

Abstract
Background
Thoracic aortic aneurysms leading to acute aortic dissections (TAAD) are the major diseases that affect the thoracic aorta. Approximately 20% of patients with TAAD have a family history of TAAD, and these patients present younger with more rapidly enlarging aneurysms than patients without a family history of aortic disease.
Methods and Results
A large family with multiple members with TAAD inherited in an autosomal dominant manner was identified. The ascending aortic aneurysms were associated with slow enlargement, a low risk of dissection, and decreased penetrance in women. Genome-wide linkage analysis was performed and a novel locus on chromosome 12 was identified for the mutant gene causing disease in this family. Of the 12 male members who carry the disease-linked microsatellite haplotype, nine had ascending aortic aneurysms with an average diameter of 4.7 cm and average age of 55 years (age range, 32-76) at the time of diagnosis; only one individual had progressed to acute aortic dissection and no other members with aortic dissections were identified. Women harboring the disease-linked haplotype did not have thoracic aortic disease, including an 84 year old woman. Sequencing of 9 genes within the critical interval at the chromosome 12 locus did not identify the mutant gene.
Conclusion
Mapping a locus for ascending thoracic aortic aneurysms associated with a low risk of aortic dissection supports our hypothesis that genes leading to familial disease can be associated with less aggressive thoracic aortic disease.
Keywords: acute aortic dissection genes, aneurysm, genome-wide analysis, mapping, risk prediction
Introduction
The natural history of ascending aortic aneurysms in the absence of surgical intervention is to progressively enlarge over time and ultimately lead to an aortic dissection (Stanford type A) or rupture. Type A aortic dissections are life-threatening events causing sudden death in approximately 40% of affected individuals, and emergency repair of these dissections are associated with a high degree of morbidity and medical expenditure. In contrast, prophylactic repair of an ascending aortic aneurysm is associated with very low morbidity and mortality, leading to the current recommendation to repair an ascending aortic aneurysm before it dissects or ruptures.1 Although medical treatment can slow the enlargement of ascending aortic aneurysms, the mainstay of treatment to prevent an aortic dissection is surgical repair when the aortic diameter expands to 5.0 – 5.5 cm.1 However, aortic dissection can occur in some patients who have little or no aortic enlargement. In fact, data from the International Registry of Aortic Dissections (IRAD) indicate that nearly 60% of aneurysms dissect at aortic diameters <5.5 cm.2 Therefore, the optimal aortic diameter when the risk of aortic dissection exceeds that of surgical repair is still debated.
Family studies indicate that up to 20% of TAAD patients who do not have a known genetic syndrome, have a first-degree relative with the disease.3;4 Familial TAAD (FTAAD) is primarily inherited in an autosomal dominant manner with decreased penetrance and variable expression, including risk for dissection at a given aortic diameter. Although FTAAD families who experience aortic dissections with minimal or no aortic enlargement have been described,5 families with thoracic aneurysms associated with a low risk of dissection have not been reported. In fact, studies have determined that TAAD patients with similarly affected first degree relatives present younger with faster enlarging aneurysms than patients without a family history, suggesting familial disease is more clinically aggressive than sporadic disease.4 Genetic heterogeneity of FTAAD is established and four genes causing the disease have been identified: TGFBR1, TGFBR2, MYH11, and ACTA2.6;7 Additionally, two disease loci have been mapped but the causative genes have yet to be identified.8;9
We describe here a large family (TAA254) with autosomal dominant inheritance of TAAD. Interestingly, aortic imaging identified aortic root aneurysms in nine family members and confirmed slow enlargement in at least one family member, but only one member had progressed to an acute aortic dissection. We hypothesize that the mutant gene at the novel chromosomal locus identified for TAAD in this family leads to stable ascending aneurysms with a slow rate of enlargement and a low risk of progression to an acute aortic dissection.
Materials and Methods
Subjects
This study was approved by the Institutional Review Board at the University of Texas Health Science Center at Houston and all subjects gave informed consent to participate in the study. Relevant medical records were obtained from hospitals and health care providers, including imaging studies, cardiology and surgical reports. Measurement of the ascending aorta was performed using 2-D echocardiography and cross-sectional diameters were obtained at four levels: the aortic annulus, the sinuses of Valsalva, the sino-tubular junction, and the ascending aorta. Individuals with aortic diameters greater than or equal to 4.2 cm were scored as affected. First degree relatives of affected individuals were imaged thoroughly for ascending aortic disease whereas peripheral artery aneurysms detected incidentally were based on report or confirmed through review of imaging studies. Physical examinations were performed on available affected family members.
Linkage Analysis
Genomic DNA from seven family members were analyzed using a 50K GeneChips Hind array from Affymetrix following manufacturer's protocol. For the fine mapping, linkage analysis using DNA from 11 additional family members was performed using microsatellite markers. Primers and map locations were based on the MAP-O-MAT (http://compgen.rutgers.edu/mapomat/) and the NCBI UniSTS (http://www.ncbi.nlm.nih.gov/sites/entrez?db=unists) databases. Fluorescent-labeled PCR products were generated using a universal fluorescent-labeled primer set following published protocols.8 The amplified products were analyzed using the ABI Prism 3130xl Genetic Analyzer and allele distribution was assigned using the Genemapper 4.0 software (Applied Biosystems).
DNA Sequencing Protocol
Bidirectional exon sequencing of ACTA2, TGFBR1, TGFBR2, MYH11, and candidate genes in the TAAD5 locus were done using intron-based, exon-specific primers. PCR amplifications were carried out using HotStar Taq™ DNA polymerase (Qiagen Inc.Valencia, CA). PCR products were treated with EXOSAP-IT (Affymetrix, Inc. OH) to digest the primers and followed with sequencing PCR using the BigDye™ sequencing reaction mix (Applied Biosystems, CA). The sequencing PCR products were purified using the BigDye XTerminator kit (Applied Biosystems, CA) and then loaded on an ABI3730xl sequencing instrument using the Rapid36 run module. DNA sequencing results were analyzed using the Mutation Surveyor software (SoftGenetics, PA).
Histoloy and Immunohistochemistry Analysis of Aortic Tissue
Hematoxylin & eosin as well as pentachrome Movat staining were carried out using standard procedures. Mouse monoclonal smooth muscle cell (SMC) ACTA2 antibody from Sigma (A5228) was used for immunohistology staining.7
Statistical Analysis
Multipoint linkage analyses of the Affymetrix 50K SNP array data was performed with the Allegro program 2.0.10 An autosomal dominant model for TAAD with a disease-gene frequency of 0.00006 was assumed. For males, four age-dependent liability classes described previously were used.8 Because the penetrance in females is extremely low, a separate liability class was constructed and a penetrance value of 0.0001 for risk genotypes was used. Multipoint non-parametric (NPL) and parametric LOD scores were calculated by a sliding window of 180-200 SNPs within the Allegro program. Linkage analysis using microsatellite markers and an extended pedigree was performed as previously described.8
Results
Clinical Data
A large family of Northern European descent with multiple members with thoracic aortic aneurysms was identified after the proband (Figure 1, III:10) presented with an acute type A dissection at the age of 32 years. On presentation, magnetic resonance (MRI) imaging revealed an enlarged aorta with a maximum diameter of 6.6 cm involving the sinuses of Valsalva, and a dissection of the ascending aorta. A paternal uncle (II:1) was reported to have been diagnosed with aortic dilatation at the age of 47 years, and aortic imaging of at risk relatives identified seven additional family members with aortic root aneurysms involving the segment of the ascending aorta from the valvular annulus to the sinotubular junction. In this family, maximal aortic diameters ranged in size from 4.1 to 6.6 cm in diameter at the time of diagnosis, which on average was 54.7 ± 15.6 years old (age range, 32 to 76 years; Table 1). Of note, none of the affected individuals in generation II have had an acute dissection but rather these individuals appear to have slow enlarging and stable aneurysms measuring less than 5.0 cm at the time of diagnosis. In addition, individual III:5 has had serial aortic imaging studies over a period of 6 years, which did not show a significant increase in the size of the aortic aneurysm. The proband (III:10) is the youngest family member to present with a thoracic aortic disease and the only member who had a dissection with no other risk factors for TAAD (e.g., hypertension or bicuspid aortic valve). Interestingly, all of the affected family members are men. None of the women have been diagnosed with aortic disease despite aortic imaging, and reduced penetrance is evident in individual II:10 who is 84 years old with no history of aortic disease but has an affected son (III:5).
Figure 1.

Pedigree of the TAA254 family indicating individuals with ascending aortic and peripheral artery aneurysms, and the haplotypes on the TAAD5 locus. Haplotype constructed for markers D12S1669, D12S1057, D12S1701, D12S1635, D12S1586, D12S1691, D12S1686, and D12S335 are presented and the TAAD-linked haplotype is boxed. Closed symbols indicate “affected” individuals; open symbols indicate unaffected or unknown disease status, and an asterisk marks the individuals who were included in the whole genome SNP linkage analysis.
Table 1.
Clinical data of affected family members.
| Individual | Age | Gender | Asc aneurysm, age at diagnosis | Type A dissection, age at diagnosis | Aortic measurements(cm)* | CAD | HTN | HLD | Smoking | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AN | SV | STJ | Asc | BSA | Age | |||||||||
| II:1 | 65 | M | +, 47 | − | 5.0** | 59 | + | + | + | + | ||||
| 4.8 | 59 | |||||||||||||
| 5.0** | 60 | |||||||||||||
| 2.9 | 5.2 | 4 | 4.3 | 2.29 | 61 | |||||||||
| II:3 | 68 | M | +, 64 | − | 4.7 | 64 | − | + | − | |||||
| II:8 | 80 | M | +, 76 | − | 4.5 | 3.4 | 3.6 | 1.92 | 76 | − | − | + | ||
| II:12 | 61 | M | +, 50 | − | 4.4** | 4.0** | 57 | − | − | − | + | |||
| 2.7 | 4.7 | 3.6 | 4.1 | 2.25 | 58 | |||||||||
| II:13 | 79 | M | +, 75 | − | 2.4 | 4.5 | 4.5 | 2.2 | 75 | − | + | + | + | |
| 4.5 | 4.2 | 2.26 | 76 | |||||||||||
| 5.3** | 3.5** | 76 | ||||||||||||
| III:4 | 44 | M | +, 40 | − | 2.8** | 4.5** | 3** | 2.02 | 40 | − | − | − | + | |
| III:5 | 50 | M | +, 44 | − | 4.1 | 2.4 | 44 | − | + | + | ||||
| 3.9 | 2.4 | 44 | ||||||||||||
| 4.3 | 2.4 | 45 | ||||||||||||
| 4.4 | 2.4 | 49 | ||||||||||||
| 2.4 | 4.2 | 3.1 | 3.1 | 2.3 | 50 | |||||||||
| III:10 | 37 | M | +, 32 | +, 32 | 6.6** | 2.21 | 32 | − | − | − | − | |||
| III:12 | 49 | M | +, 44 | − | 2.5 | 4.2 | 3.6 | 2.22 | 44 | − | − | + | ||
Symbols are as follows:
aortic measurements were obtained by 2D echocardiography;
aortic measurements were obtained by MRI or CT scan;
AN = Aortic Annulus, SV = Sinuses of Valsalva, STJ = Sinotubular junction, Asc = Ascending aorta, BSA = body surface area (m2), CAD = coronary artery disease, HTN = hypertension, HLD = hyperlipidemia, += present, − = absent, blank = not known.
In addition to TAAD, some members of this family have early onset and bilateral peripheral artery aneurysms. Individual III:4 has an aortic root aneurysm (4.5 cm) as well as bilateral dilatation of his iliac arteries that was detected incidentally on MR angiogram at the age of 40 years. A paternal uncle (II:6) was also reported to have had repair of lower peripheral artery aneurysms in his 20s although echocardiographic imaging prior to his death did not show ascending aortic dilatation. In addition, individual II:5 who passed away at the age of 55 years from multiple myocardial infarctions also had bilateral iliac aneurysms by report.
Examination of ascending aortic tissue from one affected family member (II:13) was remarkable for focal loss of smooth muscle cells (SMCs) and increased proteoglycan deposition, but only minimal degradation of the elastic fibers (Figure 2). Sequencing of the ACTA2, MYH11, TGFBR1, and TGFBR2 genes using DNA from the proband did not identify a mutation in any of these genes.
Figure 2.
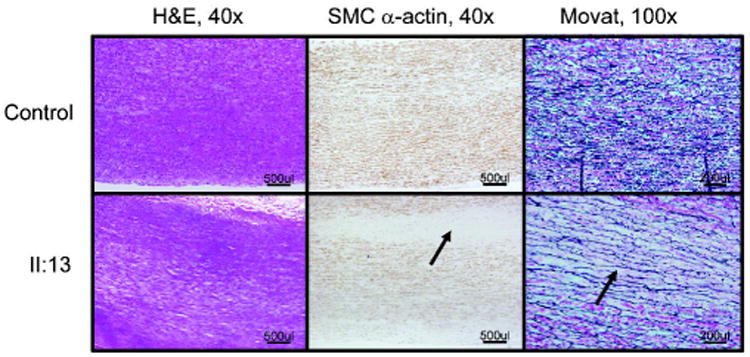
Pathology of the aortic wall from an affected individual in TAA254 (II:13). H&E, SMC-specific α-actin immunohistochemistry, and Movat staining of aorta from the patient demonstrates medial degeneration of the aortic media, characterized by increased proteoglycan deposition (blue on Movat stain, arrow) and focal loss of SMCs (SMC α- actin staining, arrow). Elastic fibers appear to be relatively intact compared with the control but there is widening between the elastic fibers (black on Movat staining, arrow).
Linkage of FTAAD to a locus on chromosome 12
To map the gene causing TAAD in this family, a genome-wide linkage analysis was performed on seven family members using the Affymetrix 50K SNP array (Figure 1). Assuming an age-dependent model of penetrance and reduced penetrance in females, parametric multipoint LOD score analysis for the TAAD phenotype yielded peaks at 1q32.1, 8p12-21, 12q13-14, and 17p12-13 with LODmax scores of more than 1.0 (Figure 3A). Fine mapping using microsatellite markers at these loci using DNA from 11 other members excluded 1q32.1, 8p12-21, and 17p12-13 as the location of the mutant gene causing TAAD (Table 2). Microsatellite genotyping however confirmed linkage to chromosome 12, designated as TAAD5 locus. To define the critical interval of this locus, 40 microsatellite markers were genotyped and microsatellite haplotypes constructed. A chromosomal recombination in individual III:12 was observed between D12S1669 and D12S1057 that defined the proximal boundary of this interval. Also, a chromosomal recombination in individuals III:4, III:5 and III:10 were observed between D12S75 and D12S335, which defined the distal boundary of this interval. Therefore, the critical interval of TAAD5 locus was defined between D12S1669 (19.5 Mb) and D12S335 (68.1 Mb) (Figure 3B). Pairwise and multipoint LOD score calculations based on the five-liability class model gave a maximum parametric LOD score of 2.7 between markers D12S1691 and D12S1726. A maximum NPL LOD score of 3.6 was also obtained in the same marker region (exact NPL P-value =0.01) (Figure 3B).
Figure 3.

Genome wide linkage analysis of family TAA254 maps to the TAAD5 locus. (A) Parametric multipoint LOD score profile for TAAD across the human genome in family TAA254 based on the Affymetrix 50K GeneChips Hind array data. Human chromosomes are concatenated from p-terminal (left) to q-terminal (right) on the x axis. The parametric LOD score is on the y axis and is correlated to physical location of human chromosome on the x axis. Four candidate loci on chromosome 1, 8, 12, and 17 were identified with a parametric multipoint LOD score over 1.0 for TAAD. (B) Multipoint linkage analysis of TAA254 demonstrates positive linkage of microsatellite markers with TAAD at the TAAD5 locus. x axis is physical distance of chromosome 12 and y axis is LOD score. The lines of non-parametric LOD and parametric LOD are as indicated in the figure legend.
Table 2. Parametric LOD Score of Markers in the Candidate Loci.
| Microsatellite marker | Chromosome | Location (Mb) | Parametric LOD score |
|---|---|---|---|
|
| |||
| D1S2622 | 1 | 200.1 | −3.12 |
| D1S2655 | 1 | 202.6 | −2.42 |
| D1S249 | 1 | 205.7 | −0.82 |
| D1S2692 | 1 | 208.0 | −1.94 |
|
| |||
| D8S298 | 8 | 21.8 | −2.67 |
| D8S1752 | 8 | 22.7 | −2.69 |
| D8S1771 | 8 | 25.4 | −3.30 |
| D8S1048 | 8 | 26.8 | −0.90 |
| D8S1820 | 8 | 28.0 | −1.91 |
| D8S1750 | 8 | 35.4 | −1.65 |
|
| |||
| D12S1301 | 12 | 44.1 | 2.68 |
| D12S85 | 12 | 47.3 | 2.41 |
| D12S2196 | 12 | 48.7 | 1.84 |
| D12S1691 | 12 | 57.5 | 2.71 |
| D12S355 | 12 | 59.3 | 2.72 |
| D12S1726 | 12 | 62.4 | 2.70 |
| D12S1686 | 12 | 65.7 | 2.15 |
|
| |||
| D17S759 | 17 | 8.8 | −1.81 |
| D17S1791 | 17 | 9.2 | −1.82 |
| D17S1148 | 17 | 10.6 | −1.81 |
| D17S1856 | 17 | 13.8 | −1.80 |
| D17S844 | 17 | 16.8 | 0.11 |
| D17S1871 | 17 | 20.8 | −1.81 |
| D17S1873 | 17 | 27.5 | −2.55 |
| D17S2163 | 17 | 29.6 | 0.11 |
Sequencing of Candidate Genes in the TAAD5 Locus
Based on current understanding of the molecular mechanisms of TAAD, we sequenced candidate genes in the TAAD5 critical interval that are involved in maintaining the smooth muscle cell contractile function including the AVIL, ITGA5, ITGA7, ITGB7, LIMA1, MYL6, MYL6B, MYO1A, and TWF1 genes. No rare variants were identified in these genes that segregated with TAAD in this family.
Discussion
A large family was identified with autosomal dominant inheritance of ascending aortic aneurysms involving the sinuses of Valsalva that is linked to a novel TAAD locus, termed TAAD5. Serial aortic imaging studies of at least one affected family member indicate a minimal rate of aortic enlargement of less than 0.1 cm per year, which is considerably slower than the 0.21 cm per year reported for patients with FTAAD.4 Only one out of nine affected family members progressed to an acute aortic dissection despite the advanced age of many affected family members. Interestingly, none of the women in this family who carry the disease-linked microsatellite haplotype are affected with TAAD, including an 84 year woman, suggesting reduced penetrance in women. Among 12 males who carried the affected microsatellite haplotype, only nine have aortic root aneurysms, also indicating reduced penetrance in men. The average diameter of the root aneurysms is 4.7 cm at the age of diagnosis and the average age at diagnosis is 55 years.
The major risk factors for aortic dissections include the diameter of TAA, rate of aneurysm enlargement, and hypertension.11;12 Despite a recommendation to repair thoracic aortic aneurysms with diameters over 5.0 cm, there is significant variability in the risk of dissection based on size, with a subset of aneurysms dissecting with no aortic enlargement while other aneurysms do not dissect even at large aortic diameters of over 10 cm.13 Another identified risk for dissection is a rapidly expanding aneurysm, defined as greater than 0.5 cm increase in a year, particularly if the patient is young or is known to have inherited a genetic predisposition for the aneurysm.14 The low risk for aortic dissection associated with the aneurysms in the family reported here was most likely the result of a slow rate of enlargement of the aneurysms and a low risk of dissection at diameters less the 5.0 cm.
Accumulating data suggest that the underlying gene predisposing an individual to thoracic aortic disease predicts the risk of aortic dissection at a given aortic diameter of an ascending thoracic aneurysm. For example, Marfan syndrome is an autosomal dominant condition resulting from mutations in FBN1, and associated with TAAD, skeletal manifestations and ocular complications.15;16 Loeys-Dietz syndrome (LDS) also predisposes individuals to TAAD and is caused by mutations in either TGFBR1 or TGFBR2.14 Although both syndromes cause aortic root aneurysms, TGFBR2 mutation patients are at a high risk of aortic dissections at relatively small aortic diameters, with reported dissections occurring with minimal enlargements (4.2 cm.).14;17 In contrast, patients with FBN1 mutations are at a low risk for dissection at aortic diameters less than 5.0 cm.16 Therefore, knowing the causative gene mutation not only makes it possible to identify family members who are at risk to develop aortic disease but also predict the aortic diameter at which a dissection can occur, thereby optimizing the timing for surgical repair of a thoracic aortic aneurysm. Based on the limited information in family TAA254 reported here, the risk of dissection appears minimal until the aortic size is greater than 6.0 cm.
Interestingly, three individuals in this family had bilateral peripheral artery aneurysms at relatively young ages, one of whom has the affected haplotype (III:4) and another is an obligate carrier (II:6). Mutations in either TGFBR1 or TGFBR2 cause TAAD, along with aneurysms of other arteries, but not specifically involving the iliac or lower peripheral arteries.14 Mutations in COL3A1 cause peripheral artery aneurysms often in the absence of aortic aneurysms, but bilateral peripheral aneurysms involving a specific artery has not been reported in these patients to our knowledge.18 We have identified a similar phenotype of thoracic aortic aneurysms and iliac aneurysms in other families in our cohort of families with two or more members with TAAD (unpublished data). Once the disease gene is identified, we will be able to assess if this gene also causes iliac aneurysms.
TAAD5 locus spans 49 Mb of chromosome 12 and encodes more than 600 genes or putative transcripts. To identify the gene mutation causing TAAD in this family, we sequenced candidate genes in this locus. Because mutations in ACTA2, MYH11, TGFBR1, and TGFBR2 result in familial TAAD, we focused on genes involved in the maintenance of SMC contractile function.19 ITGA5, ITGA7, and ITGB7 were sequenced because they encode members of the integrin receptor family, which are heterodimeric membrane glycoproteins that mediate a wide spectrum of cell-cell and cell-matrix interactions. In the arterial wall, integrins are the principal receptors for the extracellular matrix (ECM) and serve as a transmembrane link between the matrix and the actin cytoskeleton and contractile units, therefore coupling the contractile force to the extracellular matrix.20 Since ACTA2 mutations are a known cause FTAAD, TWF1 (actin-binding protein, homolog 1), LIMA1 (IM domain and actin binding 1), and AVIL (advillin) were also sequenced because they encode actin binding proteins that are involved in the regulation of actin polymerization, stabilization, and function. In addition, MYL6, MYL6B, and MYO1A were also sequenced because these genes encode proteins involved in myofibril structure and function. However, no disease-causing mutation was identified in the exons and intron-exon boundaries of these genes.
Characterization of this family with aortic root aneurysms with a slow rate of enlargement associated with a low risk for aortic dissection provides an example of further clinical heterogeneity associated with FTAAD. Mapping of a novel locus for the aortic disease in this family supports the conclusion that stable aneurysms can be due to a genetic variant at a single locus. We recommended that family members with the risk haplotype be imaged for aortic disease and the ascending aneurysms surgically repaired if the aortic root was greater than 5.0 cm. At the same time, given the low risk for aortic dissection in this family, we are not certain that this is the optimal management of this family.
Thoracic aortic aneurysms and dissections (TAAD) are a common cause of premature death and have ranked as high as the 15th leading cause of death in the United States. The natural history of ascending thoracic aortic aneurysms is to progressively enlarge over time and, if not diagnosed and treated medically and surgically, ultimately lead to life-threatening acute aortic dissections or ruptures. Although medical treatments can slow the enlargement of an aneurysm, the mainstay of treatment to prevent dissections and premature deaths is surgical repair of the TAA. Surgery is typically recommended when the aneurysm reaches 5.0 – 5.5 cm in diameter; however, studies on patients presenting with acute type A dissections indicate that up to 60% present with aneurysms smaller than 5.5 cm. Family aggregation studies indicate that approximately 20% of patients with thoracic aortic disease have a family history of TAAD (FTAAD). Patients with FTAAD tend to have early age onset of aortic disease and aneurysms that enlarge more rapidly than patients without a family history, suggesting familial disease is more clinically more aggressive than sporadic disease. In study presented, we mapped a new locus for FTAAD using a single, large family with TAAD. Aortic disease in this family was characterized by sinuses of Valsalva aneurysms with slow rate of enlargement growth and a low risk of dissection, along with decreased penetrance in women. These data provide evidence that now all familial thoracic aortic disease is characterized by rapidly progressive disease.
Acknowledgments
The authors are grateful to the family who participated in this study.
Sources of Funding: This work was supported by grants P50HL083794-01 (D.M.M.), RO1 HL62594 (D.M.M.) and UL1 RR024148 (CTSA), and grants from the Vivian Smith Foundation and the Doris Duke Charitable Trust.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dong-Chuan Guo, University of Texas Health Science Center at Houston, Houston, Texas.
Ellen S. Regalado, University of Texas Health Science Center at Houston, Houston, Texas.
Charles Minn, University of Texas Health Science Center at Houston, Houston, Texas.
Van Tran-Fadulu, University of Texas Health Science Center at Houston, Houston, Texas.
Joshua Coney, University of Texas Health Science Center at Houston, Houston, Texas.
Jiumei Cao, University of Texas Health Science Center at Houston, Houston, Texas.
Min Wang, University of Texas Health Science Center at Houston, Houston, Texas.
Robert K. Yu, University of Texas M.D. Anderson Cancer Center, Houston, Texas.
Anthony L. Estrera, University of Texas Health Science Center at Houston, Houston, Texas.
Hazim J. Safi, University of Texas Health Science Center at Houston, Houston, Texas.
Sanjay S. Shete, University of Texas M.D. Anderson Cancer Center, Houston, Texas.
Dianna M. Milewicz, University of Texas Health Science Center at Houston, Houston, Texas.
References
- 1.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, Kouchoukos NT, Lytle BW, Milewicz DM, Reich DL, Sen S, Shinn JA, Svensson LG, Williams DM. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: Executive Summary. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 2.Pape LA, Tsai TT, Isselbacher EM, Oh JK, O'Gara PT, Evangelista A, Fattori R, Meinhardt G, Trimarchi S, Bossone E, Suzuki T, Cooper JV, Froehlich JB, Nienaber CA, Eagle KA. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD) Circulation. 2007;116:1120–7. doi: 10.1161/CIRCULATIONAHA.107.702720. [DOI] [PubMed] [Google Scholar]
- 3.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–11. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 4.Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, Elefteriades JA. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–5. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 5.Milewicz DM, Chen H, Park ES, Petty EM, Zaghi H, Shashidhar G, Willing M, Patel V. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol. 1998;82:474–9. doi: 10.1016/s0002-9149(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 6.Pannu H, Fadulu V, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–20. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 7.Guo DC, Pannu H, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete S, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–93. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 8.Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, Abuelo D, Dietz HC, Basson CT, Shete SS, Milewicz DM. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation. 2001;103:2461–8. doi: 10.1161/01.cir.103.20.2461. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan CJ, Casey M, He J, Veugelers M, Henderson K, Guo D, Campagna R, Roman MJ, Milewicz DM, Devereux RB, Basson CT. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469–75. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 10.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–3. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 11.Elefteriades JA. Thoracic aortic aneurysm: reading the enemy's playbook. Yale J Biol Med. 2008;81:175–86. [PMC free article] [PubMed] [Google Scholar]
- 12.Masuda Y, Takanashi K, Takasu J, Morooka N, Inagaki Y. Expansion rate of thoracic aortic aneurysms and influencing factors. Chest. 1992;102:461–6. doi: 10.1378/chest.102.2.461. [DOI] [PubMed] [Google Scholar]
- 13.Okura T, Kitami Y, Takata Y, Fukuoka T, Arimitsu J, Hiwada K. Giant unruptured aneurysm of the thoracic aorta--a case report. Angiology. 1999;50:865–9. doi: 10.1177/000331979905001012. [DOI] [PubMed] [Google Scholar]
- 14.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–98. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 15.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995;4 Spec No:1799–809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 16.Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005;111:e150–e157. doi: 10.1161/01.CIR.0000155243.70456.F4. [DOI] [PubMed] [Google Scholar]
- 17.Tran-Fadulu V, Pannu H, Kim DH, Vick GW, III, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, Willing MC, Coselli JS, LeMaire SA, Ahn C, Byers PH, Milewicz DM. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–13. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 18.Schwarze U, Schievink WI, Petty E, Jaff MR, Babovic-Vuksanovic D, Cherry KJ, Pepin M, Byers PH. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989–1001. doi: 10.1086/324123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milewicz DM, Guo D, Tran-Fadulu V, Lafont A, Papke C, Inamoto S, Pannu H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 20.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52:372–86. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]