

Abstract
Sexually transmitted infections increase the likelihood of HIV-1 transmission. We investigated the effect of Neisseria gonorrheae (gonococcus [GC]) exposure on HIV replication in primary resting CD4+ T cells, a major HIV target cell during the early stage of sexual transmission of HIV. GC and TLR2 agonists, such as peptidylglycan (PGN), Pam3CSK4, and Pam3C-Lip, a GC-derived synthetic lipopeptide, but not TLR4 agonists including LPS or GC lipooligosaccharide enhanced HIV-1 infection of primary resting CD4+ T cells after viral entry. Pretreatment of CD4+ cells with PGN also promoted HIV infection. Anti-TLR2 Abs abolished the HIV enhancing effect of GC and Pam3C-Lip, indicating that GC-mediated enhancement of HIV infection of resting CD4+ T cells was through TLR2. IL-2 was required for TLR2–mediated HIV enhancement. PGN and GC induced cell surface expression of T cell activation markers and HIV coreceptors, CCR5 and CXCR4. The maximal postentry HIV enhancing effect was achieved when PGN was added immediately after viral exposure. Kinetic studies and analysis of HIV DNA products indicated that GC exposure and TLR2 activation enhanced HIV infection at the step of nuclear import. We conclude that GC enhanced HIV infection of primary resting CD4+ T cells through TLR2 activation, which both increased the susceptibility of primary CD4+ T cells to HIV infection as well as enhanced HIV-infected CD4+ T cells at the early stage of HIV life cycle after entry. This study provides a molecular mechanism by which nonulcerative sexually transmitted infections mediate enhancement of HIV infection and has implication for HIV prevention and therapeutics.
Sexual contact is the most common route of HIV-1 transmission. In addition to an estimated 33 million HIV-infected people worldwide in 2007, 2.7 million people have become newly infected (The Joint United Nations Programme on HIV/AIDS, report on the global HIV/AIDS epidemic 2008). In the United States, 56,300 adolescents and adults were newly infected with HIV-1 in 2006 (1). Sexually transmitted infections (STIs) are known to significantly increase the likelihood of HIV-1 transmission (2–5). STIs increase not only HIV-1 shedding in HIV-1–infected adults but also the susceptibility to HIV-1 infection in those who are HIV-1 negative (2–7). Although antibiotic chemoprophylaxis or treatment substantially decreases the incidence of STIs as well as HIV-1 shedding, it does not reduce the incidence of HIV-1 (6, 8). Understanding how STIs increase HIV transmission will provide insight into the development of new strategies to reduce the spread of HIV-1.
Gonorrhea is caused by Neisseria gonorrheae (gonococcus [GC]), a Gram-negative bacterium. The Centers for Disease Control and Prevention estimates that >700,000 persons in the United States contract new GC infections each year, with the highest reported rates of infection among sexually active teenagers, young adults, and African Americans. Several mechanisms of enhancement of HIV transmission by GC have been proposed. GC infection can enhance HIV transcription by activating HIV long-terminal repeat in transformed T cells (9). GC-infected women have more endocervical CD4+ T cells providing more targets for HIV (10). We have shown that antimicrobial peptides, such as human defensin 5 and 6, promote HIV infectivity in vitro and are induced by GC infection (11). In addition, GC infection enhances HIV infection in monocyte-derived dendritic cells (MDDCs) (12).
TLRs belong to the family of pattern recognition receptors (PRRs) and are important components of the innate immune system as the sensors of intruding pathogens (13, 14). TLR activation is known to modulate HIV infection or transmission in vitro and in vivo (15–23). Depending on specific TLR agonists and the target cells, TLR activation can either promote or inhibit HIV infection in vitro. Although TLR2 activation increases HIV infection in primary quiescent naive and memory CD4+ T cells and MDDC-mediated HIV transmission (12, 20, 21), TLR4 activation inhibits HIV infection in primary macrophages and MDDC-mediated HIV transmission (19, 20, 24, 25). TLR4 ligands, such as LPS or lipooligosaccharide (LOS), inhibit HIV infection of primary macrophages through induction of type I IFN (19, 25), whereas LPS increases the susceptibility of monocytes and PBMCs to the HIV X4 virus (26, 27). Interestingly, TLR2, 7, and 9 agonists appear to promote HIV or SIV infection in vivo (18, 28), which may be due to immune activation (18, 23, 29). GC is known to induce proinflammatory cytokines through activation of TLR2 and TLR4 (30, 31), which may play a role in increased HIV transmission. Furthermore, TLR2 is involved in GC-mediated enhancement of HIV infection in MDDCs (12).
The earliest targets during heterosexual transmission of HIV/SIV are both activated and resting CD4+ T cells in the genital mucosa (32). However, the effect of GC exposure on HIV infection of primary CD4+ T cells has not been reported. In this study, we demonstrated that GC exposure increased HIV-1 infection of primary resting CD4+ T cells, and that TLR2 was involved in this HIV enhancing effect. Furthermore, a major component of TLR2-mediated enhancement of HIV infection of primary CD4+ T cells occurred at the step of nuclear import. These results offer an additional molecular mechanism of GC-mediated increased HIV transmission, which is important for the development of preventative strategies against HIV.
Materials and Methods
Reagents
Recombinant human IL-2 was purchased from R&D Systems (Minneapolis, MN). PHA, Escherichia coli O55:B5 LPS, and PGN were from Sigma-Aldrich (St. Louis, MO). The Pam3C-Lip (from EMC Microcollections, Tuebingen, Germany), based on the N-terminal sequence of the N. gonorrheae F62 Lip protein (sequence CGGEKAAEAPAAEAS), contains a tripalmitoyl-S-glyceryl cysteine at the N-terminus. The synthetic lipopeptide Pam3CSK4 and muramyl peptides (MDP) were purchased from InvivoGen (San Diego, CA). L-731,988 was provided by Merck (Whitehouse Station, NJ). Zidovudine (AZT) was obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH. HIV-1IIIB and HIV-1BaL were purchased from Advanced Biotechnologies (Columbia, MD). Abs purchased from BD Biosciences (San Jose, CA) were PE-conjugated mouse anti-human CD25 (clone no. M-A251), FITC-conjugated mouse anti-human CD69 (FN50), APC-conjugated mouse anti-human CXCR4 (12G5), APC-conjugated mouse anti-human CCR5 (3A9), FITC-conjugated mouse IgG1, PE-conjugated mouse IgG1 (X40), and APC-conjugated mouse IgG2a (X39). Mouse anti-human TLR2 Ab (TL2.1) was purchased from eBio-science (San Diego, CA). FITC-conjugated mouse anti-human TLR2 Ab (TL2.1) was obtained from Imgenex (San Diego, CA).
Cell culture
PBMCs from normal healthy blood donors were isolated by Ficoll-Hypaque gradient centrifugation. CD4+ T cells were prepared from PBMCs by negative selection using CD4+ T cell isolation kit from Miltenyi Biotec (Auburn, CA). CD14+ cells were positively selected from PBMCs by using CD14+ cell isolation kit (Miltenyi Biotec). The purity was >98% based on flow cytometry analysis. Freshly isolated CD4+ T cells were cultured in RPMI-1640 media with 10% FBS in the absence of IL-2 overnight at 37°C before use. The cell surface levels of CD25 and CD69 decreased from ~33.8% and 13.9%, respectively, to 12.1% for CD25 and 1.1% for CD69 after overnight culture in the absence of IL-2. Cell cycle analysis by propidium iodide staining indicated that 99.9% of freshly isolated primary CD4+ T cells as well as cells after overnight culture in the absence of IL-2 were at the Go/G1 stage. To prepare PHA-activated CD4+ T cells, purified CD4+ T cells were treated with PHA at 5 μg/ml and IL-2 at 50 IU/ml for 3 d. Activated CD4+ T cells were washed three times with PBS and maintained in complete media with IL-2 before use.
N. gonorrheae
N. gonorrheae (ATCC strain 43069 and clinical isolate strains GC56, F62, and FA1090) was grown on chocolate agar for 48 h at 37°C in 5% CO2. For some experiments, phenotypic colonial variants of strain FA1090 either expressing or not expressing pilus (Pil) and Opa outer membrane proteins (Pil+/Opa+ or Pil−/Opa−, respectively) were selected by using a dissecting microscope and standard selection criteria as described previously (33). Colonies were scraped and resuspended in PBS. After bacteria were washed once with PBS, bacterial cell density was adjusted to an OD600nm equal to 0.1, corresponding to 108 CFU/ml bacteria. When fixed bacteria were used, bacteria were inactivated with 1% paraformaldehyde in PBS for 1 h at room temperature. After fixation, the bacteria were washed five times with PBS to remove residual paraformaldehyde. LOS from GC clinical isolate strain DOV was isolated by hot phenol extraction as described previously (30).
HIV-1 infection
For HIV infection, primary CD4+ T cells were cultured in RPMI-1640 media supplemented with 10% FBS and 50 IU/ml IL-2 unless otherwise described. To determine the effect of GC and various PRR ligands on HIV-1 infection after viral entry, primary CD4+ T cells (1 × 106 per sample) were first infected with replication competent CXCR4 (X4)-using virus HIV-1IIIB or CCR5 (R5)-using virus HIV-1BaL at multiplicity of infection (MOI) of 0.05 for 2 h. After washing off unbound viruses, infected cells were incubated with GC or TLR/ nucleotide-binding and oligomerization domain 2 (NOD2) ligands at 37°C in the presence of IL-2. HIV production in culture supernatants was measured by p24 ELISA kit (SAIC-Frederick, Frederick, MD).
To examine HIV-1 infection of resting CD4+ T cells with pretreatment of PGN, CD4+ T cells were incubated with PGN (20 μg/ml) in the presence of IL-2 (50 IU/ml) at 37°C for 3 d. Cells were washed with PBS and then exposed to HIV-1IIIB or HIV-1BaL at MOI of 0.05 for 2 h. The infected cells were washed and then cultured at 37°C in the presence of IL-2. HIV production in the culture supernatant was measured with ELISA.
For a single-cycle infection assay, replication-defective pseudotyped HIV-1VSV-G luciferase reporter viruses were produced as described previously (34, 35). Briefly, HEK293T cells were cotransfected with a plasmid encoding the envelope deficient HIV-1 NL4-3 virus with the luciferase reporter gene inserted into nef (pNL4-3.Luc.R-E; gift of N. Landau, NIH AIDS Research and Reference Reagent Program, Germantown, MD) and a pSV plasmid expressing the VSV-G gp (gift of D. Trono, University of Geneva, Geneva, Switzerland). The supernatant medium was collected 48 h after transfection, and filtered. HIV-1 p24 Ag concentration of viral stocks was determined by ELISA.
Primary CD4+ T cells were infected with replication-defective pseudotyped HIV-1VSV-G luciferase reporter viruses at 37°C for 2 h. After washing off unbound viruses, infected cells were treated with GC or TLR2 agonists in the presence of IL-2 at 37°C for 4 d. The cells were lysed with passive cell lysis buffer (Promega, Madison, WI). Luciferase activity (in relative light units) was measured on an EG&G Berthold MiniLumat LB 9506 luminometer (Berthold Technologies, Bad Wildbad, Germany).
Reverse transcription-PCR analysis
Total RNA was isolated from resting CD4+ T cells, PHA-activated CD4+ T cells, or CD14+ cells with RNeasy isolation kit (Qiagen, Valencia, CA). To synthesize first-strand cDNA, total RNA (500 ng), oligo d(T)16 (Invitrogen, Carlsbad, CA) at 25 μg/ml and 0.5 mM dNTP in a total volume of 12 μl were incubated at 65°C for 5 min and quick chilled on ice. Reverse transcription (RT) was performed at 42°C for 50 min using SuperScript II RT (Invitrogen) according to the manufacturer’s protocol. Primers used were listed as follows: TLR2 forward, 5′-AGGTGACACTATAGAA-TACTGGAGCCCATTGAGAAAAA-3′; TLR2 reverse, 5′-GTACGACT-CACTATAGGGACGCAGCTCTCAGATTTACCC-3′; TLR4 forward, 5′-AGGTGACACTATAGAATACCATAAAAGCCGAAAGGTGA-3′; TLR4 reverse, 5′-GTACGACTCACTATAGGGACACCTTCTGCAGGACAAT-GA-3′; NOD2 forward, 5′-AGGTGACACTATAGAATACCCTGCTCT-TCAACCTTCTG-3′; NOD2 reverse, 5′-GTACGACTCACTATAGGGA-CGCTTCCTCAGGTACAGCTC-3′; GAPDH forward, 5′-AGGTGACA-CTATAGAATACTCTCTGCTCCTCCTGTTCG-3′; GAPDH reverse, 5′-GTACGACTCACTATAGGGAACGACCAAATCCGTTGACTC-3′; β-actin forward, 5′-GTGGACTTGGGAGAGGACTG-3′; β-actin reverse, 5′-ACTGGAACGGTGAAGGTGAC-3′. The PCR reaction contained Qiagen Tag master mix, 0.2 μM each primer set, and 3 μl RT reaction. After an initial incubation at 94°C for 3 min, 35 cycles of amplification were performed as follows: denaturation for 30 s at 94°C, annealing for 30 s at 56°C, and 30 s at 72°C, and a final extension cycle of 7 min at 72°C in a DNA thermal cycler (Perkin Elmer 480). PCR products were separated by electrophoresis on a 2% agarose gel and analyzed with the FluroChem 8800 imaging system (Alpha Innotech, San Leandro, CA).
Flow cytometry
Cells were stained with flurochrome-conjugated mAbs specific for T cell activation markers and HIV-1 coreceptors. Appropriate isotype-matched mAbs conjugated with PE, FITC, or APCs were used as negative controls. Results were acquired with CellQuest software (BD Biosciences, San Jose, CA) on a FACScalibur (BD Biosciences) and analyzed with Flow Jo (Tree Star, Ashland, OR).
Quantitative real-time PCR analysis of HIV-1 DNA
DNA was extracted from resting CD4+ T cells with infection by pseudo-typed HIV-1VSV-G luciferase reporter viruses using the DNeasy Blood and Tissue Kit (Qiagen). The level of HIV RT DNA products was determined by quantitative real-time PCR analysis. Each PCR reaction contained 100 ng genomic DNA, each primer at 0.2 μM, SYBR Green Master Mix (Qiagen). The primer sequences for HIV-1 RT DNA products were as follows: R/U5 forward, M667 (5′-GGCTAACTAGGGAACCCACTG-3′); R/U5 reverse, AA55 (5′-CTGCTAGAGATTTTCCACACTGAC-3′); R/ gag forward, M667 (above); R/gag reverse, M661 (5′-CCTGCCTCGA-GAGAGCTCCACACTGAC-3′) (36). β-actin was amplified to normalize the DNA input, the primer sequences were as follows: actin-forward (5′-TGCGTGACATTAAGGAGAAG-3′); actin-reverse (5′-GCTCGTAG-CTCTTCTCCA-3′). Plasmids pNL4-3.Luc was used to generate standard curves for HIV RT DNA products. In every experiment, a standard curve for RT products derived from serial dilution of plasmids containing the target sequence and ranging from 101 to 108 copies was measured in triplicate. The level of HIV closed 2-long terminal repeat (c2-LTR) circles was determined by using primers LR31: 5′-CTTGCCTTGAGTGCTT-CAAG-3′; and LR32: 5′-TGCCAATCAGGGAAGTAGCC-3′ (37). Plasmid pTA2LTR (38) was used to generate a standard curve. The detection limit was 10 copies. PCR cycling conditions included a 95°C denaturation for 10 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Reactions were carried out and analyzed using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems).
Results
GC exposure enhances HIV-1 replication in primary resting CD4+ T cells
To determine the effect of GC exposure on HIV-1 replication in primary CD4+ T cells, CD4+ T cells were infected with R5 HIV-1BaL at an MOI of 0.05 for 2 h, washed to remove unbound virus, and then exposed to live or fixed GC ATCC strain 43069 at various MOIs in the presence of IL-2. HIV-1 production was determined by measuring virus particles released into media with HIV-1 p24 ELISA. At day 14 postinfection, live GC at an MOI of 100 increased HIV-1 production in primary CD4+ T cells by 1.7-fold (Fig. 1A), whereas fixed GC at an MOI of 100 enhanced virus production by 5.8-fold (Fig. 1B). To determine whether GC exposure could affect proliferation of CD4+ T cells, primary resting CD4+ T cells were exposed to GC for 3 d in the presence of IL-2. Cell proliferation was determined by MTS assay. No significant difference in proliferation of primary resting CD4+ T cells was observed between cells with or without exposure to either live or fixed GC (Fig. 1C). These results demonstrated that GC exposure increased HIV-1 production after viral entry but did not induce proliferation in CD4+ T cells.
FIGURE 1.
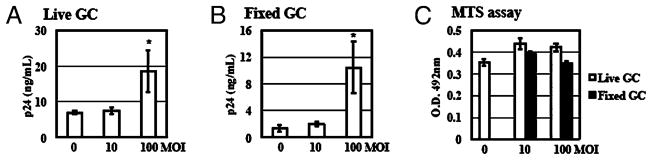
GC exposure enhances HIV-1 infection of primary resting CD4+ T cells. Primary resting CD4+ T cells were infected with HIV-1BaL at an MOI of 0.05 at 37°C for 2 h. After washing off unbound virus, infected cells were exposed to live (A) or fixed (B) GC (ATCC strain 43069) at various MOIs in the presence of IL-2. HIV production was determined by measuring the level of HIV capsid protein p24 at day 14 after viral infection using HIV p24 ELISA. C, Primary resting CD4+ T cells (1 × 104 cells per well) were exposed to live GC or fixed GC at 37°C for 72 h in the presence of IL-2. Cell proliferation was examined by the MTS assay (Promega CellTiter96 aqueous one solution cell proliferation assay). Data are means ± SD of triplicate sample. Difference in HIV infection between samples without GC exposure and GC-exposed samples at an MOI of 100 is significant as calculated by the two-tailed, paired Student t test. *p < 0.05. There is no difference in cell proliferation between samples with or without GC exposure (p > 0.05). Data represent three independent experiments.
To determine whether the HIV enhancing effect of GC was strain specific, the effect of three clinical GC isolates (GC56, F62, and FA1090) on HIV infection was studied using a single-cycle infection assay, measuring the steps of the HIV life cycle from entry to gene expression. A replication-defective HIV-1 luciferase reporter virus pseudotyped with VSV-G protein, which is not dependent on HIV-1 coreceptors for virus entry, was used to maximize HIV infection efficiency. HIV-infected cells were exposed to fixed GCs at various MOIs in the presence of IL-2 for 4 d before measurement of luciferase activity. Similar to the strain in Fig. 1, these three GC clinical isolates also enhanced HIV infection of primary resting CD4+ T cells (Fig. 2A). N. gonorrheae strains F62 and FA1090 at an MOI of 10 enhanced HIV infection of resting CD4+ T cells by 34.2- and 30.7-fold, respectively, whereas a higher MOI of 100 of N. gonorrheae strain GC56 was required to reach the similar enhancement of HIV (26-fold).
FIGURE 2.

Multiple GC strains enhanced HIV-1 infection of primary resting CD4+ T cells. A, Primary resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus at 37°C for 2 h. After washing off unbound virus, infected cells were exposed to various strains of fixed GC in the presence of IL-2 for 4 d before measuring luciferase activity. There is a significant difference between samples with and without GC-exposure. *p < 0.05. B, Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus at 37°C for 2 h, washed, and then treated with FA1090 Pil+/Opa+ or FA1090 Pil−/Opa− in the presence of IL-2. Luciferase activity was measured at 4 d postinfection. The difference between cells treated with FA1090 Pil+/Opa+ and FA1090 Pil−/Opa− is significant. #p < 0.05. C, Resting CD4+ T cells (1 × 104 cells per well) were exposed to various strains of fixed GC at MOIs of 1, 10, and 100 at 37°C for 72 h in the presence of IL-2. Cell proliferation was examined by the MTS assay. Data are means ± SD of triplicate sample. Data represent two independent experiments.
Pil and Opa are surface-exposed structures of GC that play a role in gonococcal adherence to and invasion of host cells. Both components are involved in GC-mediated activation and proliferation of CD4+ T cells in response to TCR signaling (39, 40). However, a recent report demonstrates that, in conjunction with anti-CD3 Abs and IL-2 treatment, both Opa+ and Opa− bacteria induce T cell proliferation to a similar extent (41). To determine the role of Pil and Opa in GC-mediated HIV enhancement, the effect of FA1090 Pil+/Opa+ and FA1090 Pil−/Opa− on HIV infection was examined using single-cycle infection assay. Both bacteria promoted HIV infection despite their distinct phenotypes (Fig. 2B); however, the degree of HIV enhancement by FA1090 Pil−/Opa− was significantly reduced, suggesting that Pil and Opa could modulate the extent of the HIV enhancing effect.
To delineate whether GC exposure affected T cell proliferation, which in turn facilitated HIV infection, primary resting CD4+ T cells were exposed to various fixed GC strains at MOIs of 1, 10, and 100 for 3 d in the presence or absence of IL-2. Cell proliferation was determined by MTS assay. We did not observe significant differences in proliferation of resting CD4+ T cells between cells with or without GC exposure (Fig. 2C). In addition, IL-2 did not affect cell proliferation of GC-exposed CD4+ T cells (Fig. 2C). These results indicated that GC exposure did not affect resting CD4+ T cell proliferation, even in the presence of IL-2 signaling.
TLR2 activation promotes HIV-1 infection of primary resting CD4+ T cells
GC expresses ligands for multiple PRRs, including TLR2, TLR4, and NOD2 (30, 42, 43). To investigate the role of PRR signaling in GC-induced enhancement of HIV-1 infection, we first examined the effect of TLR2, TLR4, and NOD2 agonists on HIV-1 infection of primary resting CD4+ T cells. CD4+ T cells were infected with X4 HIV-1IIIB (Fig. 3A) or R5 HIV-1BaL (Fig. 3B) for 2 h. After washing off unbound virus, infected cells were treated with various agonists for TLR2 (PGN), TLR4 (LPS and GC LOS), or NOD2 (MDP) and cultured in complete media with IL-2 for 7 d. HIV replication was determined by HIV p24 ELISA. The TLR2 agonist PGN at a concentration of 20 μg/ml significantly increased the replication of both X4 virus (12.8-fold) and R5 virus (13-fold) after viral entry. Although the degree of the HIV enhancing effect of PGN ranged from 2- to 46-fold depending on CD4+ T cell donor, TLR2 activation consistently increased HIV infection of primary CD4+ T cells. The NOD2 ligand MDP at 20 μg/ml exhibited a moderate effect on replication of X4 virus and no effect for that of R5 virus. In contrast, TLR4 ligands, such as LPS (1 μg/ml) and GC LOS (0.1 μg/ ml), did not exhibit any effect on HIV-1 replication in CD4+ T cells from any donor.
FIGURE 3.

TLR2 activation promotes HIV replication in primary resting CD4+ T cells. A and B, Primary resting CD4+ T cells were infected with X4 HIV-1IIIB or R5 HIV-1BaL at an MOI of 0.05 at 37°C for 2 h. After washing off unbound virus, infected cells were then treated with LPS (1 μg/ml), MDP (20 μg/ml), PGN (20 μg/ml), or GC DOV LOS (0.1 μg/ml) in the presence of IL-2 at 37°C. Infected cells without treatment were included as a control. The p24 level in the culture supernatant was determined on day 7 postinfection. C, Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus at 37°C for 2 h and then treated with Pam3CSK4 at 5 μg/ml with IL-2 for 4 d before measuring luciferase assay. D, Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus and then exposed to Pam3C-Lip at various concentrations with IL-2 for 4 d before measuring luciferase assay. E, Resting CD4+ T cells were treated with PGN (20 μg/ml) for 3 d, followed by washing. PGN-treated primary CD4+ cells were infected with HIV-1IIIB or HIV-1BaL. The p24 level in the culture supernatant was determined on day 7 postinfection. Data are means ±SD of triplicate sample and represent seven independent experiments. The difference in HIV infection between samples with and without TLR2 agonists treatment is significant as calculated by the two-tailed, paired Student t test. *p < 0.05.
To further investigate the effect of other TLR2 agonists in addition to PGN on HIV infection of primary resting CD4+ T cells, CD4+ T cells were exposed to pseudotyped HIV-1vsv luciferase reporter virus for 2 h, washed, and then treated with synthetic TLR2 agonists Pam3CSK4 and Pam3C-Lip, a lipopeptide derived from the GC Lip protein (44). Both synthetic TLR2 agonists promoted HIV infection of resting CD4+ T cells after viral entry (Fig. 3C, 3D). Taken together, these results indicated that TLR2 signaling but not TLR4 or NOD2 played a role in GC-mediated enhancement of HIV-1 infection after viral entry.
Pretreatment of quiescent naive and memory CD4+ T cells with the synthetic TLR2 agonist Pam3CSK4 has been reported to increase the susceptibility of cells to infection of both X4 virus and R5 viruses (21). Pam3CSK4 and PGN activate TLR2 in association with TLR1 and TLR6, respectively (45–47), although hetero-dimerization of TLR2 with TLR1 or TLR6 does not lead to differential signaling (48, 49). We examined whether pretreatment of resting CD4+ T cells with PGN could enhance HIV infection. Primary resting CD4+ T cells were pretreated with PGN at 20 μg/ ml for 3 d in the presence of IL-2. Cells were washed and then infected with HIV-1IIIB or HIV-1BaL. Similar to the previous report using a TZM-bl cell reporter system (21), we found that pre-treatment of primary CD4+ T cells with PGN increased viral infection for both X4 virus (by 2.4-fold) and R5 virus (by 2.1-fold) (Fig. 3E). However, the enhancing effect on HIV production was moderate when cells were pretreated with PGN compared with viral production from cells that were treated with PGN after HIV entry (Fig. 3A, 3B). These results indicated that TLR2 activation could enhance HIV-1 infection of primary CD4+ T cells through multiple mechanisms.
HIV enhancing effect of GC is mediated through TLR2 signaling
We observed differential effects of TLR2, TLR4, and NOD2 agonists on HIV infection of primary resting CD4+ T cells (Fig. 3). To investigate whether the levels of these PRRs contributed to their differential HIV enhancing effect, we examined gene expression of TLR2, TLR4, and NOD2 in primary resting CD4+ T cells by RT-PCR analysis. In agreement with previous reports (50, 51), gene expression of TLR2 and NOD2 was detectable in primary resting CD4+ T cells (Fig. 4A). After PHA activation, expression of TLR2 and NOD2 was elevated. In contrast, gene expression of TLR4 was not detectable in both primary resting and PHA-activated CD4 T cells, whereas it was expressed in monocytic CD14+ cells that are known to express TLR4 (Fig. 4A).
FIGURE 4.

Enhancement of HIV infection of primary resting CD4+ T cells by GC is mediated through TLR2 signaling pathway. A, Gene expression of NOD2, TLR2, and TLR4 in resting CD4+ T cells and PHA-activated CD4+ T cells was determined by RT-PCR analysis. Total RNA from CD14+ cells was included as a control for analysis of TLR4 gene expression. B, To determine the role of TLR2 in GC-mediated enhancement of HIV infection, resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus at 37°C for 2 h. After washing off unbound virus, infected cells were treated with anti-TLR2 Ab or isotype control Ab at 10 μg/ml for 1 h. Cells were exposed to fixed GC (ATCC strain 43069) at an MOI of 50 with IL-2 and cultured for 4 d before measurement of luciferase activity. C, Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus and treated with anti-TLR2 Ab or isotype control Ab as described previously. Cells were then exposed to Pam3C-Lip at various concentrations in the presence of IL-2 for 4 d. Data are means ±SD of triplicate sample and represent two independent experiments. There is a significant difference between samples with and without GC or Pam3C-Lip treatment (*p < 0.05) as well as between stimulated cells in the presence or absence of anti-TLR2 Ab (#p < 0.05). There is no difference between stimulated samples in the presence or absence of isotype control Ab (**p < 0.05).
To establish the involvement of TLR2 in GC-mediated enhancement of HIV infection, we used Abs against TLR2 to block the HIV enhancing effect of GC- and GC-derived lipopeptide Pam3C-Lip using a single-cycle infection assay. HIV-infected resting CD4+ T cells were treated with anti-TLR2 Abs for 1 h before GC exposure. An isotype control Ab was included as a negative control. After 4 d incubation, cells were lysed, and the luciferase activity was measured. Fixed GC promoted HIV-1 infection by 8.2-fold in a single-cycle infection assay (Fig. 4B). Anti-TLR2 or isotype control Abs did not have any effect on HIV infection in the absence of GC exposure (data not shown). Anti-TLR2 Abs completely abolished the HIV enhancing effect of GC, whereas isotype control Abs did not exhibit any significant effect (Fig. 4B). Similarly, Pam3C-Lip at 0.2 and 1 μg/ml enhanced HIV infection by 1.8-fold and 5.6-fold, respectively (Fig. 4C). This enhancing effect was blocked by anti-TLR2 Ab but not isotype control Ab (Fig. 4C). These results demonstrated that TLR2 was involved in GC-mediated enhancement of HIV infection of primary resting CD4+ T cells after viral entry.
TLR2 activation does not enhance postentry HIV-1 infection in activated CD4+ T cells and the HIV enhancing effect is dependent on IL-2
To examine whether TLR2 activation exerted any effect on HIV infection of activated CD4+ T cells, PHA-activated CD4+ T cells were exposed to pseudotyped HIV-1VSV-G luciferase reporter virus for 2 h before treatment with PGN. Primary resting CD4+ T cells from the same donor were included as a comparison. We observed enhancement of HIV-1 infection by TLR2 activation in resting but not PHA-activated CD4+ T cells, despite our finding that activated CD4+ T cells, expressing TLR2, were highly susceptible to HIV-1 infection (Fig. 5A).
FIGURE 5.
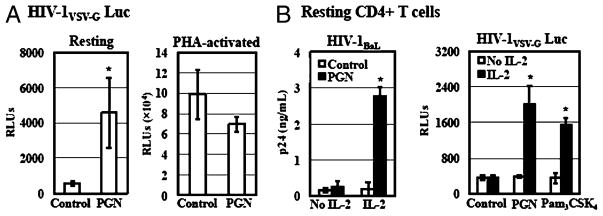
TLR2-mediated HIV enhancement occurs in resting but not PHA-activated CD4+ T cells and IL-2 is required for the HIV enhancing effect. A, Resting CD4+ T cells were activated with or without PHA and IL-2 for 3 d. Resting CD4+ T cells with or without PHA activation were infected with pseudotyped HIV-1VSV-G luciferase reporter virus for 2 h. Infected cells were then treated with PGN at 20 μg/ml in the presence of IL-2 and cultured for 4 d before measurement of luciferase activity. B, Resting CD4+ T cells were infected with HIV-1BaL at an MOI of 0.05 or pseudotyped HIV-1VSV-G luciferase reporter virus. Infected cells were treated with Pam3CSK4 at 5μg/ml or PGN at 20 μg/ml in the presence or absence of IL-2. HIV-infected cells without treatment were included as a control. Viral production by primary CD4+ T cells infected with HIV-1BaL was determined by measuring the p24 level in the culture supernatant at day 7 postinfection. Luciferase activity was determined at day 4 postinfection for the single-cycle infection assay. Data are means ±SD of triplicate sample and represent three independent experiments. *p < 0.05.
IL-2 is an important factor for T cell proliferation and survival as well as HIV replication in primary CD4+ T cells (52, 53). In addition, IL-2 is required for the TLR2-mediated induction of IFN-γ by Th1 effector cells (54). To investigate the role of IL-2 in TLR2-mediated enhancement of HIV infection, the HIV enhancing effect of PGN and Pam3CSK4 was determined in the presence or absence of IL-2. IL-2 treatment alone did not increase HIV infection. In the absence of IL-2, PGN or Pam3CSK4 did not enhance HIV infection of primary resting CD4+ T cells in both multiple-round infection assays and single-cycle infection assays (Fig. 5B). Enhancement of HIV infection was observed only in the presence of both IL-2 and TLR2 agonists. This result indicated that IL-2 signaling was required to achieve enhancement of HIV infection in response to TLR2.
GC and TLR2 agonists induce expression of T cell activation markers and HIV-1 coreceptors CCR5 and CXCR4 on CD4+ T cells
The synthetic TLR2 agonist Pam3CSK4 has been shown to induce T cell activation markers including CD69, CD25, ICAM-1, and HLA-DR on both quiescent naive and memory CD4+ T cells (21). Plant et al. demonstrated that piliated live GC exposure increased the TCR signaling-mediated activation and proliferation of primary CD4+ T cells (39). Because activation of T cells facilitates productive HIV-1 infection, we examined the effect of GC or PGN on expression of T cell activation markers. Primary resting CD4+ T cells were incubated with fixed GC (ATCC strain 43069) at an MOI of 50 or PGN at 20 μg/ml for 3 d in the presence of IL-2 before measuring cell surface expression of CD25 and CD69 by FACS analysis. PHA-activated CD4+ T cells were included as a positive control. As expected, PHA upregulated CD25 and CD69 expression significantly (Table I). In agreement with the previous reports using a synthetic TLR2 agonist (21) or GC (39), PGN and GC increased the level of cell surface CD25 and CD69 on primary resting CD4+ T cells (Table I).
Table I.
Effect of TLR2 activation on cell activation and coreceptor expression
| Activation Markers (%)
|
Coreceptors (%)
|
CXCR4 (MFI) | ||||
|---|---|---|---|---|---|---|
| Isotypes | CD25 | CD69 | CXCR4 | CCR5 | ||
| Medium | 1.6 ± 0.7 | 11.7 ± 0.6 | 0.8 ± 0.4 | 91.0 ± 0.2 | 2.7 ± 0.4 | 78.4 ± 4.1 |
| PGN | 1.1 ± 0.8 | 16.6 ± 2.8a | 11.0 ± 1.1a | 90.4 ± 1.0 | 9.1 ± 5.2a | 102.6 ± 4.0a |
| Fixed GC | 3.0 ± 1.0 | 19.6 ± 3.0a | 13.1 ± 0.8a | 92.9 ± 0.5 | 9.5 ± 2.3a | 112.9 ± 6.5a |
| PHA | 2.7 ± 1.3 | 97.6 ± 0.6a | 95.9 ± 1.7a | 96.5 ± 3.1 | 2.0 ± 1.4 | 116.7 ± 5.5a |
Primary resting CD4+ T cells were incubated with PGN or fixed GC for 3 d in the presence of IL-2. PHA-activated cells and mock-treated cells were included as controls. The percentage of cells expressing activation markers or HIV coreceptors is determined by FACS analysis. The data shown are mean ± SD of three independent experiments.
There is a significant difference between ligand-treated and mock-treated samples, p < 0.05.
Pretreatment of CD4+ T cells with PGN led to enhancement of HIV-1 infection using both X4 and R5 viruses (Fig. 3E). We hypothesized that PGN upregulates CXCR4 and CCR5 expression that in turn results in increased HIV-1 susceptibility of target cells. The effect of GC or PGN on expression of cell surface CXCR4 and CCR5 on CD4+ T cells was determined by FACS analysis. PHA-activated CD4+ T cells were included as a comparison. GC and PGN treatments increased the number of CCR5+ cells and the level of cell surface CXCR4 (Table I). These results suggest that pretreatment of cells with GC and PGN may promote HIV infection through induction of HIV coreceptors. It is noted that Pam3CSK4 also induces cell surface expression of CCR5 on naive and memory T cells (21).
The maximal HIV enhancing effect is achieved when TLR2 agonist was added at an early stage of the HIV life cycle
To dissect the stages of HIV infection that were enhanced by TLR2 activation after viral entry, we first studied the kinetics of the HIV life cycle in CD4+ T cells in the presence of PGN using a single-cycle infection assay. HIV-infected resting CD4+ T cells were cultured in presence of IL-2 and treated with PGN at 2, 24, 48, and 72 h after viral infection. The luciferase activity was determined at day 4 after viral infection. The maximal HIV-1 enhancing effect was observed when PGN was added at 2 h postinfection (Fig. 6A). The degree of the HIV enhancing effect decreased when PGN was added at later time points of the HIV life cycle. PGN did not affect HIV infection when added at 72 h postinfection, possibly because of degradation of viral DNA. To determine whether prolonged incubation could increase the level of HIV infection in samples with PGN treatment at later time points, infected cells were treated with PGN at 48 and 72 h postinfection and luciferase activity was determined on day 6 and day 7 postinfection, respectively. There was an increase in the level of HIV enhancement when infected cells with PGN treatment at 48 h postinfection were cultured for an additional 2 d, although this HIV enhancing effect was not as pronounced compared with that observed in cells with PGN treatment at 2 h postinfection. Prolonged culture of cells with PGN treatment at 72 h postinfection did not restore the level of enhancement of HIV infection. To ensure that resting CD4+ T cells at later time points were still viable in the presence of IL-2, the viability of resting CD4+ T cells at different time points postinfection was determined by MTS assay. There was no significant difference among CD4+ T cells at 2, 24, 48, or 72 h after HIV exposure in the presence of IL-2 (Fig. 6B). This result indicated that the lack of HIV enhancement by TLR2 at 72 h postinfection was not due to decreased cell viability.
FIGURE 6.
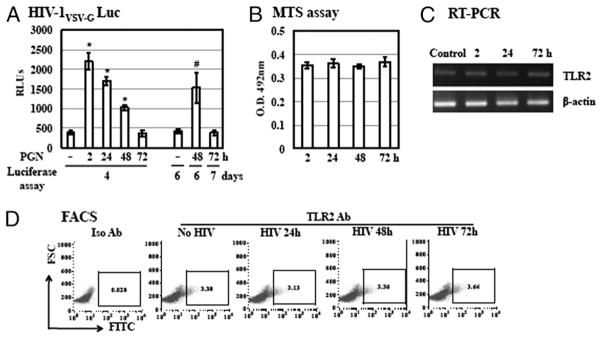
The maximal HIV enhancing effect of the TLR2 activation is achieved when PGN is added at an early stage of the HIV life cycle. A, Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus for 2 h. Infected cells were cultured with IL-2 and then treated with PGN at 20 μg/ml at 2, 24, 48, or 72 h postinfection before measurement of luciferase activity at day 4, 6, or 7 postinfection. There is a significant difference between samples without PGN and with PGN at 2, 24, or 48 h postinfection (*p < 0.05). When samples were harvested at day 6 postinfection the difference between cells with PGN at 48 h postinfection and without treatment is significant (#p < 0.05). B, The viability of resting CD4+ T cells postinfection with pseudotyped HIV-1VSV-G luciferase reporter virus was assessed by MTS assay at 2, 24, 48, and 72 h after viral infection. C, Total RNA from resting CD4+ T cells with infection by pseudotyped HIV-1VSV-G reporter virus for 2, 24, or 72 h was prepared. TLR2 gene expression of TLR2 was analyzed by RT-PCR. Samples from mock-infected resting CD4+ T cells were also included as a control. D, Cell surface expression of TLR2 on HIV-infected resting CD4+ T cells was determined by FACS analysis. Data are means ±SD of triplicate sample and represent three independent experiments.
We further investigated whether the lack of responsiveness to TLR2 stimulation at 72 h after viral infection was due to down-regulation of TLR2 expression by HIV. The mRNA level of TLR2 expression from HIV-infected CD4+ T cells at 2, 24, and 72 h after viral infection was first determined by RT-PCR analysis. There was no change in the level of TLR2 gene expression on HIV infection (Fig. 6C). We then examined cell surface expression of TLR2 on HIV-infected resting CD4+ T cells by FACS analysis. HIV infection did not alter the expression level of cell surface TLR2 (Fig. 6D). These results indicated that the lack of HIV enhancement by TLR2 activation at 72 h after viral infection was not due to downregulation of cell surface TLR2.
TLR2 activation promotes HIV infection after RT
To dissect the molecular mechanism by which TLR2 activation enhanced HIV infection after viral entry, we first studied the effect of TLR2 activation in the presence of inhibitors, AZT and L-731,988, that specifically blocked the steps of RT and integration, respectively, using a single-cycle cycle infection assay. AZT was added to primary CD4+ T cells at 1 h before infection, during infection, or at 2, 8, and 24 h postinfection. PGN was added at 2 h postinfection. AZT blocked TLR2-mediated enhancement of HIV-1 infection when the inhibitor was added before infection, during infection, and at 2 h postinfection. AZT had no effect on the HIV enhancing effect when added at 8 and 24 h postinfection (Fig. 7A). L-731,988 blocked the HIV enhancing effect of PGN when added at 2 and 24 h postinfection (Fig. 7B). The intergrase inhibitor did not exhibit any effect on TLR2-mediated HIV enhancement when added at 48 h postinfection, suggesting that HIV integration was completed at this time point. Because there was no productive HIV infection in primary resting cells without TLR2 activation, even in the presence of IL-2, the levels of luciferase activity from samples treated with or without inhibitors were the same as those from uninfected cells (data not shown).
FIGURE 7.
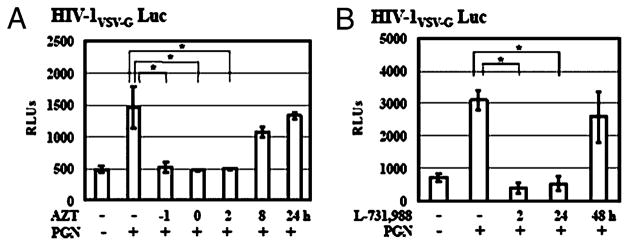
The kinetics of HIV infection in TLR2-activated resting CD4+ T cells in the presence of inhibitors for reverse transcriptase and integrase. A, Resting CD4+ T cells were infected with pseudotyped HIV-VSV-G luciferase reporter virus for 2 h. Infected cells were treated with AZT at 5 μM at 1 h before infection (−1 h), at the same time during HIV exposure (0 h) or at 2, 8, and 24 h postinfection. PGN at 20 μg/ml was added at 2 h after viral infection. Infected cells were cultured in the presence of IL-2 for 4 d before measurement of luciferase activity. B, In a single-cycle infection assay, resting CD4+ T cells were infected with pseudotyped HIV-1 reporter virus for 2 h, treated with PGN at 20 μg/ml, and cultured in complete media with IL-2. L-731,988 at 10 μM was added to cell culture at 2, 24, and 48 h postinfection. Luciferase activity was measured on day 4 postinfection. Data are means ± SD of triplicate sample and represent three independent experiments.
To further delineate the specific stages of the HIV-1 life cycle altered by TLR2 activation, we analyzed HIV DNA products representing HIV RT and nuclear import during HIV infection. Samples with GC exposure were also included as a comparison. DNA was prepared from HIV-infected primary resting CD4+ T cells with or without exposure to fixed GC or TLR2 agonists, Pam3CSK4 and PGN, at various time points postinfection. The levels of HIV early and late RT products were determined by quantitative real-time PCR analysis. Results in Fig. 8A revealed that there was no significant difference in the level of HIV early or late RT products from cells with or without exposure to GC or TLR2 agonists (data not shown for PGN).
FIGURE 8.

TLR2 activation and GC exposure promote HIV infection at the step of nuclear import. Resting CD4+ T cells were infected with pseudotyped HIV-1VSV-G luciferase reporter virus and then treated with Pam3CSK4 at 5 μg/ml or fixed GC (ATCC strain 43069) at an MOI of 50 in the presence of IL-2. DNA was prepared from TLR2-activated or GC-exposed HIV-infected cells at 0, 6, 12, 24, and 48 h postinfection. DNA from mock-infected primary resting CD4+ T cells was also isolated as a control. A, Quantitation of HIV-1 early strong-stop (R/U5) and late full-length (R/gag) RT products was performed by quantitative real-time PCR analysis. Data are means ± SD of triplicate sample and represent three independent experiments. Differences in the levels of early and late RT products between samples with or without exposure to GC or TLR2 agonists are not significant as calculated by the two-tailed, paired Student t test, p < 0.05. B, To assess whether TLR2 activation or GC exposure affected HIV-1 nuclear import, quantitative real-time PCR analysis was performed to measure c2-LTR circles in infected CD4+ T cells with or without Pam3CSK4 or fixed GC treatment for 12, 24, and 48 h. Plasmid pTA2LTR at different copy numbers was used to generate a standard for c2-LTR circles detection. The detection limit of the PCR analysis was 10 copies. There is a significant difference between samples with or without exposure to TLR2 agonists or GC (*p < 0.05) at 24 and 48 h postinfection.
To examine whether TLR2 activation promoted HIV replication at the step of nuclear import, we analyzed the level of c2-LTR circles, a marker of nuclear import (55). Primary resting CD4+ T cells are known to have low copy numbers of c2-LTR circles (56). At 12 h after viral infection, there were no detectable c2-LTR circles in samples with or without exposure to GC or TLR2 agonists, such as Pam3CSK4 and PGN (Fig. 8B, data not shown for PGN). At 24 h postinfection, the level of c2-LTR circles in HIV-exposed cells was below the detection limit (10 copies), whereas ~27 and 21 copies of c2-LTR circles per 100 ng total DNA was detected in primary resting CD4+ T cells with treatment of Pam3CSK4 and GC, respectively (Fig. 8C). At 48 h after viral infection, the level of c2-LTR circles in HIV-infected cells with exposure to Pam3CSK4 and GC was increased by 4.3- and 3.1-fold 1 over nontreated control, respectively. Similarly, the level of c-2LTR circles was increased in PGN-treated infected cells (data not shown). This result demonstrated that GC exposure and TLR2 activation promoted HIV infection at the step of nuclear import in resting CD4+ T cells.
Discussion
In this study, we demonstrated that GC enhanced HIV-1 infection of primary resting CD4+ T cells through TLR2 activation. Pre-treatment of uninfected CD4+ T cells as well as stimulation of HIV-infected primary CD4+ cells after entry with the TLR2 agonist PGN increased HIV replication, suggesting that multiple mechanisms were involved in TLR2-mediated enhancement of HIV infection of primary resting CD4+ T cells. GC exposure and TLR2 stimulation activated resting CD4+ T cells and induced cell surface expression of HIV coreceptors, resulting in an increase in the susceptibility of primary CD4+ T cells to HIV infection. STIs, such as GC and Chlamydia trachomatis, are known to activate TLR2 and TLR4 (30, 57, 58). Although there is a discrepancy in the literature with respect to gene expression of TLR4 in primary CD4+ T cells (51, 59, 60), we did not detect TLR4 mRNA in primary CD4+ T cells by RT-PCR nor did we find an HIV enhancing effect of TLR4 agonists LPS and GC LOS. Importantly, anti-TLR2 Abs completely abolished the enhancement of HIV infection induced by GC or GC-derived lipopeptide Pam3C-Lip. These results suggest that TLR2 may play a major role in GC-mediated enhancement of HIV transmission, particularly if bacterial products come in contact with intraepithelial resting T cells. Indeed, TLR2 activation is also involved in GC-mediated enhancement of HIV transmission by dendritic cells to T cells (20) as well as induction of HIV LTR gene expression (9) and cytokine production by cervicovaginal fluid from women with bacterial vaginosis (61). Interestingly, bacterial vaginosis is also associated with increased HIV acquisition and HIV shedding (15, 62), suggesting common pathways for HIV enhancement. Our results also show that components on GC surface may affect the efficiency of TLR2-activation. Although both Pil+/Opa+ as well as Pil−/Opa−GC enhanced HIV infection, the Pil+/Opa+ variant was more active presumably due to the known interactions of Pil and Opa with T cells that may bring the bacterial membrane into closer or more prolonged proximity of TLR2 receptors (40, 63).
Our studies presented a novel mechanism demonstrating that TLR2 activation promoted HIV infection of primary resting CD4+ T cells after viral entry. The maximal effect of PGN was achieved when PGN was added to HIV-infected cells immediately after viral infection, indicating that TLR2 activation acted on an early stage of the HIV life cycle. Analysis of HIV DNA products revealed that TLR2 activation significantly enhanced the step of nuclear import. Several viral proteins, including Vpr, integrase, and Gag, are reported to play a role in nuclear import (64, 65). Some viral proteins, particularly Vpr, influence cell cycle progression, which modulates nuclear import (66). Our results showed that GC and TLR2 agonists promoted HIV infection in a single-cycle infection assay using pseudotyped HIV-1 luciferase reporter virus that does not contain the Vpr gene, suggesting that TLR2-mediated enhancement of HIV infection of resting CD4+ T cells is not dependent on HIV Vpr. Nuclear import of SIV and HIV requires signaling pathways induced by both TCRs and CD28 receptors in resting T cells (67, 68). IL-2 can replace CD28 ligation to enhance the level of HIV 2-LTR circles, a marker of nuclear import, in CD3-activated T cells (67). Although the synthetic TLR2 agonist Pam3CSK4 has been shown to enhance HIV infection of quiescent naive and memory T cells in the absence of TCR cross-linking or IL-2, HIV infection was significantly increased in the presence of IL-2 or OKT3 Ab (21). Our studies indicated that IL-2 signaling was essential for TLR2-mediated enhancement of HIV infection of primary resting CD4+ T cells using both PGN and Pam3CSK4. It is not clear whether purification of specific subsets of T cells or the use of TZM-bl reporter gene system contributes to the different outcome with respect to IL-2 dependency. Nevertheless, we are currently investigating the effect of GC and TLR2 agonists using subsets of T cells in the presence or absence of IL-2.
HIV 2-LTR circle formation is blocked at G0/G1 transition (55, 69, 70). In agreement with the previous report using naive and memory CD4 T cells in response to Pam3CSK4 (21), we found that GC and PGN induced T cell activation, although the effect on cell cycle in primary resting CD4+ T cells remains to be determined. It is possible that, in conjunction with TLR2 signaling pathways, IL-2–dependent T cell activation and G0/G1 transition promote nuclear import of HIV DNA. After nuclear import and integration, TLR2 signaling may further enhance HIV transcription because TLR2 activation is known to activate HIV LTR-driven gene expression through NF-κB activation and induction of TNF-α and IL-8 (15, 61).
In vitro, HIV enters primary resting CD4+ T cells and remains latent in the absence of T cell stimulation (71–73), although HIV infection has been reported in resting CD4+ T cells in HIV-infected individuals and SIV-infected macaque monkeys (32, 74, 75). Barriers blocking completion of HIV infection in resting CD4+ T cells include host restriction factors, such as Trim5α (76), the blockage of nuclear import of preintegration complex (77), nuclear retention of multiply spliced HIV RNA in resting CD4+ T cells (78), inhibition of NF-κB activation by Murr1 (79), and cellular microRNA that inhibits HIV gene expression (80). Although it remains to be determined the effect of TLR2 activation on other HIV inhibitory factors in resting CD4+ T cells, our results showed that TLR2 activation overcame a block at the level of nuclear import of HIV DNA.
HIV-1 infection in resting CD4+ T cells generally leads to preintegration or postintegration latency in the absence of T cell activation (81, 82). In resting CD4+ T cells, HIV-1 DNA in pre-integration latency decays rapidly but HIV infection can be rescued when T cells are activated at early stages of HIV infection (77, 83). When PGN was added at 72 h after viral infection, there was no enhancement of HIV infection of primary CD4+ T cells in a single-cycle infection assay (Fig. 6). We excluded the possibility that the lack of responsiveness was due to decreased cell viability (Fig. 6B) or reduced TLR2 expression (Fig. 6C, 6D). This lack of responsiveness to TLR2 activation could be due to degradation of HIV DNA in resting CD4+ T cells at 72 h postinfection. We demonstrated that TLR2 signaling also induced HIV infection of primary resting CD4+ T cells when added 48 h postinfection (Fig. 6A). The specific stage of the HIV life cycle affected by TLR2 signaling at 48 h postinfection and the effect of TLR2 activation on the steps of postintegration remain to be determined.
An increase in the number of endocervical CD4+ T cells has been found in GC and C. trachomatis-infected women (10, 84, 85). The endocervical cell infiltrate from C. trachomatis-infected women is dominated by activated effector memory T cells with high CCR5 expression (85). Interestingly, studies on phenotypes of endocervical T cells from C. trachomatis-infected women after treatment indicate a decrease in T cell activation but not the number of CCR5+ cells posttreatment (85). In agreement with the previous report (21), our studies showed that TLR2 activation resulted in T cell activation and elevation of CCR5 on resting CD4+ T cells. In addition, we found the level of CXCR4 was upregulated in response to GC or PGN. PHA treatment is known to downregulate IL-2–mediated induction of CCR5 gene expression in CD4+ T cells (86). Similar to previous findings (86, 87), we did not observe an increase in the CCR5 level on PHA-activated primary CD4+ T cells (Table I). Our results suggest that induction of CCR5 by GC or PGN is not simply due to T cell activation. As antibiotic chemoprophylaxis does not reduce the incidence of HIV-1 (6, 8), it is important to dissect underlying mechanisms to develop an effective strategy to prevent HIV spread. Our studies provide additional mechanisms by which GC increases HIV transmission that may be important for the development of novel prevention strategies by targeting TLR2 signaling pathways.
Acknowledgments
This work was supported by National Institutes of Health Grants AI073205 (to T.L.C.) and AI063927 (to G.A.J.).
We thank Flora Samaroo for providing N. gonorrhoeae ATCC strain 43069 and Arevik Mosoian for helpful discussions.
Abbreviations used in this paper
- c2-LTR
HIV closed 2-long terminal repeat
- GC
gonococcus
- LOS
lipooligosaccharide
- MDDC
monocyte-derived dendritic cell
- MDP
muramyl peptide
- MOI
multiplicity of infection
- NOD2
nucleotide-binding and oligomerization domain 2
- PGN
peptidylglycan
- PRR
pattern recognition receptor
- STI
sexually transmitted infection
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Hall HI, Song R, Rhodes P, Prejean J, An Q, Lee LM, Karon J, Brookmeyer R, Kaplan EH, McKenna MT, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300:520–529. doi: 10.1001/jama.300.5.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen MS, Miller WC. Sexually transmitted diseases and human immunodeficiency virus infection: cause, effect, or both? Int J Infect Dis. 1998;3:1–4. doi: 10.1016/s1201-9712(98)90087-x. [DOI] [PubMed] [Google Scholar]
- 3.Plummer FA, Simonsen JN, Cameron DW, Ndinya-Achola JO, Kreiss JK, Gakinya MN, Waiyaki P, Cheang M, Piot P, Ronald AR, et al. Cofactors in male-female sexual transmission of human immunodeficiency virus type 1. J Infect Dis. 1991;163:233–239. doi: 10.1093/infdis/163.2.233. [DOI] [PubMed] [Google Scholar]
- 4.Cameron DW, Simonsen JN, D’Costa LJ, Ronald AR, Maitha GM, Gakinya MN, Cheang M, Ndinya-Achola JO, Piot P, Brunham RC, et al. Female to male transmission of human immunodeficiency virus type 1: risk factors for seroconversion in men. Lancet. 1989;2:403–407. doi: 10.1016/s0140-6736(89)90589-8. [DOI] [PubMed] [Google Scholar]
- 5.Ghys PD, Fransen K, Diallo MO, Ettiègne-Traoré V, Coulibaly IM, Yeboué KM, Kalish ML, Maurice C, Whitaker JP, Greenberg AE, Laga M. The associations between cervicovaginal HIV shedding, sexually transmitted diseases and immunosuppression in female sex workers in Abidjan, Côte d’Ivoire. AIDS. 1997;11:F85–F93. doi: 10.1097/00002030-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Kaul R, Kimani J, Nagelkerke NJ, Fonck K, Ngugi EN, Keli F, MacDonald KS, Maclean IW, Bwayo JJ, Temmerman M Kibera HIV Study Group. Monthly antibiotic chemoprophylaxis and incidence of sexually transmitted infections and HIV-1 infection in Kenyan sex workers: a randomized controlled trial. JAMA. 2004;291:2555–2562. doi: 10.1001/jama.291.21.2555. [DOI] [PubMed] [Google Scholar]
- 7.Corbett EL, Steketee RW, Oter Kuile F, Latif AS, Kamali A, Hayes RJ. HIV-1/AIDS and the control of other infectious diseases in Africa. Lancet. 2002;359:2177–2187. doi: 10.1016/S0140-6736(02)09095-5. [DOI] [PubMed] [Google Scholar]
- 8.Cohen MS, I, Hoffman F, Royce RA, Kazembe P, Dyer JR, Daly CC, Zimba D, Vernazza PL, Maida M, Fiscus SA, et al. Reduction of concentration of HIV-1 in semen after treatment of urethritis: implications for prevention of sexual transmission of HIV-1. Lancet. 1997;349:1868–1873. doi: 10.1016/s0140-6736(97)02190-9. [DOI] [PubMed] [Google Scholar]
- 9.Chen A, I, Boulton C, Pongoski J, Cochrane A, Gray-Owen SD. Induction of HIV-1 long terminal repeat-mediated transcription by Neisseria gonorrhoeae. AIDS. 2003;17:625–628. doi: 10.1097/00002030-200303070-00019. [DOI] [PubMed] [Google Scholar]
- 10.Levine WC, Pope V, Bhoomkar A, Tambe P, Lewis JS, Zaidi AA, Farshy CE, Mitchell S, Talkington DF. Increase in endocervical CD4 lymphocytes among women with nonulcerative sexually transmitted diseases. J Infect Dis. 1998;177:167–174. doi: 10.1086/513820. [DOI] [PubMed] [Google Scholar]
- 11.Klotman ME, Rapista A, Teleshova N, Micsenyi A, Jarvis GA, Lu W, Porter E, Chang TL. Neisseria gonorrhoeae-induced human defensins 5 and 6 increase HIV infectivity: role in enhanced transmission. J Immunol. 2008;180:6176–6185. doi: 10.4049/jimmunol.180.9.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Li G, Bafica A, Pantelic M, Zhang P, Broxmeyer H, Liu Y, Wetzler L, He JJ, Chen T. Neisseria gonorrhoeae enhances infection of dendritic cells by HIV type 1. J Immunol. 2005;174:7995–8002. doi: 10.4049/jimmunol.174.12.7995. [DOI] [PubMed] [Google Scholar]
- 13.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Equils O, Schito ML, Karahashi H, Madak Z, Yarali A, Michelsen KS, Sher A, Arditi M. Toll-like receptor 2 (TLR2) and TLR9 signaling results in HIV-long terminal repeat trans-activation and HIV replication in HIV-1 transgenic mouse spleen cells: implications of simultaneous activation of TLRs on HIV replication. J Immunol. 2003;170:5159–5164. doi: 10.4049/jimmunol.170.10.5159. [DOI] [PubMed] [Google Scholar]
- 16.Scheller C, Ullrich A, McPherson K, Hefele B, Knöferle J, Lamla S, Olbrich AR, Stocker H, Arasteh Vter Meulen K, et al. CpG oligodeoxynucleotides activate HIV replication in latently infected human T cells. J Biol Chem. 2004;279:21897–21902. doi: 10.1074/jbc.M311609200. [DOI] [PubMed] [Google Scholar]
- 17.Sundstrom JB, Little DM, Villinger F, Ellis JE, Ansari AA. Signaling through Toll-like receptors triggers HIV-1 replication in latently infected mast cells. J Immunol. 2004;172:4391–4401. doi: 10.4049/jimmunol.172.7.4391. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Abel K, Lantz K, Krieg AM, McChesney MB, Miller CJ. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intra-vaginally to rhesus macaques. J Virol. 2005;79:14355–14370. doi: 10.1128/JVI.79.22.14355-14370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Mosoian A, Li-Yun Chang T, Zerhouni-Layachi B, Snyder A, Jarvis GA, Klotman ME. Gonococcal lipooligosaccharide suppresses HIV infection in human primary macrophages through induction of innate immunity. J Infect Dis. 2006;194:751–759. doi: 10.1086/506360. [DOI] [PubMed] [Google Scholar]
- 20.Thibault S, Fromentin R, Tardif MR, Tremblay MJ. TLR2 and TLR4 triggering exerts contrasting effects with regard to HIV-1 infection of human dendritic cells and subsequent virus transfer to CD4+ T cells. Retrovirology. 2009;6:42. doi: 10.1186/1742-4690-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thibault S, Tardif MR, Barat C, Tremblay MJ. TLR2 signaling renders quiescent naive and memory CD4+ T cells more susceptible to productive infection with X4 and R5 HIV-type 1. J Immunol. 2007;179:4357–4366. doi: 10.4049/jimmunol.179.7.4357. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa Y, Kawamura T, Kimura T, Ito M, Blauvelt A, Shimada S. Gram-positive bacteria enhance HIV-1 susceptibility in Langerhans cells, but not in dendritic cells, via Toll-like receptor activation. Blood. 2009;113:5157–5166. doi: 10.1182/blood-2008-10-185728. [DOI] [PubMed] [Google Scholar]
- 23.Báfica A, Scanga CA, Schito M, Chaussabel D, Sher A. Influence of coinfecting pathogens on HIV expression: evidence for a role of Toll-like receptors. J Immunol. 2004;172:7229–7234. doi: 10.4049/jimmunol.172.12.7229. [DOI] [PubMed] [Google Scholar]
- 24.Equils O, Salehi KK, Cornataeanu R, Lu D, Singh S, Whittaker K, Baldwin GC. Repeated lipopolysaccharide (LPS) exposure inhibits HIV replication in primary human macrophages. Microbes Infect. 2006;8:2469–2476. doi: 10.1016/j.micinf.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Simard S, Maurais E, Gilbert C, Tremblay MJ. LPS reduces HIV-1 replication in primary human macrophages partly through an endogenous production of type I interferons. Clin Immunol. 2008;127:198–205. doi: 10.1016/j.clim.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Juffermans NP, Paxton WA, Dekkers PE, Verbon A, de Jonge E, Speelman P, van Deventer SJ, van der Poll T. Up-regulation of HIV coreceptors CXCR4 and CCR5 on CD4(+) T cells during human endotoxemia and after stimulation with (myco)bacterial antigens: the role of cytokines. Blood. 2000;96:2649–2654. [PubMed] [Google Scholar]
- 27.Moriuchi M, Moriuchi H, Turner W, Fauci AS. Exposure to bacterial products renders macrophages highly susceptible to T-tropic HIV-1. J Clin Invest. 1998;102:1540–1550. doi: 10.1172/JCI4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Báfica A, Scanga CA, Schito ML, Hieny S, Sher A. Cutting edge: in vivo induction of integrated HIV-1 expression by mycobacteria is critically dependent on Toll-like receptor 2. J Immunol. 2003;171:1123–1127. doi: 10.4049/jimmunol.171.3.1123. [DOI] [PubMed] [Google Scholar]
- 29.Cummins JE, Jr, Doncel GF. Biomarkers of cervicovaginal inflammation for the assessment of microbicide safety. Sex Transm Dis. 2009;36(3, Suppl):S84–S91. doi: 10.1097/OLQ.0b013e3181994191. [DOI] [PubMed] [Google Scholar]
- 30.Pridmore AC, Jarvis GA, John CM, Jack DL, Dower SK, Read RC. Activation of toll-like receptor 2 (TLR2) and TLR4/MD2 by Neisseria is independent of capsule and lipooligosaccharide (LOS) sialylation but varies widely among LOS from different strains. Infect Immun. 2003;71:3901–3908. doi: 10.1128/IAI.71.7.3901-3908.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. Cutting edge: Immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J Immunol. 2002;168:1533–1537. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 33.Griffiss JM, Lammel CJ, Wang J, Dekker NP, Brooks GF. Neisseria gonorrhoeae coordinately uses Pili and Opa to activate HEC-1-B cell microvilli, which causes engulfment of the gonococci. Infect Immun. 1999;67:3469–3480. doi: 10.1128/iai.67.7.3469-3480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen BK, Saksela K, Andino R, Baltimore D. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994;68:654–660. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1—infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 37.Zazzi M, Romano L, Catucci M, Venturi G, De Milito A, Almi P, Gonnelli A, Rubino M, Occhini U, Valensin PE. Evaluation of the presence of 2-LTR HIV-1 unintegrated DNA as a simple molecular predictor of disease progression. J Med Virol. 1997;52:20–25. doi: 10.1002/(sici)1096-9071(199705)52:1<20::aid-jmv4>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 38.Cara A, Cereseto A, Lori F, Reitz MS., Jr HIV-1 protein expression from synthetic circles of DNA mimicking the extrachromosomal forms of viral DNA. J Biol Chem. 1996;271:5393–5397. doi: 10.1074/jbc.271.10.5393. [DOI] [PubMed] [Google Scholar]
- 39.Plant LJ, Jonsson AB. Type IV pili of Neisseria gonorrhoeae influence the activation of human CD4+ T cells. Infect Immun. 2006;74:442–448. doi: 10.1128/IAI.74.1.442-448.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulton IC, Gray-Owen SD. Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat Immunol. 2002;3:229–236. doi: 10.1038/ni769. [DOI] [PubMed] [Google Scholar]
- 41.Youssef AR, van der Flier M, Estevão S, Hartwig NG, van der Ley P, Virji M. Opa+ and Opa- isolates of Neisseria meningitidis and Neisseria gonorrhoeae induce sustained proliferative responses in human CD4+ T cells. Infect Immun. 2009;77:5170–5180. doi: 10.1128/IAI.00355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 43.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 44.Fisette PL, Ram S, Andersen JM, Guo W, Ingalls RR. The Lip lipoprotein from Neisseria gonorrhoeae stimulates cytokine release and NF-kappaB activation in epithelial cells in a Toll-like receptor 2-dependent manner. J Biol Chem. 2003;278:46252–46260. doi: 10.1074/jbc.M306587200. [DOI] [PubMed] [Google Scholar]
- 45.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 46.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawai T, Takeuchi O, Fujita T, Inoue J, Mühlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 48.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 49.Farhat K, Riekenberg S, Heine H, Debarry J, Lang R, Mages J, Buwitt-Beckmann U, Röschmann K, Jung G, Wiesmüller KH, Ulmer AJ. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol. 2008;83:692–701. doi: 10.1189/jlb.0807586. [DOI] [PubMed] [Google Scholar]
- 50.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, Fernandez-Luna JL. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 51.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdörfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 52.Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- 53.Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian EL, Permutter RM, Taniguchi T. Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell. 1995;81:223–231. doi: 10.1016/0092-8674(95)90332-1. [DOI] [PubMed] [Google Scholar]
- 54.Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, Saito T. Cutting edge: TLR2 directly triggers Th1 effector functions. J Immunol. 2007;178:6715–6719. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 55.Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, Stevenson M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc Natl Acad Sci USA. 1992;89:6580–6584. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plesa G, Dai J, Baytop C, Riley JL, June CH, O’Doherty U. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J Virol. 2007;81:13938–13942. doi: 10.1128/JVI.01745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heine H, Müller-Loennies S, Brade L, Lindner B, Brade H. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur J Biochem. 2003;270:440–450. doi: 10.1046/j.1432-1033.2003.03392.x. [DOI] [PubMed] [Google Scholar]
- 58.Bas S, Neff L, Vuillet M, Spenato U, Seya T, Matsumoto M, Gabay C. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J Immunol. 2008;180:1158–1168. doi: 10.4049/jimmunol.180.2.1158. [DOI] [PubMed] [Google Scholar]
- 59.Xu D, Komai-Koma M, Liew FY. Expression and function of Toll-like receptor on T cells. Cell Immunol. 2005;233:85–89. doi: 10.1016/j.cellimm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 60.Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner-Fischer M, Lider O, Cohen IR. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol. 2007;179:41–44. doi: 10.4049/jimmunol.179.1.41. [DOI] [PubMed] [Google Scholar]
- 61.Mares D, Simoes JA, Novak RM, Spear GT. TLR2-mediated cell stimulation in bacterial vaginosis. J Reprod Immunol. 2008;77:91–99. doi: 10.1016/j.jri.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS. 2008;22:1493–1501. doi: 10.1097/QAD.0b013e3283021a37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hansen JK, Demick KP, Mansfield JM, Forest KT. Conserved regions from Neisseria gonorrhoeae pilin are immunosilent and not immuno-suppressive. Infect Immun. 2007;75:4138–4147. doi: 10.1128/IAI.02015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aida Y, Matsuda G. Role of Vpr in HIV-1 nuclear import: therapeutic implications. Curr HIV Res. 2009;7:136–143. doi: 10.2174/157016209787581418. [DOI] [PubMed] [Google Scholar]
- 65.Piller SC, Caly L, Jans DA. Nuclear import of the pre-integration complex (PIC): the Achilles heel of HIV? Curr Drug Targets. 2003;4:409–429. doi: 10.2174/1389450033490984. [DOI] [PubMed] [Google Scholar]
- 66.Romani B, Engelbrecht S. Human immunodeficiency virus type 1 Vpr: functions and molecular interactions. J Gen Virol. 2009;90:1795–1805. doi: 10.1099/vir.0.011726-0. [DOI] [PubMed] [Google Scholar]
- 67.Sun Y, Pinchuk LM, Agy MB, Clark EA. Nuclear import of HIV-1 DNA in resting CD4+ T cells requires a cyclosporin A-sensitive pathway. J Immunol. 1997;158:512–517. [PubMed] [Google Scholar]
- 68.Polacino PS, Liang HA, Firpo EJ, Clark EA. T-cell activation influences initial DNA synthesis of simian immunodeficiency virus in resting T lymphocytes from macaques. J Virol. 1993;67:7008–7016. doi: 10.1128/jvi.67.12.7008-7016.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polacino PS, Pinchuk LM, Sidorenko SP, Clark EA. Immunodeficiency virus cDNA synthesis in resting T lymphocytes is regulated by T cell activation signals and dendritic cells. J Med Primatol. 1996;25:201–209. doi: 10.1111/j.1600-0684.1996.tb00017.x. [DOI] [PubMed] [Google Scholar]
- 70.Stevenson M, Brichacek B, Heinzinger N, Swindells S, Pirruccello S, Janoff E, Emerman M. Molecular basis of cell cycle dependent HIV-1 replication. Implications for control of virus burden. Adv Exp Med Biol. 1995;374:33–45. doi: 10.1007/978-1-4615-1995-9_4. [DOI] [PubMed] [Google Scholar]
- 71.Chou CS, Ramilo O, Vitetta ES. Highly purified CD25- resting T cells cannot be infected de novo with HIV-1. Proc Natl Acad Sci USA. 1997;94:1361–1365. doi: 10.1073/pnas.94.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scales D, Ni H, Shaheen F, Capodici J, Cannon G, Weissman D. Nonproliferating bystander CD4+ T cells lacking activation markers support HIV replication during immune activation. J Immunol. 2001;166:6437–6443. doi: 10.4049/jimmunol.166.10.6437. [DOI] [PubMed] [Google Scholar]
- 73.Zack JA, Haislip AM, Krogstad P, Chen IS. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol. 1992;66:1717–1725. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blaak H, van’t Wout AB, Brouwer M, Hooibrink B, Hovenkamp E, Schuitemaker H. In vivo HIV-1 infection of CD45RA(+)CD4(+) T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4(+) T cell decline. Proc Natl Acad Sci USA. 2000;97:1269–1274. doi: 10.1073/pnas.97.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ostrowski MA, Chun TW, Justement SJ, Motola I, Spinelli MA, Adelsberger J, Ehler LA, Mizell SB, Hallahan CW, Fauci AS. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J Virol. 1999;73:6430–6435. doi: 10.1128/jvi.73.8.6430-6435.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 77.Bukrinsky MI, Stanwick TL, Dempsey MP, Stevenson M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science. 1991;254:423–427. doi: 10.1126/science.1925601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lassen KG, Ramyar KX, Bailey JR, Zhou Y, Siliciano RF. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2006;2:e68. doi: 10.1371/journal.ppat.0020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LW, Wijmenga C, Duckett CS, Nabel GJ. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature. 2003;426:853–857. doi: 10.1038/nature02171. [DOI] [PubMed] [Google Scholar]
- 80.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–1247. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 81.Williams SA, Greene WC. Regulation of HIV-1 latency by T-cell activation. Cytokine. 2007;39:63–74. doi: 10.1016/j.cyto.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pierson T, McArthur J, Siliciano RF. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. 2000;18:665–708. doi: 10.1146/annurev.immunol.18.1.665. [DOI] [PubMed] [Google Scholar]
- 83.Pierson TC, Zhou Y, Kieffer TL, Ruff CT, Buck C, Siliciano RF. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J Virol. 2002;76:8518–8531. doi: 10.1128/JVI.76.17.8518-8531.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mittal A, Rastogi S, Reddy BS, Verma S, Salhan S, Gupta E. Enhanced immunocompetent cells in chlamydial cervicitis. J Reprod Med. 2004;49:671–677. [PubMed] [Google Scholar]
- 85.Ficarra M, Ibana JS, Poretta C, Ma L, Myers L, Taylor SN, Greene S, Smith B, Hagensee M, Martin DH, Quayle AJ. A distinct cellular profile is seen in the human endocervix during Chlamydia trachomatis infection. Am J Reprod Immunol. 2008;60:415–425. doi: 10.1111/j.1600-0897.2008.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patterson BK, Czerniewski M, Andersson J, Sullivan Y, Su F, Jiyamapa D, Burki Z, Landay A. Regulation of CCR5 and CXCR4 expression by type 1 and type 2 cytokines: CCR5 expression is downregulated by IL-10 in CD4-positive lymphocytes. Clin Immunol. 1999;91:254–262. doi: 10.1006/clim.1999.4713. [DOI] [PubMed] [Google Scholar]
- 87.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]