

Abstract
The μ-opioid receptor is the site of action of many endogenous opioids as well as opiates. We hypothesize that differences in DNA methylation of specific CpG dinucleotides between former severe heroin addicts in methadone maintenance treatment and control subjects will depend, in part, upon ethnicity. DNA methylation analysis of the μ-opioid receptor gene (OPRM1) promoter region was performed on African-Americans (118 cases, 80 controls) and Hispanics (142 cases, 61 controls) and these were compared with a similar Caucasian cohort from our earlier study. In controls, a higher methylation level was found in the African-Americans compared with the Hispanics or Caucasians. Significant experiment-wise differences in methylation levels were found at the −25 and +12 CpG sites in the controls among the three ethnicities. The overall methylation level of the CpG sites were significantly higher in the former heroin addicts when compared with the controls (point-wise P = 0.0457). However, in the African-Americans, the degree of methylation was significantly decreased experiment-wise in the former heroin addicts at the +12 CpG site (P = 0.0032, Bonferroni corrected general estimating equations). In Hispanics, the degree of methylation was increased in the former heroin addicts at the −25 (P < 0.001, experiment-wise), −14 (P = 0.001, experiment-wise), and +27 (P < 0.001, experiment-wise) CpG sites. These changes in methylation of the OPRM1 promoter region may lead to altered expression of the μ-opioid receptor gene in the lymphocytes of former heroin addicts who are stabilized in methadone maintenance treatment.
Introduction
Epigenetic factors, such as DNA methylation, and histone modifications, have been shown to be altered by illicit drugs or in drug addiction (Kumar et al. 2005; Nielsen et al.2009; Novikova et al. 2008; Renthal et al. 2007, 2008; Zhang et al. 2007) and may play a role in an individual’s vulnerability to develop drug addiction, or to the response to pharmacotherapy. In genomic DNA, cytosine residues may be methylated at cytosine:guanine (CpG) dinucleotides by DNA methyltransferases. When CpG sites are methylated in promoter regions, in general, gene expression is decreased (Heller et al. 2008). A number of transcription binding sites contain CpG dinucleotides that, when methylated, alter transcription factor binding (e.g. Alikhani-Koopaei et al. 2004; Douet et al. 2007; Michelotti et al. 2007; Zhang et al. 2007).
Recently, we found hypermethylation of two CpG sites in the μ-opioid receptor (OPRM1) gene promoter in DNA from peripheral lymphocytes from Caucasian methadone maintained former heroin addicts (Nielsen et al. 2009). The OPRM1 gene codes for the μ-opioid receptor, the receptor to which morphine, methadone, and β-endorphin bind and exert their actions (Kreek et al. 2005). We hypothesized that this hypermethylation may alter the transcriptional regulation of the OPRM1 gene in these individuals. It is not known whether the hypermethylation of these two CpG sites was due to heroin use, methadone maintenance pharmacotherapy, imprinting, or major life events prior to using heroin. Other studies have shown that DNA methylation is higher in genomic DNA from lymphocytes of alcoholics than from those of controls (Bleich et al. 2006; Bonsch et al. 2004, 2006). Increased DNA methylation was found in the promoter regions of the α-synuclein SNCA gene (Bonsch et al. 2005), the homocysteine induced endoplasmic reticulum protein HERP gene (Bleich et al. 2006), and the vasopressin AVP gene (Hillemacher et al. 2009) and decreased methylation of the promoter region of the atrial natriuretic peptide gene ANP (Hillemacher et al. 2009) in alcoholics. In women, but not men, overall DNA methylation of the monoamine oxidase A MAOA gene promoter was found to be associated point-wise with nicotine dependence and alcohol dependence (Philibert et al. 2008). Maternal cocaine exposure in mice caused decreased global methylation at P3 and increased global methylation at P30 in hippocampal pyramidal neurons (Novikova et al. 2008) and caused increased methylation of the protein kinase C epsilon (PKCε) PRKCE gene promoter (Zhang et al. 2007). Several genes that were linked to a change in methylation had their expression increased or decreased by as much as 19-fold.
A few studies have demonstrated ethnic diversity in the overall DNA methylation levels. Methylation of leukocyte DNA levels are highest in Hispanics, lowest in Blacks, and of intermediate levels in Whites (Terry et al. 2008). Ethnic differences have also been observed in the overall methylation of tumor DNA. For example, methylation of the IGTBP-3 gene in malignant mesothelioma was higher in Japanese patients than patients from the USA (Tomii et al. 2006) and the TMS1/ASC gene is more methylated in prostate cancer cases than controls in White patients, but not in Black patients (Das et al. 2006).
CpG methylation may occur rapidly and may persist for decades. For instance, when rats were exposed to fear conditioning, methylation of the protein phosphatase 1 gene pp1 was increased and methylation of the reelin gene reln was decreased in 1 h (Miller and Sweatt 2007). The methylation of these genes returned to baseline within 24 h. Methylation status of the promoter of the imprinted insulin-like growth factor 2 IGF2 gene, a gene involved in energy metabolism, was examined in subjects prenatally exposed to famine during the Dutch Hunger Winter of 1944–1945 (Heijmans et al. 2008). It was found that the promoter of the IGF2 gene was hypomethylated compared with controls, presumably reflecting events that occurred 60 years ago.
In this study, we have extended our earlier work in Caucasians (Nielsen et al. 2009) by examining in subjects of two ethnicities, African-Americans and Hispanics, methylation levels at 16 CpG sites in the OPRM1 promoter region. Subjects were former severe heroin addicts stabilized in methadone maintenance pharmacotherapy and non-drug using controls. OPRM1 methylation levels of these subjects were compared to methylation levels in Caucasians that were recently reported (Nielsen et al. 2009).
Materials and methods
Subjects and phenotyping
Our sample consisted of 198 African-American and 203 Hispanic subjects (Table 1). We have reported methylation levels of 329 Caucasian subjects, used in this study as a comparison group for the extent of methylation (Nielsen et al. 2009). The 401 former severe heroin addicts and control subjects of African-American and Hispanic ethnicity were drawn from consecutive volunteers (January 1995-May 2007) in genetic studies conducted by the Laboratory of the Biology of Addictive Diseases at The Rockefeller University who met the inclusion criteria defined below (same criteria for the Caucasians reported earlier). Subjects were recruited from clinical resources in New York City, and from newspaper advertisements, referrals, and posted notices. Ethnicity was based on the ethnic/cultural background of the subjects, their parents, grandparents, and great-grandparents. All subjects gave specific consent for genetic studies and signed an informed consent approved by The Rockefeller University Hospital Institutional Review Board.
Table 1.
Sample categorization
| Classification | Starting sample |
Samples for analysis of CpG methylationa |
||||
|---|---|---|---|---|---|---|
| Male | Female | Total | Male | Female | Total | |
| African-Americans | ||||||
| MMTPb | 57 | 61 | 118 | 48 | 52 | 100 |
| Controls | 30 | 50 | 80 | 23 | 47 | 70 |
| Total African-American | 87 | 111 | 198 | 71 | 99 | 170 |
| Hispanics | ||||||
| MMTP | 91 | 51 | 142 | 74 | 46 | 120 |
| Controls | 24 | 37 | 61 | 21 | 32 | 53 |
| Total Hispanic | 115 | 88 | 203 | 95 | 78 | 173 |
| Caucasiansc | ||||||
| MMTP | 117 | 77 | 194 | 99 | 68 | 167 |
| Controls | 66 | 69 | 135 | 50 | 53 | 103 |
| Total Caucasian | 183 | 146 | 329 | 149 | 121 | 270 |
| Total | 385 | 345 | 730 | 315 | 298 | 613 |
Samples for analysis of CpG methylation are those which had sequencing/ESME analysis performed in both directions and had a correlation between these analyses ≥0.7
Former severe heroin addicts stabilized in methadone maintenance treatment
Caucasian data previously reported (Nielsen et al. 2009)
The Addiction Severity Index (McLellan et al. 1980) was administered to all subjects and urine analyses were performed for multiple drugs of abuse. Former severe heroin addicts (African-American, N = 118; Hispanic, N = 142) were long-term heroin addicts who met Federal guidelines for methadone maintenance treatment (1 year or more of daily multiple injections of heroin or other opiates) (Rettig and Yarmolinsky 1995). Subjects in the control group (African-American, N = 80; Hispanic, N = 61) had (1) no illicit drug use for more than 6 months (except cannabis), (2) no excessive use of cannabis (3 or more times per week for more than 4 years), (3) no previous history of alcohol drinking to intoxication (3 or more times per week for 6 months or more), and (4) no alcohol intoxication or illicit drug use (except cannabis) in the last 30 days.
In the study sample that was used to determine DNA methylation levels, those subjects with a correlation cutoff ≥0.7 (see below), 24% of the African-American former severe heroin addicts also had a history of alcohol dependence (24 of 100) and 21% of the Hispanic former severe heroin addicts had a history of alcohol dependence (25 of 120).
Isolation of lymphocyte DNA and the determination of percent methylated cytosine
DNA was extracted from peripheral blood lymphocytes and percent cytosine methylation was determined as previously described (Nielsen et al. 2009). Briefly, genomic DNA (300 ng) was sodium bisulfite treated using the EZ-96 DNA Methylation Kit D5004 (Zymo Research, Orange, CA) and amplified (Nielsen et al. 2009). DNA was sequenced at GENEWIZ, Inc. (South Plainfield, NJ) on an ABI 3730 XL sequencer (Applied Biosystems, Foster City, CA). Trace files (.ab1) were analyzed using the ESME version 3.2.1 software from Epigenomics AG (Berlin, Germany) (Lewin et al. 2004). The percent methylation calls by the ESME were reviewed by two independent researchers who visually inspected all the methylation calls using the associated electropherograms generated by the ESME software.
Sequence analysis
The OPRM1 promoter region was analyzed for predicted transcription factor binding sites using TESS: Transcription Element Search System (Schug and Overton 1977).
Statistical analysis
For each sample, for which a forward and reverse sequence file was obtained, a correlation value was calculated between the arcsine of methylation frequency in the forward and reverse directions. Samples with a correlation ≥0.7 (African-American, N = 170; Hispanic, N = 173) were included in the analysis of percent methylation at each CpG site (Table 1). This was the same cut off used in our earlier study of Caucasians (Nielsen et al. 2009).
The mean of the forward and reverse values was calculated for each CpG site in each sample. Within each ethnicity, Welch’s two-tailed t tests were used to determine if the mean level of methylation at each CpG dinucleotide differed between sexes. Within controls, Welch’s two-tailed t tests were used to determine if there was a significant difference between African-Americans and Hispanics in overall methylation for all 16 CpG dinucleotides, and, also, using our earlier data to compare African-Americans with Caucasians, and Hispanics with Caucasians (Table 2).
Table 2.
Ethnic differences in the overall degree of methylation in the controls among ethnicities
| Ethnicity | Methylation (%) | Ethnicity | Methylation (%) |
P valuea |
|
|---|---|---|---|---|---|
| Point-wise | Experiment-wise | ||||
| African-American | 19.6 | Hispanic | 16.1 | 1.16 × 10−6 | 3.48 × 10−6 |
| African-American | 19.6 | Caucasianb | 17.4 | 0.0010 | 0.0031 |
| Hispanic | 16.1 | Caucasian | 17.4 | 0.0434 | n.s.c |
Welch two sample t test
Caucasian data previously reported (Nielsen et al. 2009)
n.s.: P value > 0.05
To determine if there was a significant difference in the mean level of methylation at each CpG dinucleotide among the three ethnicities, we performed an ANOVA (Table 3). Using those CpG sites that were significantly different point-wise between cases and controls within any ethnicity (Table 4), we used a one-tailed t test to see if the extent of methylation was higher in cases than controls.
Table 3.
Ethnic differences in the degree of methylation at each CpG site in the controls among ethnicities
| CpG site | Methylation (%) |
P value |
|||
|---|---|---|---|---|---|
| African-American (N = 70) |
Hispanic (N = 53) |
Caucasiana (N = 103) |
Point-wise | Experiment-wise | |
| −93 | 13.1 | 14.0 | 12.5 | n.s. | |
| −90 | 14.4 | 15.9 | 13.1 | n.s. | |
| −80 | 25.2 | 24.2 | 19.8 | 0.0274 | n.s. |
| −71 | 21.4 | 18.8 | 17.6 | n.s. | |
| −60 | 14.9 | 12.9 | 13.8 | n.s. | |
| −50 | 10.6 | 6.6 | 8.6 | n.s. | |
| −32 | 13.6 | 11.5 | 13.6 | n.s. | |
| −25 | 33.9 | 23.4 | 32.7 | 0.0016 | 0.0249 |
| −18 | 23.7 | 19.8 | 21.8 | n.s. | |
| −14 | 22.6 | 15.5 | 18.1 | 0.0135 | n.s. |
| −10 | 16.2 | 10.3 | 13.3 | 0.0154 | n.s. |
| 12 | 12.2 | 4.7 | 6.6 | 0.0005 | 0.0080 |
| 23 | 19.7 | 14.3 | 17.2 | n.s. | |
| 27 | 10.9 | 7.7 | 11.9 | n.s. | |
| 53 | 62.1 | 58.5 | 60.3 | n.s. | |
| 84 | 6.0 | 5.6 | 5.5 | n.s. | |
Caucasian data previously reported (Nielsen et al. 2009)
Table 4.
Pairwise comparisons of mean methylation levels at each CpG site in the controls between ethnicities
| CpG site | African-American versus Hispanic (P value) |
Hispanics versus Caucasian (P value) |
African-American versus Caucasian (P value) |
|||
|---|---|---|---|---|---|---|
| Point-wise | Experiment-wisea | Point-wise | Experiment-wise | Point-wise | Experiment-wise | |
| −93 | n.s. | n.s. | n.s. | |||
| −90 | n.s. | n.s. | n.s. | |||
| −80 | n.s. | n.s. | 0.0141 | n.s. | ||
| −71 | n.s. | n.s. | n.s. | |||
| −60 | n.s. | n.s. | n.s. | |||
| −50 | 0.0230 | n.s. | n.s. | n.s. | ||
| −32 | n.s. | n.s. | n.s. | |||
| −25 | 0.0005 | 0.0086 | 0.0008 | 0.0128 | n.s. | |
| −18 | n.s. | n.s. | n.s. | |||
| −14 | 0.0052 | n.s. | n.s. | n.s. | ||
| −10 | 0.0013 | 0.0210 | n.s. | n.s. | ||
| 12 | 0.0028 | 0.0447 | n.s. | 0.0047 | n.s. | |
| 23 | 0.0325 | n.s. | n.s. | |||
| 27 | 0.0314 | 0.0241 | n.s. | n.s. | ||
| 53 | n.s. | n.s. | n.s. | |||
| 84 | n.s. | n.s. | n.s. | |||
Experiment-wise P value
Because there were two measurements for each individual, logistic regression using generalized estimating equations, which accounts for the correlation between forward and reverse measurements, was performed to determine if methylation at each CpG dinucleotide differed between cases and controls (Halekoh and Hojsgaard 2006) (Table 5).
Table 5.
Percent methylation of the 16 CpG dinucleotide in the OPRM1 promoter region in former severe heroin addicts and control in African-Americans and Hispanics
| Site | African-Americans |
Hispanics |
||||||
|---|---|---|---|---|---|---|---|---|
| Methylation |
P value |
Methylation |
P value |
|||||
| Cases | Controls | GEE | Experiment-wise | Cases | Controls | GEE | Experiment-wise | |
| A | ||||||||
| −93 | 17.1 | 13.1 | 0.0361 | n.s. | 11.4 | 14.0 | n.s. | |
| −90 | 18.4 | 14.4 | 0.0406 | n.s. | 14.9 | 15.9 | n.s. | |
| −80 | 28.3 | 25.2 | n.s. | 23.1 | 24.2 | n.s. | ||
| −71 | 22.6 | 21.4 | n.s. | 15.9 | 18.8 | n.s. | ||
| −60 | 14.2 | 14.9 | n.s. | 10.0 | 12.9 | 0.025 | n.s. | |
| −50 | 10.7 | 10.6 | n.s. | 8.1 | 6.6 | 0.024 | n.s. | |
| −32 | 12.3 | 13.6 | n.s. | 13.7 | 11.5 | n.s. | ||
| −25 | 31.4 | 33.9 | n.s. | 30.5 | 23.4 | <0.001 | <0.001 | |
| −18 | 25.8 | 23.7 | n.s. | 20.7 | 19.8 | n.s. | ||
| −14 | 21.0 | 22.6 | n.s. | 20.4 | 15.5 | <0.001 | 0.001 | |
| −10 | 14.7 | 16.2 | n.s. | 12.8 | 10.3 | 0.045 | n.s. | |
| 12 | 6.6 | 12.2 | 0.0002 | 0.0032 | 3.6 | 4.7 | n.s. | |
| 23 | 17.8 | 19.7 | n.s. | 18.9 | 14.3 | 0.004 | n.s. | |
| 27 | 10.4 | 10.9 | n.s. | 12.9 | 7.7 | <0.001 | <0.001 | |
| 53 | 65.3 | 62.1 | n.s. | 57.5 | 58.5 | n.s. | ||
| 84 | 7.6 | 6.0 | n.s. | 6.7 | 5.6 | n.s. | ||
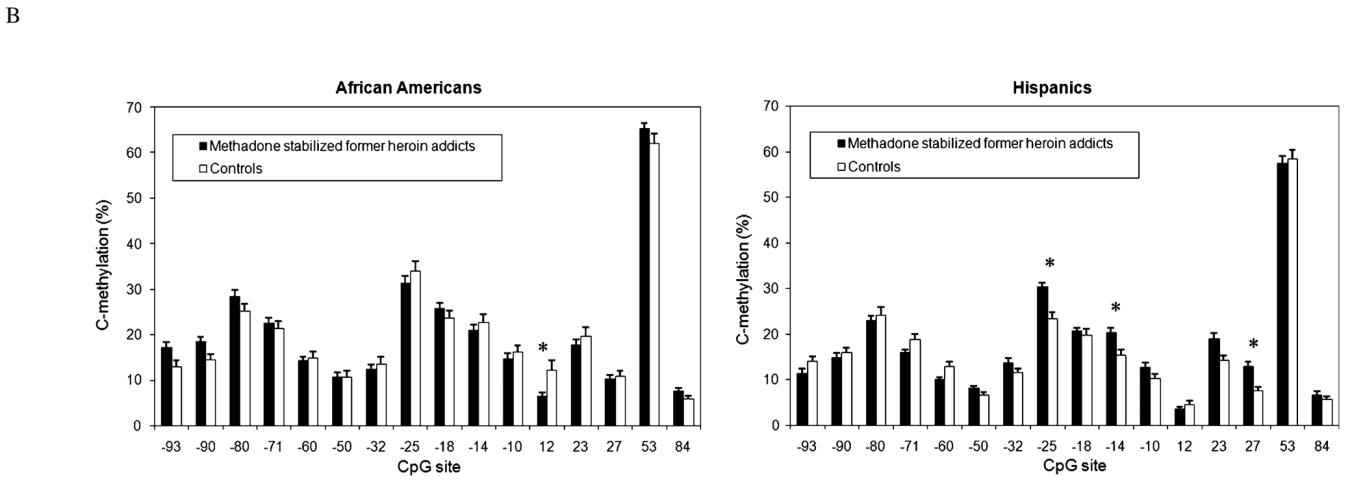
| ||||||||
P < 0.01 experiment-wise significance Error bars represent SEM
To correct for multiple testing issues, we determined the experiment-wise significance by the Bonferroni method (Tables 2, 3). We also calculated experiment-wise significance within each ethnicity (Tables 4, 5).
Results
We determined the methylation state of the upstream CpG island of the μ-opioid receptor OPRM1 promoter region in 198 African-American and 203 Hispanic subjects. The upstream OPRM1 CpG island is located from nucleotides −93 to +27 (relative to the A of the ATG translation start site). Sixteen CpG dinucleotide sites are located within this region at −93, −90, −80, −71, −60, −50, −32, −25, −18, −14, −10, +12, +23, +27, +53, and +84. There are three potential Sp1-binding sites within this amplified region. One potential Sp1-binding site spans the −18 and the −14 CpG sites, another spans the +12 CpG site, and a third spans the +84 CpG dinucleotide site.
Differences in the degree of methylation between ethnicities in controls
In this study, we have determined the degree of methylation at each of the 16 CpG sites in the upstream CpG island of the OPRM1 gene in 70 African-American and 53 Hispanic controls. We have previously reported the degree of methylation at each of these CpG sites in 103 Caucasian controls (Nielsen et al. 2009). When we compared the methylation levels of all the CpG sites in the controls of each ethnicity (Table 2), the African-Americans had a higher mean methylation level (19.6%) than was found in the Hispanics (16.1%) with an experiment-wise P = 3.48 × 10−6 or in Caucasians (17.4%) with an experiment-wise P = 0.0031. There was no experiment-wise difference in mean methylation levels between Hispanic controls and Caucasian controls.
Differences in the degree of methylation at each of the CpG sites in controls among the three ethnicities
Next, we compared, among the ethnicities, the degree of methylation at each of the CpG sites. We found significant experiment-wise differences in the degree of methylation for the −25 and the +12 CpG sites in the controls among the three ethnicities (Table 3). For the −25 CpG site, the percent methylation was similar in African-American and the previously reported Caucasian controls (33.9, 32.7%, respectively), and was higher than that found in Hispanic controls (23.4%) (experiment-wise P = 0.0249). The highest level of methylation at the +12 CpG site in the controls was in the African-Americans (12.2%) and was found to be lowest in the Hispanics (4.7%) (experiment-wise P = 0.0080); it was intermediate in the Caucasians (6.6%).
Differences in the degree of methylation at each of the CpG sites between ethnicities in controls
Comparisons between each pair of ethnicities were performed with the methylation levels in the controls at each CpG site (Table 4). There was an experiment-wise significant difference in the level of methylation between the African-American and Hispanic controls for the −25 (P = 0.0086), −10 (P = 0.0210), and the +12 CpG sites (P = 0.0447). Between the Hispanic controls and our previously reported Caucasian controls, an experiment-wise significant difference was found for the −25 CpG site (P = 0.0128). No experiment-wise significant difference was found between the African-American and the Caucasian controls.
Relationship of methylation to age in both cases and controls
No experiment-wise significant correlation was found in the level of methylation of all 16 CpG sites combined with age in either the African-American cases or controls or the Hispanic cases or controls. In the African-American controls (age range of 19–70 years, median 32, mean 33), the only CpG site with a point-wise significant correlation of methylation with age was at the −10 site (r2 = 0.2310, 95% CI 0.0009–0.4378, P = 0.049). In the African-American cases (age range of 20–65 years, median 46, mean 45), no CpG site was found to have a point-wise significant correlation of DNA methylation level with age. In the Hispanic controls (age range of 18–54 years, median 28, mean 31), there was a significant point-wise correlation of DNA methylation with age at the −60 CpG site (r2 = 0.31, 95% CI 0.03–0.54, P = 0.030). In the Hispanic cases (age range of 18–60 years, median 34, mean 35), there was a significant point-wise correlation of DNA methylation with age at the −18 CpG site (r2 = 0.19, 95% CI 0.00–0.36, P = 0.047). None of these correlations were significant after adjusting for multiple testing. Thus, although several CpG sites were found to be correlated with age with point-wise significance, there was no significance found with all 16 CpG sites combined.
Relationship of methylation to alcohol dependence in the severe former heroin addicts
To determine whether comorbid alcohol dependence may have influenced our results, we compared DNA methylation levels at each of the sixteen CpG sites and at all CpG sites combined between the former severe heroin addicts without a history of alcohol dependence and former severe heroin addicts with a history of alcohol dependence. No point-wise significant association was found in the level of DNA methylation of all 16 CpG sites combined with history of alcoholism in either the African-American or the Hispanic former severe heroin addicts. No significant point-wise association was found for DNA methylation level at any of specific CpG sites with history of alcoholism in the African-American former severe heroin addicts. In the Hispanic former severe heroin addicts, there was a significant point-wise association of history of alcoholism with DNA methylation level at the −93 and the −60 CpG sites (11.8% without alcoholism, 8.6% with alcoholism, P = 0.020; 10.5% without alcoholism, 8.0% with alcoholism, P = 0.028, respectively). Neither of these sites had a significant association of DNA methylation levels with history of alcoholism after adjusting for multiple testing.
Relationship of methylation to sex in controls
To determine whether there were differences between the sexes in the levels of DNA methylation, we examined the controls of each ethnicity stratified by sex. No point-wise significant difference of methylation level at any CpG site by sex was found in controls in any of the three ethnicities (African-American, Hispanic, or Caucasian). Assuming a significance level of 0.05 where the between-group variance is 1/10 of within-group variance, we had a 99, 95, and 99% power to detect a difference between sexes in African-Americans, Hispanics, and, Caucasians, respectively.
CpG methylation in former severe heroin addicts versus controls
Differences in methylation were determined between methadone stabilized former severe heroin addicts and controls within each ethnicity. In 170 African-Americans, significant point-wise differences in methylation levels were found between the former heroin addicts and the controls at the −93 (P = 0.0361), −90 (P = 0.0406), and +12 (P = 0.0002) CpG sites (Table 5a, b). At two of these three CpG sites, cases were hypermethylated compared with controls. When corrected for multiple testing, the only difference in methylation levels between cases and controls was at the +12 CpG site (experiment-wise P = 0.0032). The +12 CpG site was hypomethylated in the former heroin addicts (6.6%) compared to that found in the controls (12.2%).
In 173 Hispanics, methylation levels at the −60 (P = 0.025), −50 (P = 0.024), −25 (P < 0.001), −14 (P < 0.001), −10 (P = 0.045), +23 (P = 0.04), and the +27 (P < 0.001) CpG sites were significantly different point-wise between the cases and controls (Table 5). At six of these seven CpG sites, cases were hypermethylated compared with the controls. After correcting for multiple testing, the −25 (P < 0.001), −14 (P = 0.001), and the +27 (P < 0.001) CpG sites had experiment-wise significant differences in methylation levels. These three CpG sites were hypermethylated in the former heroin addicts as compared to the controls (−25 CpG site 30.5 vs. 23.4%; −14 CpG site 20.4 vs. 15.5%; +27 CpG site: 12.9 vs. 7.7%).
We examined all the CpG sites (−93, −90, −60, −50, −25, −18,−14, −10, +12, +23, +27, and +84) that had a significant difference in methylation levels between cases and controls in any of the three ethnicities (Table 5; Nielsen et al. 2009). The level of methylation was significantly higher in the cases than in the controls (14.6% cases, 13.9% controls; P = 0.0457). When we did this test separately for each ethnicity, there was no significant difference between cases and controls.
Resequencing of the OPRM1 promoter region
To determine if common variants in the OPRM1 promoter region may be contributing to our findings, we resequenced DNA from a subset of our cohort. One hundred and fortyeight subjects (47 African-American, 51 Hispanic, and 50 Caucasian) were resequenced across the OPRM1 promoter region, which contained the 16 CpG sites we examined above. The allele frequency of rs17174638 was 0.09 in African-Americans. This variant was not found in the Hispanics or Caucasians. Variant rs9282815 was found with an allele frequency of 0.02 in both the African-American and the Hispanic subjects, and was absent in the Caucasian subjects. Variant rs1799972 (C17T), which substitutes a valine for an alanine at amino acid 6, was found with an allele frequency of 0.22 in the African-Americans, 0.04 in the Hispanics, and 0.01 in the Caucasians. None of the variants we identified in the OPRM1 promoter region alters any of the CpG sites that we have studied above.
Discussion
This study extends our previous findings on the methylation levels at 16 CpG dinucleotide sites in the OPRM1 promoter region in Caucasians (Nielsen et al. 2009) to Africans American and Hispanics. We found ethnic differences in the methylation levels at specific CpG dinucleotides. Although the methylation levels at each site differed depending upon ethnicity, we found that the methadone stabilized former heroin addicts had an overall hypermethylation of their OPRM1 CpG sites as compared to controls. In cell culture studies of undifferentiated and differentiated P19 carcinoma cells, it has been reported that OPRM1 gene expression increases in concert with the demethylation of the OPRM1 promoter during differentiation or by the action of the demethylating agent azacitidine (Hwang et al. 2007). In NS20Y neuroblastoma cells that normally do not express OPRM1, the expression of OPRM1 can be induced by the demethylating agent azacitidine (Hwang et al. 2008).
When we compared ethnic differences in DNA methylation in the controls, the average methylation level of all 16 CpG sites was significantly higher in the African-Americans than in the Hispanics or the Caucasians. There was no difference in overall methylation level between the Hispanic and the Caucasian controls. However, when we compared ethnic differences in DNA methylation at individual CpG sites, methylation levels at the majority of the CpG sites (14 of 16) were not different. Methylation at the −25 and the +12 CpG sites showed the biggest difference among ethnicities. The −25 CpG site was methylated less in the Hispanics when compared with the African-Americans and the Caucasians, both of which had similar methylation levels. The +12 CpG site had the lowest level of methylation in the Hispanics, an intermediate level of methylation in the Caucasians, and the highest level in the African-Americans. At this time, the mechanisms regulating methylation at specific CpG sites are not known.
We examined the sequence of the OPRM1 promoter region for genetic variants that may alter CpG sites, and, therefore, may influence our results. The dbSNP database (www.ncbi.nlm.nih.gov/SNP/index.html) was searched for SNPs (single-nucleotide polymorphisms) located in this region. We found nine SNPs (rs17174638, rs9282814, rs9282815, rs9282816, rs41292890, rs35174096, rs1799972, rs62436460, and rs1799973). Of these, three SNPs (rs41292890, rs1799973, and rs9282816) could alter CpG sites. rs41292890 is a C to a T transition at the −25 CpG, and rs1799973 is a G to A transition at the +23 CpG. Neither of these SNPs has been validated and they are extremely rare. rs9282816 is a G-T transversion at the −32 CpG site. This SNP is rare; it has not been found in Caucasians or Hispanics, and has a minor allele frequency of 0.021 in African-Americans. This SNP does not affect our findings, as we did not find any significant results with the −32 CpG site.
In a small subset of our cohort in each of the three ethnicities resequenced, no variants were found that altered any of the 16 CpG sites that we examined. The only common variant observed was rs1799972 (C17T). This variant codes for an amino acid substitution at residue 6 of a valine for an alanine.
Our results of ethnic differences in DNA methylation were not unexpected. Ethnic diversity in overall DNA methylation has been observed in leukocyte DNA (Terry et al. 2008) and in studies on the development of cancers (e.g. Tomii et al. 2006). In contrast to our finding in a specific genomic region, the Terry et al. (2008) study found whole genome methylation of leukocyte DNA was highest in Hispanics, intermediate in Whites, and lowest in Blacks. Ethnic differences in the frequency and distribution of genetic polymorphisms also are well documented (Lao et al. 2006). The frequency of variants in genes whose products are involved in the methylation of DNA differs among ethnicities, such as in the genes coding for DNA methyltransferase 2 (Franchina and Kay 2001) and 3B (Zhao et al. 2009). Some of these variants may alter the expression and function of the enzymes involved in DNA methylation and demethylation.
Ethnic specificity was also found at specific CpG sites that were significantly different between former heroin addicts and controls. In African-Americans, the +12 CpG site was hypomethylated in the former heroin addicts. The +12 CpG site was not found to differ significantly, point-wise or experiment-wise, by age, sex, or history of alcoholism in the African-American cases or controls. In Hispanics, a different set of CpG sites had methylation levels that differed between cases and controls. Three sites (−25, −14, and +27) were hypermethylated in the Hispanic former heroin addicts compared to controls. None of these three CpG sites were found to have methylation levels that differed significantly, point-wise or experiment-wise, with age, sex, or history of alcoholism in the Hispanic cases or controls. Overall, three (−25, −14, and +12) of five (−80, −25, −14, −10, and +12) CpG sites with point-wise significant differences in methylation levels in the controls among the ethnicities had experiment-wise significant differences between cases and controls in either the African-Americans or Hispanics.
Methylation differences at the +12 CpG site found in this study may have several effects. The +12 CpG site is located in a Sp1 transcription binding site. In general, methylation of CpG sites in promoter regions decreases gene expression. In studies of Sp1 transactivation of genes with methylated and unmethylated Sp1 transcription binding sites, most studies show decreased binding of Sp1 to its cognate binding site (Alikhani-Koopaei et al. 2004; Douet et al. 2007; Iguchi-Ariga and Schaffner 1989); however, an increase in affinity for Sp1 with methylated DNA has also been observed (Jane et al. 1993). Methylation of Sp1 transcription binding sites has been shown to decrease Sp1 transactivation (Douet et al. 2007; Iguchi-Ariga and Schaffner 1989); however, increases in gene expression have also been reported (Cheng et al. 2005).
The treatment of cell lines with azacitidine has been shown to both up-regulate specific genes and to down-regulate other genes, with the up-regulation predominating (Heller et al. 2008). The latter effect may be due to decreased DNA methylation of these genes, direct inhibition by azacitidine, or to down-regulation of repressors of these genes. Therefore, methylation at the +12 CpG site of the OPRM1 gene may decrease its rate of transcription.
We cannot tell at this time whether the differences found in DNA methylation between the former heroin addicts and controls were due to altered methylation at specific CpG sites within the OPRM1 promoter region or to a global increase in DNA methylation, and whether either has a functional role in heroin addiction. The role of global methylation has been hypothesized to repress transcription of repetitive elements (Maksakova et al. 2008). Altered DNA methylation at specific CpG sites has been shown to alter the expression of various genes (e.g. Iguchi-Ariga and Schaffner 1989; Zhang et al. 2010). At the level of a single gene, methylation at specific CpG sites may prevent the binding of transcriptional activators (Tate and Bird 1993). Alternatively, methyl-CpG binding domains of transcriptional repressors may bind methylated CpG sites to either block the interaction of transcriptional activators with DNA (Tate and Bird 1993) or through the recruitment of histone modifying enzymes, such as histone deacetylases, that may alter surrounding chromatin (Ballestar and Wolffe 2001). In addition, DNA methylation can increase the compaction and rigidity of the nucleosome, which may further repress transcription (Choy et al. 2010).
Aging has been shown to increase DNA methylation globally and at specific genes (Fraga et al. 2005; Ronn et al. 2008; Sarter et al. 2005). We found several CpG sites with point-wise significant increases in DNA methylation with age. These were at the −10 CpG site in African-American controls, the −60 CpG site in Hispanic controls, and the −18 CpG site in Hispanic cases. When these results were adjusted for multiple testing, no significant experiment-wise correlation was found for DNA methylation levels at any of the CpG sites in the OPRM1 promoter region with subject age, and therefore, do not confound our analyses of ethnic or drug related differences in methylation.
Increased global DNA methylation has been found in lymphocytes of alcoholics (Bleich et al. 2006; Bonsch et al. 2004, 2006). Altered DNA methylation at specific CpG sites has been shown to alter the expression of various genes (e.g. Iguchi-Ariga and Schaffner 1989; Zhang et al. 2010). In our cohort, we found point-wise significant decreases in DNA methylation in the Hispanic, but not the African-American, severe former heroin addicts with a history of alcoholism at the −93 and the −60 CpG sites. However, these differences were not significant after adjusting for multiple testing.
In our study, we found no evidence that DNA methylation of the OPRM1 promoter region differs by sex, although some studies have reported males to have higher DNA methylation at specific genes than females (Durcova-Hills et al. 2004, 2006; El-Maarri et al. 2007; Sarter et al. 2005).
A limitation of our study is that we have not performed an analysis of population structure. In other studies conducted in our laboratory (Levran et al. 2008, 2009; Nielsen et al. 2008), 91% of the African-American controls and 95% of the Caucasian controls were analyzed for population structure. Those analyses have demonstrated that the self-identified ethnicities are quite accurate. Furthermore, the Risch group found that self-identification of race/ethnicity of subjects was similar to that found using microsatellite markers and genetic cluster membership; only five of 3,363 subjects were miscategorized (Tang et al. 2005). Similar findings were reported by Liu et al. using 1,334 subjects self-identified as African-American, European American, or Hispanic (Liu et al. 2006).
The methylation differences found between cases and controls could be due to several factors (Nielsen et al. 2009). Methylation levels at specific CpG sites in the OPRM1 promoter region may be a predisposing factor for vulnerability to develop heroin addiction. Although methylation may be inherited through genomic imprinting, examination of our published data (Nielsen et al. 2009), in which we determined methylation on individual strands of cloned DNA, showed that the number of methylated CpG sites in each individual had a unimodal distribution. Imprinted genes show a bimodal methylated CpG distribution (e.g. Durcova-Hills et al. 2006; El-Maarri et al. 2007); therefore, it is unlikely that genomic imprinting occurs in the OPRM1 promoter region. The alterations in DNA methylation may be due to major life events occurring prior to heroin addiction, to chronic heroin use, or to long-term methadone maintenance pharmacotherapy.
This study demonstrates ethnic diversity of DNA methylation globally and at specific CpG sites. Several of the CpG sites that we analyzed were significantly different experiment-wise among the ethnicities. This finding has implications for future studies on the role of DNA methylation. Population stratification has been found to produce false-positive and false-negative findings in genetic studies (Enoch et al. 2006). Possible confounding factors, such as population structure, must be addressed in future DNA methylation studies. We have demonstrated significant differences in DNA methylation between former heroin addicts and controls specific to each ethnicity. This extends our previous study on Caucasians and demonstrates that DNA methylation may play a role in heroin addiction or pharmacotherapy in African-Americans and Hispanics.
Acknowledgments
We thank all of our clinical staff for recruiting, screening, and assessing study subjects. We are grateful to C. D. Allis for his helpful advice prior to the start of this project and to O. Levran for critical review of this manuscript. This study was supported by National Institutes of Health (NIH) Grants R03-DA022266 (D.A.N.), P60-DA005130 (M.J.K.), R01-MH79880 (M.J.K.), RR UL1RR024143 (B.C.), and NSFC Grants 30730057 and 30700442 from the Chinese Government (J.O.). The experiments described in this manuscript comply with the current laws of the USA.
Footnotes
Conflict of interest statement The authors declare that they have no conflict of interest.
Contributor Information
David A. Nielsen, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA; Menninger Department of Psychiatry and Behavioral Sciences, Baylor College of Medicine, Houston, TX, USA; Michael E. DeBakey V.A. Medical Center, Houston, TX, USA
Sara Hamon, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA.
Vadim Yuferov, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA.
Colin Jackson, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA.
Ann Ho, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA.
Jurg Ott, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Mary Jeanne Kreek, Laboratory of the Biology of Addictive Diseases, The Rockefeller University, Box 171, 1230 York Avenue, New York 10065, USA kreek@rockefeller.edu.
References
- Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114:1146–1157. doi: 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem. 2001;268:1–6. doi: 10.1046/j.1432-1327.2001.01869.x. [DOI] [PubMed] [Google Scholar]
- Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res. 2006;30:587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Reulbach U, Kornhuber J, Bleich S. Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm. 2004;111:1611–1616. doi: 10.1007/s00702-004-0232-x. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Kornhuber J, Bleich S. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport. 2005;16:167–170. doi: 10.1097/00001756-200502080-00020. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Fiszer R, Frieling H, Kornhuber J, Bleich S. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm. 2006;113:1299–1304. doi: 10.1007/s00702-005-0413-2. [DOI] [PubMed] [Google Scholar]
- Cheng CK, Chow LW, Loo WT, Chan TK, Chan V. The cell cycle checkpoint gene Rad9 is a novel oncogene activated by 11q13 amplification and DNA methylation in breast cancer. Cancer Res. 2005;65:8646–8654. doi: 10.1158/0008-5472.CAN-04-4243. [DOI] [PubMed] [Google Scholar]
- Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee TH. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PM, Ramachandran K, Vanwert J, Ferdinand L, Gopisetty G, Reis IM, Singal R. Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Mol Cancer. 2006;5:28. doi: 10.1186/1476-4598-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douet V, Heller MB, Le Saux O. DNA methylation and Sp1 binding determine the tissue-specific transcriptional activity of the mouse Abcc6 promoter. Biochem Biophys Res Commun. 2007;354:66–71. doi: 10.1016/j.bbrc.2006.12.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcova-Hills G, Burgoyne P, McLaren A. Analysis of sex differences in EGC imprinting. Dev Biol. 2004;268:105–110. doi: 10.1016/j.ydbio.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Durcova-Hills G, Hajkova P, Sullivan S, Barton S, Surani MA, McLaren A. Influence of sex chromosome constitution on the genomic imprinting of germ cells. Proc Natl Acad Sci USA. 2006;103:11184–11188. doi: 10.1073/pnas.0602621103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Maarri O, Becker T, Junen J, Manzoor SS, Diaz-Lacava A, Schwaab R, Wienker T, Oldenburg J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet. 2007;122:505–514. doi: 10.1007/s00439-007-0430-3. [DOI] [PubMed] [Google Scholar]
- Enoch MA, Shen PH, Xu K, Hodgkinson C, Goldman D. Using ancestry-informative markers to define populations and detect population stratification. J Psychopharmacol. 2006;20:19–26. doi: 10.1177/1359786806066041. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchina M, Kay PH. Novel nucleotide substitutions within the coding region of DNMT2 are in strong linkage disequilibrium in Caucasians and Japanese. Hum Hered. 2001;52:210–216. doi: 10.1159/000053378. [DOI] [PubMed] [Google Scholar]
- Halekoh U, Hojsgaard S. The R package geepack for generalized estimating equations. J Stat Softw. 2006;15:1–11. [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller G, Schmidt WM, Ziegler B, Holzer S, Mullauer L, Bilban M, Zielinski CC, Drach J, Zochbauer-Muller S. Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008;68:44–54. doi: 10.1158/0008-5472.CAN-07-2531. [DOI] [PubMed] [Google Scholar]
- Hillemacher T, Frieling H, Luber K, Yazici A, Muschler MA, Lenz B, Wilhelm J, Kornhuber J, Bleich S. Epigenetic regulation and gene expression of vasopressin and atrial natriuretic peptide in alcohol withdrawal. Psychoneuroendocrinology. 2009;34:555–560. doi: 10.1016/j.psyneuen.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, Law PY, Wei LN, Loh HH. Evidence of endogenous mu opioid receptor regulation by epigenetic control of the promoters. Mol Cell Biol. 2007;27:4720–4736. doi: 10.1128/MCB.00073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, Law PY, Wei LN, Loh HH. Epigenetic programming of mu opioid receptor gene in mouse brain is regulated by MeCP2 and brg1 chromatin remodelling factor. J Cell Mol Med. 2008;13:3591–3615. doi: 10.1111/j.1582-4934.2008.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989;3:612–619. doi: 10.1101/gad.3.5.612. [DOI] [PubMed] [Google Scholar]
- Jane SM, Gumucio DL, Ney PA, Cunningham JM, Nienhuis AW. Methylation-enhanced binding of Sp1 to the stage selector element of the human gamma-globin gene promoter may regulate development specificity of expression. Mol Cell Biol. 1993;13:3272–3281. doi: 10.1128/mcb.13.6.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreek MJ, Bart G, Lilly C, LaForge KS, Nielsen DA. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol Rev. 2005;57:1–26. doi: 10.1124/pr.57.1.1. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Lao O, van Duijn K, Kersbergen P, de Knijff P, Kayser M. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am J Hum Genet. 2006;78:680–690. doi: 10.1086/501531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Londono D, O’Hara K, Nielsen DA, Peles E, Rotrosen J, Casadonte P, Linzy S, Randesi M, Ott J, Adelson M, Kreek MJ. Genetic susceptibility to heroin addiction: a candidate gene association study. Genes Brain Behav. 2008;7:720–729. doi: 10.1111/j.1601-183X.2008.00410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Londono D, O’Hara K, Randesi M, Rotrosen J, Casadonte P, Linzy S, Ott J, Adelson M, Kreek MJ. Heroin addiction in African Americans: a hypothesis-driven association study. Genes Brain Behav. 2009;8:531–540. doi: 10.1111/j.1601-183X.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin J, Schmitt AO, Adorjan P, Hildmann T, Piepenbrock C. Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinformatics. 2004;20:3005–3012. doi: 10.1093/bioinformatics/bth346. [DOI] [PubMed] [Google Scholar]
- Liu X-Q, Paterson AD, John EM, Knight JA. The role of self-defined race/ethnicity in population structure control. Ann Human Genet. 2006;70:496–505. doi: 10.1111/j.1469-1809.2005.00255.x. [DOI] [PubMed] [Google Scholar]
- Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008;65:3329–3347. doi: 10.1007/s00018-008-8494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan AT, Luborsky L, Woody GE, O’Brien CP. An improved diagnostic evaluation instrument for substance abuse patients: the addiction severity index. J Nerv Ment Dis. 1980;168:26–33. doi: 10.1097/00005053-198001000-00006. [DOI] [PubMed] [Google Scholar]
- Michelotti GA, Brinkley DM, Morris DP, Smith MP, Louie RJ, Schwinn DA. Epigenetic regulation of human alpha1d-adrenergic receptor gene expression: a role for DNA methylation in Sp1-dependent regulation. FASEB J. 2007;21:1979–1993. doi: 10.1096/fj.06-7118com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Nielsen DA, Ji F, Yuferov V, Ho A, Chen A, Levran O, Ott J, Kreek MJ. Genotype patterns that contribute to increased risk for or protection from developing heroin addiction. Mol Psychiatry. 2008;13:417–428. doi: 10.1038/sj.mp.4002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, Kreek MJ. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873. doi: 10.1038/npp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova SI, He F, Bai J, Cutrufello NJ, Lidow MS, Undieh AS. Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS One. 2008;3:e1919. doi: 10.1371/journal.pone.0001919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:565–570. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, 3rd, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig RA, Yarmolinsky A. Federal regulation of methadone treatment. National Academy Press; Washington, DC: 1995. [PubMed] [Google Scholar]
- Ronn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, Tuomi T, Isomaa B, Groop L, Vaag A, Ling C. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51:1159–1168. doi: 10.1007/s00125-008-1018-8. [DOI] [PubMed] [Google Scholar]
- Sarter B, Long TI, Tsong WH, Koh WP, Yu MC, Laird PW. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum Genet. 2005;117:402–403. doi: 10.1007/s00439-005-1317-9. [DOI] [PubMed] [Google Scholar]
- Schug J, Overton GC. TESS: transcription element search software on the www. computational biology and informatics laboratory. School of Medicine, University of Pennsylvania; Philadelphia, PA: 1977. www. computational [Google Scholar]
- Tang H, Quertermous T, Rodriguez B, Kardia SL, Zhu X, Brown A, Pankow JS, Province MA, Hunt SC, Boerwinkle E, Schork NJ, Risch NJ. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet. 2005;76:268–275. doi: 10.1086/427888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, Gamble MV, Susser E. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev. 2008;17:2306–2310. doi: 10.1158/1055-9965.EPI-08-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomii K, Tsukuda K, Toyooka S, Kote H, JHanafusa T, Asano H, Naitou M, Doihara H, Kisimoto T, Katayama H, Pass HI, Kate H, Shimizu N. Aberant promoter methylation of insulin-like growth factor binding protein-3 gene in human cancers. Int J Cancer. 2006;120:566–573. doi: 10.1002/ijc.22341. [DOI] [PubMed] [Google Scholar]
- Zhang H, Darwanto A, Linkhart TA, Sowers LC, Zhang L. Maternal cocaine administration causes an epigenetic modification of protein kinase C epsilon gene expression in fetal rat heart. Mol Pharmacol. 2007;71:1319–1328. doi: 10.1124/mol.106.032011. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wu M, Xiao H, Lee MT, Levin L, Leung YK, Ho SM. Prostate. 2010. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Du W, Gu D, Wang D, Xue F, Ge J, Sui T, Yang R. DNMT3B 579G>T promoter polymorphism and the risk for idiopathic thrombocytopenic purpura in a Chinese population. Acta Haematol. 2009;122:31–35. doi: 10.1159/000235616. [DOI] [PubMed] [Google Scholar]