertility. Reprod Biomed Online. 2010;20:711–23. doi: 10.1016/j.rbmo.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Chavarro JE, Toth TL, Wright DL, Meeker JD, Hauser R. Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertil Steril. 2010;93:2222–31. doi: 10.1016/j.fertnstert.2009.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kort HI, Massey JB, Elsner CW, Mitchell-Leef D, Shapiro DB, et al. Impact of body mass index values on sperm quantity and quality. J Androl. 2006;27:450–2. doi: 10.2164/jandrol.05124. [DOI] [PubMed] [Google Scholar]
- Pauli EM, Legro RS, Demers LM, Kunselman AR, Dodson WC, et al. Diminished paternity and gonadal function with increasing obesity in men. Fertil Steril. 2008;90:346–51. doi: 10.1016/j.fertnstert.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigueras-Villaseñor RM, Rojas-Castañeda JC, Chávez-Saldaña M, Gutiérrez-Pérez O, García-Cruz ME, et al. Alterations in the spermatic function generated by obesity in rats Acta Histocheme-pub ahead of print 8 February 2010; doi: 10.1016/j.acthis.2009.10.004. [DOI] [PubMed]