

Abstract
Rationale
Primary ciliary dyskinesia (PCD) is an autosomal recessive, genetically heterogeneous disorder characterized by oto-sino-pulmonary disease and situs abnormalities (Kartagener syndrome) due to abnormal structure and/or function of cilia. Most patients currently recognized to have PCD have ultrastructural defects of cilia; however, some patients have clinical manifestations of PCD and low levels of nasal nitric oxide, but normal ultrastructure, including a few patients with biallelic mutations in DNAH11.
Objectives
In order to test further for mutant DNAH11 as a cause of PCD, we sequenced DNAH11 in patients with a PCD clinical phenotype, but no known genetic etiology.
Methods
We sequenced 82 exons and intron/exon junctions in DNAH11 in 163 unrelated patients with a clinical phenotype of PCD, including those with normal ciliary ultrastructure (n=58), defects in outer ± inner dynein arms (n=76), radial spoke/central pair defects (n=6), and 23 without definitive ultrastructural results, but who had situs inversus (n=17), or bronchiectasis and/or low nasal nitric oxide (n=6). Additionally, we sequenced DNAH11 in 13 patients with isolated situs abnormalities to see if mutant DNAH11 could cause situs defects without respiratory disease.
Results
Of the 58 unrelated PCD patients with normal ultrastructure, 13 (22%) had two (biallelic) mutations in DNAH11; plus, 2 PCD patients without ultrastructural analysis had biallelic mutations. All mutations were novel and private. None of the patients with dynein arm or radial spoke/central pair defects, or isolated situs abnormalities, had mutations in DNAH11. Of the 35 identified mutant alleles, 24 (69%) were nonsense, insertion/deletion or Ioss-of-function splice-site mutations.
Conclusions
Mutations in DNAH11 are a common cause of PCD in patients without ciliary ultrastructural defects; thus, genetic analysis can be used to ascertain the diagnosis of PCD in this challenging group of patients.
Keywords: Cilia, Dynein, Kartagener syndrome, Dextrocardia, Heterotaxy
INTRODUCTION
Primary ciliary dyskinesia (PCD) is a rare, genetically heterogeneous disorder. Defective ciliary and/or flagellar function underlies the clinical manifestations, which include chronic oto-sino-pulmonary disease. Situs inversus totalis occurs in ~50% of patients (Kartagener syndrome) and situs ambiguus occurs in at least 6%.[1–4]
The diagnosis of PCD is important for the initiation of clinical care. The diagnosis largely relies on demonstration of ciliary ultrastructural defects by transmission electron microscopy (EM), but this test fails to support the diagnosis of PCD in patients with normal ultrastructure. Genetic testing holds promise as a diagnostic approach in patients with a clinical phenotype compatible with PCD, as >30% of PCD can be accounted for by biallelic mutations in 12 genes.[5–23] Mutations in two genes (DNA11 and DNAH5) that code for ciliary outer dynein arm proteins are the most common genetic causes of PCD (18–30% of PCD),[9, 10, 13, 14] and mutations in the remaining genes are relatively uncommon.
DNAH11 (dynein axonemal heavy chain 11) encodes a ciliary outer dynein arm (ODA) protein. Mutations in DNAB11 were originally described in a patient with a genetic diagnosis of cystic fibrosis, but who also had features of PCD, but normal ciliary ultrastructure.[19] Subsequent reports conclusively demonstrated that mutant DNAH11 causes PCD in patients with normal ultrastructure.[19] DNAH11-mutant cilia have a reduced waveform amplitude and hyperkinetic beating pattern.[20, 21] Based on these findings, a European consensus conference modified the diagnostic algorithm for PCD, and highlighted the importance of high-speed videomicroscopy analysis to evaluate ciliary beat pattern.[24]
To estimate the mutation frequency in DNAH11 in PCD, we undertook a large study of 163 unrelated PCD patients displaying a variety of ciliary EM findings, including patients with a compatible PCD phenotype, but without ciliary ultrastructural defects.
MATERIALS AND METHODS
Subjects Evaluation
The study included 195 patients with PCD from 163 unrelated families of which 137 were simplex families with only one affected, 25 were multiplex families with two or more affected siblings and a family with 3 affected individuals from an isolated population and 13 unrelated subjects with isolated situs abnormalities (Supplement, Table E1). The majority were evaluated at the University of North Carolina (n=98) or University Hospital, Freiburg (n=38). The remaining were evaluated at sites in the Genetic Disorders of Mucociliary Clearance Consortium and other specialized PCD centers in Europe, Australia and Israel (see Supplement). Evaluations included medical and family history, physical examination, spirometry, sputum microbiology, chest radiograph and/or CT scan, and nasal nitric oxide (nNO) measurement in most patients, as described.[8, 25] The diagnosis of PCD in patients with a compatible phenotype was assessed by ciliary ultrastructure (see below). When ciliary ultrastructure by EM analysis or immunofluorescence was normal, a presumptive diagnosis was made by adjunct tests (ciliary waveform analysis, and/or nNO measurements; see Supplement).[11–13, 25, 26] Patients with isolated situs abnormalities (n=13) had normal ciliary ultrastructure and nNO, and no clinical features of PCD (Supplement, Figure E1). This study was approved by the committee for the protection of the rights of human subjects at participating institutions, and written consent was obtained.
Ciliary Ultrastructural and Waveform Analysis
Epithelial cells were obtained by nasal curettage from the inferior turbinate, processed for EM, and >= 20 cilia with adequate images were interpreted at UNC by 3 blinded observers (JLC, MRK, MWL, and/or SUM), as described.[8, 25, 27, 28] Videomicroscopy was performed as previously described.[20, 29, 30] (details in Supplement).
Mutation Profiling
DNA was extracted from blood, buccal swabs, or lymphoblastoid cell lines from proband and available relatives, as described (details in Supplement).[8, 25, 31] For the evaluation of mutation frequency amongst unrelated families, one patient with PCD per family was used for the full DNAH11 sequencing and analysis. The majority of sequencing 82 exons and splice junctions was performed by NHLBI Genotyping and Resequencing Services in Seattle (http://rsng.nhlbi.nih.gov/scripts/index.cfm) using Sanger sequencing. The remainder of sequencing was performed by Sanger sequencing at UNC (see details and primer sequences in Supplement, Methods and Table E2). Estimates of allele frequencies for missense variants were obtained using either direct sequencing or restriction endonuclease digestion (Supplement, Methods) in at least 104 chromosomes from anonymized non-PCD subjects (hemophilia patients) of Caucasian ethnicity. Additionally, 1000 Genomes (http://www.1000genomes.org/), and dbSNP public databases were queried (http://www.ncbi.nlm.nih.gov/proiects/SNP/).
cDNA Analysis
To determine the effect of splice-site variants on transcripts, RT-PCR was employed, using RNA from nasal epithelial cells or transformed lymphoblastoid cell lines, as described.[25, 27] (See details and primer sequences in Supplement, Methods and Table E3.)
RESULTS
Clinical Phenotype of Study Subjects
PCD patients
There were 195 subjects (163 families) with PCD (or presumed PCD), including 90 males (46%) and 105 females (54%) between the ages of 2 months and 75 years. Parental consanguinity was present in 21 (13%) families. The majority of families were of Caucasian origin (79%), and remaining families represented a broad mixture of ethnicities (Supplement, Table El). Situs inversus and situs ambiguus were present in 80 (41%) and 15 (8%) patients, respectively. Most patients had neonatal respiratory distress (70%), recurrent otitis media (82%), sinusitis (95%), and bronchiectasis (70%) by chest CT scan (Supplement, Table El). Of the 101 patients who had nNO measured, the values were low (24.6±22.6 nl/min; mean±SD) compared to values (376±124 nl/min; mean±SD) seen in healthy controls. [24] Other details of the clinical features and nNO levels are available (Supplement, Table El). Patients with normal ciliary ultrastructure, either by EM (Supplement, Figure El) or immunofluorescence staining techniques, were considered to have a presumptive diagnosis of PCD, based on a compatible clinical phenotype (including bronchiectasis in most) and/or situs abnormalities, as well as low levels of nNO and dyskinetic/hyperkinetic waveform and/or increased beat frequency in videomicroscopy studies, consistent with previous reports.[20]
Subjects with isolated situs abnormalities
There were 13 unrelated subjects with situs abnormalities, but no clinical features of PCD, and all who were tested (n=10) had normal nNO levels. Thus, these 13 subjects were considered to have isolated situs abnormalities unrelated to PCD (Supplement, Table El). These subjects were included because mouse models of DNAH11 ortholog [32–34] were originally reported to have isolated situs abnormalities without the respiratory phenotype.
Mutation Profiling
There were 58 unrelated patients used from mutation profiling who had a clinical phenotype, nNO levels, and/or ciliary waveform or situs abnormalities compatible with PCD, but the diagnosis couid not be confirmed either in the patients or their affected sibling by demonstration of a defect in ciliary ultrastructure. Of these 58 unrelated patients with a presumptive diagnosis of PCD, 20 had at least one mutation in DNAH11, and the clinical demographics, nNO levels, situs status, ciliary phenotype and mutations are summarized (Tables 1 and 2). Of these 20 patients, 15 had two (biallelic) mutations, including 3 homozygotes, and 12 compound heterozygotes (Table 1). Seven of the 15 patients with biallelic mutations had an affected sibling with the identical mutations (Table 2). Most of the 15 families with biallelic mutations had a PCD patient with situs abnormalities (13/15) (Table 2), which probably represents an ascertainment bias. As with PCD patients with ultrastructural defects, there was an age-related distribution of bronchiectasis in patients with biallelic mutations. Three of the 6 patients without bronchiectasis were ≤ 8 years old (Table 2).
Table 1.
Details of DNAH11 mutations in 20 unrelated patients with Primary Ciliary Dyskinesia††
| Patient Number | Family Number | Sex | Situs Status | Ciliary EM | Allele 1 | Allele 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Ex/Int | Base Change (cDNA) | Amino-Acid Change | Seg* | Ex/Int | Base Change | Amino-Acid Change | Seg* | |||||
| Homozygous Mutations | ||||||||||||
| PCD623† | UNC101 | F | SS | Normal | Ex 25 | 4438C>T | R1480X | Pat | Ex 25 | 4438C>T | R1480X | Mat |
| PCD1022† | UNC177 | M | SS | Normal | Ex 24 | 4333C>T | R1445X | Pat | Ex 24 | 4333C>T | R1445X | Mat |
| OP20-II:1‡ | OP20 | M | SI | na | Ex 71 | 11663G>A | R3888H | na | Ex 71 | 11663G>A | R3888H | na |
| Compound Heterozygous Mutations | ||||||||||||
| PCD108† | UNC14 | M | SI | Normal | Ex 26 | 4516_4517delCT | L1506SfsX10 | Mat | Int 44 | 7266+1G>A§ | T2379_Q2422del | Pat |
| PCD157 | UNC21 | F | SI | Normal | Ex 37 | 6244C>T | R2082X | Mat | Ex 73 | 11929G>T | E3977X | Pat |
| PCD761 | UNC126 | F | SI | Normal | Int 13 | 2275-1G>C§ | Y759_E889del | Mat | Ex 81 | 13213dC | R4405AfsX1 | Pat |
| PCD919† | UNC147 | M | SA | Normal | Ex 80 | 13065_67delCCT | 4356delL | Mat | Ex 80 | 13075C>T | R4359X | Pat |
| OP98-II:1† | OP98 | M | SI | Normal | Ex 48 | 7914G>C§ | W2604X | Pat | Ex 82 | 13333_34insACCA | I4445NfsX3 | Mat |
| OP406-II:1† | OP406 | M | SI | Normal | Int 23 | 4254+5G>T§ | E1366_G1418del | Mat | Int 26 | 4726-1G>A§ | E1576AfsX4 | Pat |
| PCD565 | UNC90 | M | SI | Normal | Int 33 | 5778+1G>A§ | V1821TfsX7 | Pat | Ex 80 | 13061T>A | L4354H | Mat |
| PCD1077 | UNC199 | F | SI | Normal | Ex 21 | 3901G>T | E1301X | Pat | Ex 72 | 11804C>T | P3935L | Mat |
| PCD1126 | UNC222 | F | SS | Normal | Ex 74 | 12064G>C | A4022P | na | Ex 82 | 13504_05insGAAGA | T4502RfsX14 | na |
| OP235-II:2† | OP235 | F | SI | Normal | Ex 77 | 12697C>T | Q4233X | Pat | Ex 79 | 12980T>C | L4327S | Mat |
| OP41-II:1 | OP41 | M | SI | Normal | Ex 1 | 350A>T‡ | E117V | na | Ex 44 | 7148T>C | L2383P | na |
| PCD812 | UNC128 | M | SI | na | Ex 34 | 5815G>A | G1939R | Pat | Ex 82 | 13373C>T | P4458L | Mat |
| Heterozygous Mutations | ||||||||||||
| PCD998 | UNC174 | M | SS | Normal | Ex 56 | 9113_16delAAGA | K3038TfsX13 | Pat | - | Unknown | Unknown | - |
| PCD1033 | UNC179 | F | SA | Normal | Ex 63 | 10324C>T | Q3442X | Pat | - | Unknown | Unknown | - |
| PCD1174 | UNC256 | F | SS | na | Ex 14 | 2569C>T | R857X | Mat | - | Unknown | Unknown | - |
| PCD974 | UNC162 | F | SS | Normal | Ex 60 | 9764T>C | L3255S | Mat | - | Unknown | Unknown | - |
| PCD545 | UNC-O | M | SS | Normal | Ex 33 | 5643A>T | Q1881H | na | - | Unknown | Unknown | - |
Additional demographic information in Supplement Table E1.
Mutant allele shown to segregates from either the father (Paternal) or mother's (maternal) side of the family.
Patients have affected siblings who also carry same biallelic familial mutations.
Consanguineous family.
Splice site mutations, see details in Table 3.
Abbreviations:
M = Male, F = Female, SI = Situs inversus, SA = Situs ambiguus, SS = Situs solitus, na = Not available, DA = dynein arms, EM = Electron microscopy Ex/Int = exon/intron, Mat = Maternal, Pat = Paternal
Table 2.
Clinical, demographic, and ciliary features of 20 unrelated families carrying DNAH11 mutations
| PCD patient # | Family # | Sex | Age in yrs | Ethnicity | nNO nl/min* | Situs Status | Ciliary Videomicroscopy Wave form†† | CBF (Hz)‡‡ | Neo RDS | Otitis Media | Bxsis | Sinusitis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Homozygous Mutations | ||||||||||||
| PCD623 | UNC101 | F | 24 | Caucasian | 9.7 | SS | dys-/hyperkinetic | - | yes | yes | yes | yes |
| PCD627† | F | 26 | 24.1 | SS | - | - | yes | yes | no | yes | ||
| PCD1022 | UNC177 | M | 4 | Caucasian | 12.5 | SS | - | - | yes | yes | no | yes |
| PCD1023† | M | 7.5 | 12.6 | SI | - | - | yes | yes | no | yes | ||
| OP20-II:1‡ | OP-20 | M | 12 | Turkish | na | SI | - | - | no | yes | yes | no |
|
| ||||||||||||
| Compound Heterozygous Mutations | ||||||||||||
| PCD106† | UNC14 | M | 29 | Caucasian | 14 | SS | - | - | no | yes | no | yes |
| PCD108 | M | 24 | 20 | SI | dys-/hyperkinetic | - | yes | yes | no | yes | ||
| PCD157 | UNC21 | F | 12 | Caucasian | 2.1 | SI | dys-/hyperkinetic | - | yes | yes | yes | yes |
| PCD761 | UNC126 | F | 30 | Caucasian | 24.5 | SI | dys-/hyperkinetic | 15.2 | yes | yes | yes | yes |
| PCD918† | UNC147 | F | 10 | Asian | 19.4 | SS | - | - | yes | yes | yes | yes |
| PCD919 | M | 8 | 25.5 | SA | dys-/hyperkinetic | 7.9 | yes | yes | yes | yes | ||
| OP98-II:1 | OP98 | M | 20 | Caucasian | na | SI | dys-/hyperkinetic | - | no | yes | yes | yes |
| OP98-II:2† | M | 15 | na | SS | dys-/hyperkinetic | - | na | yes | yes | yes | ||
| OP406-II:1 | OP406 | M | 1 | Caucasian | na | SI | dys-/hyperkinetic | - | na | na | na | na |
| OP406- II:2† | F | 7 | na | SS | dys-/hyperkinetic | - | yes | na | na | yes | ||
| PCD565 | UNC90 | M | 7 | Caucasian | 23.5 | SI | dys-/hyperkinetic | 10.2 | yes | yes | yes | yes |
| PCD1077 | UNC199 | F | 2 | Caucasian | 16.9 | SI | - | - | yes | yes | na | yes |
| PCD1126 | UNC222 | F | 42 | Asian | 16.2 | SS | dys-/hyperkinetic | 13.7 | no | no | yes | yes |
| OP235-II:1† | OP235 | F | 24 | Caucasian | na | SS | dys-/hyperkinetic | - | no | yes | yes | yes |
| OP235-II:2 | F | 21 | na | SI | dys-/hyperkinetic | - | yes | yes | yes | yes | ||
| OP41-II:1 | OP41 | M | 13 | Caucasian | na | SI | dys-/hyperkinetic | - | yes | yes | na | yes |
| PCD812 | UNC128 | M | 8 | Caucasian | 9 | SI | - | - | yes | yes | no | yes |
|
| ||||||||||||
| Heterozygous Mutations | ||||||||||||
| PCD998 | UNC174 | M | 29 | Caucasian | 70.4 | SS | dys-/hyperkinetic | 7.1 | no | yes | yes | yes |
| PCD1033 | UNC179 | F | 10 | Caucasian | 34.8 | SA | dys-/hyperkinetic | 10.5 | yes | yes | no | yes |
| PCD1174 | UNC256 | F | 35 | Caucasian | 32.1 | SS | dys-/hyperkinetic | 6.9 | yes | yes | yes | yes |
| PCD974 | UNC162 | F | 12 | Caucasian | 40 | SS | dys-/hyperkinetic | 14.0 | no | yes | yes | yes |
| PCD545 | UNC-O | M | 25 | Lebanese | na | SS | - | - | no | no | yes | yes |
normal nNO levels were 376±124 nl/min (mean±SD), calculated from 27 healthy subjects.[25]
affected sibling (only tested for targeted mutation).
consanguineous family (parents of the patients were related).
dyskinetic/hyperkinetic: dyskinetic means non-flexible beating pattern with reduced range of motion, especially at mid-shaft of the cilia; hyperkinetic means many fields with increased ciliary activity, particularly in the distal 1/3 of the ciliary shaft.
Abbreviations:
SI = Situs inversus, SA = Situs ambiguus, SS = Situs solitus, M = Male, F = Female, na = Not avaliable, nNO = Nasal nitric oxide, Bxsis = Bronchiectasis, Neo RDS = Neonatal respiratory distress in full term birth CBF= ciliary beat frequency, Hz= Hertz, dys = dyskinetic.
We identified 35 mutant alleles, not previously observed.[19–21] These included nonsense mutations (n=1 1), small insertions-deletions (n=6), splice-site mutations (n=7), and missense mutations (n=1 1). Except for 3 patients with homozygous mutations, each mutation appeared only once, which demonstrates extensive allelic heterogeneity (see all 32 unique mutant alleles and their corresponding protein domain in Figure 1 and Supplement, Table E4). Carrier studies in families showed that mutations were inherited in trans, and segregation analysis was consistent with an autosomal recessive trait. Selected pedigrees illustrate the segregation analysis (Figure 2), and additional families where segregation analysis was possible with either biallelic mutations (Supplement, Figure E2) or with only monoallelic mutation (Supplement, Figure E3) are presented in online Supplement.
Figure 1. Schematic representation of DNAH11 (not to the scale) showing AAA 1–6 domains, four P-loop, Microtubule binding domain (MTB) and Helix-1 and 2.
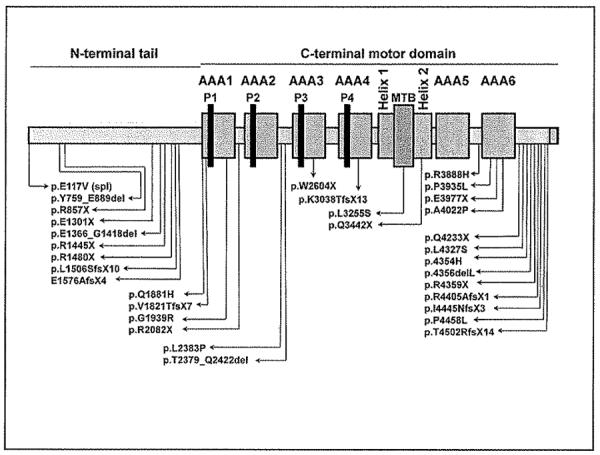
Positions of the all the mutations are shown.
Figure 2. Representative pedigrees showing autosomal recessive mode of inheritance for DNAH11 mutations.
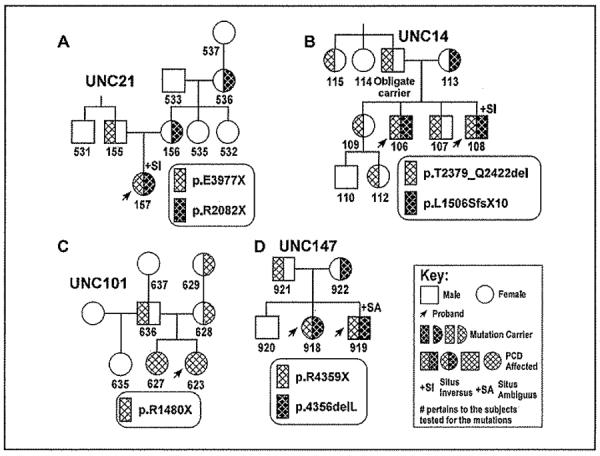
Segregation analysis from the parents, siblings and the extended family members demonstrates that mutations were inherited in trans (A–D), and there was no bias for gender or situs status. Additional pedigrees are presented in supplemental data.
cDNA Analysis of Splice-site Mutations
RNA was available for transcript studies for 6 of the 7 splice-site mutations. Three of these splice mutations (c.2275−1G>C; c.4254+5G>T; c.7266+1G>A) caused in-frame deletions of exon 14 (131 amino acids), exon 23 (53 amino acids), and exon 44 (44 amino acids), respectively (Table 3, Figure 3). Additionally, three mutations (c.4726−1G>A; c.5778+1G>A; c.7914G>C) caused out-of-frame deletions of exon 27, exons 32–35 and exon 48, respectively, leading to premature stop signals (Table 3, Figure 3).
Table 3.
Effect of DNAH11 splice mutations on cDNA transcript using Reverse Transcriptase PCR (RT-PCR) in patients with PCD
| Sample # | Intron/Exon Location | Genomic Mutations and Predicted Amino-Acid Change | cDNA Transcript after RT-PCR | Comments |
|---|---|---|---|---|
| OP41-II:1 | Exon I | c.350A>T (p.E117V) Splice defect? |
r.(spl?) RNA not available |
Second last base in exon 1 on conserved canonical splice donor site. Population studies: 0/216 control alleles and 1/326 PCD alleles |
| PCD761 | Intron 13 | c.IVS13-1G>C (c.2275-1G>C) Splice defect |
r.2275_2667del p.Y759_JE889del |
Inframe deletion of exon 14 consisting of 131 amino-acid residues Wild type amplification product: 1089 bp Mutant amplification product: 696 bp |
| OP406-II:2 | Intron 23* | c.IVS23+5G>T (c.4254+5G>T) Splice defect |
r.4096_4254del p.E1366_G1418del |
Inframe deletion of exon 23 consisting of 53amino-acid residues Wild type amplification product: 741 bp Mutant amplification product: 582 bp |
| OP406-II:2 | Intron 26 | c.IVS26-1G>A (c.4726-1G>A) Splice defect |
r.4726_4817del p.E1576AfsX4 |
Out-of-frame deletion of exon 27 leading to premature translation termination signal Wild type amplification product: 992 bp Mutant amplification product: 900 bp |
| PCD653† | Intron 33* | c.IVS33+1G>A (c.5778+1G>A) Splice defect |
r.5461_6041del p.V1821TfsX7 |
Out-of-frame deletion of exons 32–35 leading to premature translation termination signal Wild type amplification product: 1013 bp Mutant amplification product: 432 bp |
| PCD108 | Intron 44 | c.IVS44+1G>A (c.7266+1G>A) Splice defect |
r.7135_7266del p.T2379_Q2422del |
Inframe deletion of exon 44 consisting of 44 amino-acid residues Wild type amplification product: 918 bp Mutant amplification product: 786 bp |
| OP98-II:1 | Exon 48 | c.7914G>C (p.Q2638H) Splice defect |
r.7812_7914del p.W2604X |
Last base in exon 48 on conserved canonical splice donor site. Out-of-frame deletion of exon 48 leading to premature translation termination signal Wild type amplification product: 1090 bp Mutant amplification product: 987 bp |
Intron 23 and Intron 33 analysis showed the absence of last 15 bases (5 amino-acid residues) in exon 22 and 6 bases of exon 32 (2 amino-acid residues) respectively, in multiple controls depicting error in published sequence.
RNA from affected individual PCD565 was not available hence cDNA analysis was done on the carrier father (PCD653).
Figure 3. Effect of splice-site mutations on the DNAH11 transcript using Reverse Transcriptase-polymerase chain reaction (RT-PCR).
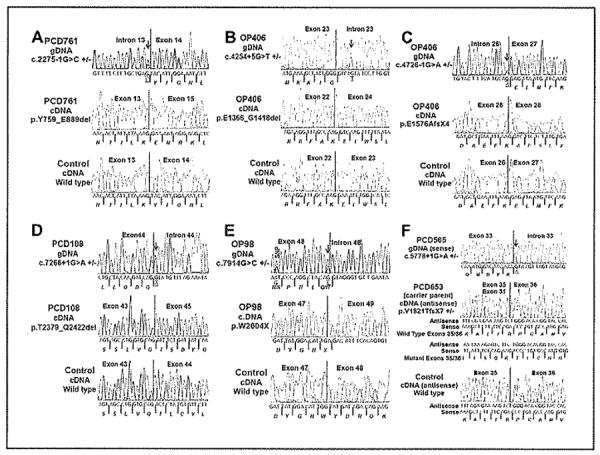
(A) Splice-acceptor site mutation in intron 13 (c.2275-1 G>C) in patient PCD761 led to the in-frame deletion of exon 14 that consisted of 131 amino-acid residues. (B) Splice-donor site mutation in intron 23 (c.4254+5G>T) in patient OP406-II:2 led to the in-frame deletion of exon 23 that consisted of 53 amino-acid residues. (C) Splice-acceptor site mutation in intron 26 (c.4726-1G>A) in patient OP406-II:2 led to out-of-frame deletion of exon 27, and resulted in a premature stop signal. (D) Splice-donor site mutation in intron 44 (c.7266+1G>A) in patient PCD 108 led to the in-frame deletion of exon 44 that consisted of 44 amino-acid residues. (E) Splice-donor site mutation in exon 48 (c.7914G>C) in patient OP98-II:1 led to out-of-frame deletion of exon 48, and resulted in a premature stop signal. (F) Splice-donor site mutation in intron 33 (c.5778+1G>A) in patient PCD565 led to out-of-frame deletion of exons 32–35, and resulted in a premature stop signal. The cDNA was available only from the carrier parent of the patient PCD565, which was used to check the transcript. All of the six panels with three electropherograms each shows the genomic location of the mutation (top) with a red arrow and bases underlined, mutant cDNA transcript (middle) and wild type transcript (bottom). Amino-acid residues are italicized and the protein product due to the out-of frame mutation is shown with the red fonts. Genomic base change for the mutation is shown with underline. A known single nucleotide polymorphism (SNP) was observed in OP98-II:1 and its location is shown. Further details on RT-PCR are shown in Table 3 (primer sequences shown in Supplement, Table E3).
Correlation of Genotype with Ultrastructure and Ciliary Waveform
The genetics of PCD involves locus, allelic, and ultrastructural heterogeneity; thus, we studied patients with different ciliary EM findings, including patients with normal ultrastructure, but compatible clinical phenotype. Mutations in DNAH11 were exclusively seen in patients with a clinical phenotype of PCD and normal ciliary ultrastructure. Each of the 14 patients (11 families) with biallelic mutations in DNAH11 that were tested by videomicroscopy had the characteristic hyperkinetic beating pattern and reduced waveform amplitude, as previously reported (see Table 2 and Supplement, Movies E1 and E2).[20] None of the other groups carried mutations, including patients with isolated situs abnormalities. In total, we identified biallelic DNAH11 mutations in 13 (22%) of the 58 unrelated families with compatible clinical phenotype, low nasal NO and confirmed normal ciliary ultrastructure and/or abnormal videomicroscopy. Despite full gene (coding region) sequencing, we found only one mutant allele in 5 patients (4 with confirmed normal ultrastructure), which could reflect either a second mutation in DNAH11 (introns or promoter regions, or large indels), or a heterozygous mutation in a different ciliary gene (which would represent digenic mode of inheritance), or biallelic mutations in a PCD gene other than DNAH11.
Population Studies
There were 10 unique missense variants, one possible single nucleotide polymorphism, 2 splice mutations, and one amino-acid deletion that were studied to examine its role as pathogenic or benign. Due to the nature of sequence based assay, certain amplicons (exons 33, 44 and 80) harbored splice and nonsense mutations in addition to variants of interest, and they were interrogated as well. Each of these variants was identified in only 1 of the 163 unrelated PCD patients tested, and never identified in 13 patients with isolated situs abnormalities. Additionally, these missense variants were not observed in at least 104 alleles tested in non-PCD individuals, ethnically matched when possible (ethnically matched controls were not available for 3 subjects). In addition, they were predicted to be deleterious based on in-silico program “Mutation Taster” (http://neurocore.charite.de/MutationTaster/). Furthermore, none of these missense variants or loss-of-function or splice mutations were seen in 1000 Genomes http://www.1000genomes.org/ or dbSNP <http://www.ncbi.nlm.nih.gov/projects/SNP/> databases, except for having been listed from this current study in dbSNP. Taken together, these data suggest that these variants are not benign polymorphisms (Supplement, Table E5).
Polymorphisms and Variants of Unknown Significance
DNAH11 is a large gene, and we identified 310 novel and/or known polymorphisms. The polymorphisms and corresponding SNP database number (http://www.ncbi.nlm.nih.gov/SNP/) are available (Supplement, Table E6). The novel variants that are not present in SNP database were considered benign, due to high minor allele frequency in the PCD patients (footnotes of Supplement, Table E6). One rare variant (c.11059A>G; p.K3687E) was seen on only one allele of a PCD patient with an ODA defect, and was never seen in either control or isolated situs abnormalities groups. This was a non-synonymous substitution, conserved (80%) across species, and present at the third last base of exon 67 near the splice-donor site. Due to the unavailability of RNA, we could not check the effect of this variant on splicing. We classified this substitution as a variant of uncertain significance, because mutations in DNAH11 are seen (otherwise) exclusively in patients with normal ciliary ultrastructure; plus, a second mutation was not identified, despite full gene sequencing.
Errors in Published Sequence of DNAH11
During analysis of cDNA from nasal epithelial cells and lymphoblastoid cell lines from two unrelated control subjects, we observed errors in the ensemble database (http://uswest.ensembl.org/index.html), and published sequence of DNAH11[19] The last 15 bases of exon 22 (and 5 amino-acid residues) are not present in the DNAH11 transcript from multiple control subjects (details in bottom panel of Figure 3B, and Supplement, Figure E4A). These 5 amino acids were previously shown in the human DNAH11,[19] but not in other species, which is congruent with sequence error. Additionally, 6 bases in exon 32 of the ensembl database (and 2 amino-acid residues) are not present in the DNAH11 transcript from multiple control subjects (correct cDNA sequence for exons 22 and 32, and multiple sequence alignment in Supplement, Figure E4), Due to errors in the publicly available sequences, the full-length DNAH11 will contain 4216 amino-acids and the mutation nomenclature for all the previously published mutations (and variants/SNPs) will change (see Supplement, Table E7 for mutation nomenclature that corresponds with the current and formerly published sequenced information).
DISCUSSION
It is challenging to confirm a diagnosis of PCD in patients with a compatible clinical phenotype, but who do not have hallmark defects in ciliary ultrastructure. Some specialized centers use nasal NO measurement as an aid to diagnosis. A few centers use videomicroscopy to evaluate ciliary waveform to confirm the diagnosis, but this assay is difficult, and limited in availability.
Mutations in DNAH11 have been reported in 4 families where PCD-affected patients have normal ciliary ultrastructure.[19–21] However, the prevalence of DNAH11 mutations, and genotype-ciliary phenotype correlations, are not well-defined. In this study, we tested the hypothesis that mutations in DNAH11 are a relatively common cause of PCD in patients with normal ciliary ultrastructure. We studied a large number of well-characterized PCD patients with different ciliary ultrastructural phenotypes to determine the frequency of DNAH11 mutations in each group.[25] In patients where ciliary ultrastructure was normal, the clinical phenotype was typical of PCD, including a high prevalence of respiratory distress in full term neonates, chronic otitis media and sinusitis, productive cough, bronchiectasis, situs abnormalities, and infertility (Supplement, Table E1). In addition, these patients had low nNO and/or abnormal immunofluorescence with ciliary antibodies and/or abnormal ciliary waveform with limited range of motion and hyperkinesis, which are compatible with PCD (Tables 1 and 2).[20]
We determined that biallelic mutations in DNAH11 are relatively common (22%) in PCD patients without a defined ciliary ultrastructural defect (Table 1). None of the PCD patients with ultrastructural defects had mutations in DNAH11. Thus, disease-causing mutations in DNAH11 appear specific for PCD patients with normal ciliary ultrastructure. It is difficult to determine the proportion of all PCD patients carrying biallelic mutations in DNAH11, since the fraction of PCD patients with normal ciliary ultrastructure is not known. However, several studies, and the experience of our centers, estimate that at least 30% of PCD patients have normal axonemal ultrastructure [2]; thus, DNAH11 mutations may occur in ~6–7% of all PCD patients.
Segregation analysis in families was consistent with trans allelic inheritance of the mutation as an autosomal recessive trait (Table 2, Figure 2 and Supplement, Figures E2 and E3). Pedigree analysis showed horizontal transmission, and carrier analysis showed that parents carried the mutation, but were clinically unaffected; hence, autosomal dominant inheritance was ruled out (Supplement, Figure E2). In the 5 patients where a second mutation was not identified, it is likely that a second mutation in DNAH11 is present in most of these patients, but not discovered by sequence analysis (e.g., promoter, intronic or large insertions-deletions).[15] Alternatively, a few of these patients may only be a carrier of a DNAH11 mutation, and the actual biallelic PCD-causing mutations are present in a different gene. Finally, there might be a heterozygous mutation in another axonemal gene, and (together with the identified DNAH11 mutation), would represent a digenic mode of inheritance; however, digenic inheritance has never been reported in PCD.
Of the 20 unrelated patients carrying mutations, there were 35 mutant alleles, including 7 splice-site mutations (Table 1). These splice-site mutations abrogated splicing in all 6 cases tested, which resulted in shorter DNAH11 transcripts (Table 3, Figure 3). We also concluded that the p.E117V splice-donor site variant (where RNA was not available) and 10 missense variants were likely disease-causing, because: (a) each variant was seen only once, and not seen in dbSNP and 1000 genomes databases; (b) variants were absent in control subjects who were tested; (c) the majority of missense mutations had a loss-of-function mutation on the trans allele; (d) the amino-acid affected by the missense mutations was highly conserved across species, and in-silico analyses predicted it to be deleterious; and (e) the majority of missense mutations were in a conserved AAA module or was on a microtubule binding domain (Table 1 and Figure 1). We also discovered some errors in the published sequence of DNAH11; thus, the mutation nomenclature needs to be updated, based on the currently revised sequence (Supplement, Table E7, Figure E4).
The ability to establish (or rule-out) a diagnosis of PCD by a genetic test in patients with a compatible phenotype and normal ciliary ultrastructure is significant at several levels. For example, several reports suggest that the vast majority (~ 90%) of patients with PCD have defined ultrastructural defects.[2, 3, 35, 36] However, this perspective may greatly underestimate the number of PCD patients with normal ciliary ultrastructure, particularly in patients with normal situs status. At an individual case level, the importance of being able to establish (or exclude) PCD by a genetic test is demonstrated by the situation in one of our families (UNCI01; Figure 2C), where one female patient (#623) had a compatible clinical phenotype and low levels of nasal NO consistent with PCD, but no situs abnormalities. Her sister (#627) also had some clinical features of PCD, as did an 8 year old paternal half-sister (#635). Before genetic testing was possible, we were unable to clarify the diagnosis of PCD in this family. Subsequently, we defined biallelic nonsense mutations in DNAH11 in the proband and the full sibling, but the half-sibling did not carry any mutation.
There are some instructive genotype-phenotype correlations in Chlamydomonas and murine orthologs of mutant DNAH11. The Chlamydomonas reinhardtii ortholog of DNAH11 is β-dynein heavy chain (β-DHC), and Chlamydomonas mutants of β-DHC can assemble outer arm subunits into the flagellar axoneme, but swimming velocity and/or beat frequency are reduced.[37–40] In humans, immunofluorescence studies show normal distribution of ODA proteins (DNAH9 and DNAH5) in a patient with biallelic DNAH11 mutations.[29] Thus, mutant DNAH11 does not cause defective ODA assembly, but causes defective ciliary function.[2, 20] The mouse ortholog of DNAH11 (Dnahc11) is left-right-dynein (lrd), and lrd null mice have situs defects.[32, 34] The spontaneously occurring mouse model of Dnahc11 (inversus viscerum mutant; iv/iv) has situs defects and recent work shows these mice have no detectable ciliary beat frequency, and suffer otitis media and rhinitis, even though they have normal ciliary ultrastructure.[32, 33,41]
In conclusion, our large scale mutation analysis indicates that biallelic mutations in DNAH11 occur in 22% of patients with a clinical phenotype of PCD, but normal ciliary ultrastructure, and is consistent with autosomal recessive mode of inheritance. Transcript analysis of six splice-site mutations revealed abrogation of normal splicing. These data clearly establish that clinical disease (PCD) occurs in patients with normal ciliary ultrastructure. This study also demonstrates that genetic analysis of DNAH11 can be useful to assist in the diagnosis of PCD, and supports the concept to search for additional genetic origins of PCD.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to all the PCD patients and the family members for their participation in this research work. The authors would like to thank Ms. Michele Manion, who founded the US PCD Foundation. We are indebted to other investigators and the coordinators of the “Genetic Disorders of Mucociliary Clearance Consortium” that is part of the Rare Disease Clinical Research Network (url: http://rarediseasesnetwork.epi.usf.edu/gdmcc/index.htm). including Dr. Jeffrey Krischer (Data Management and Coordinating Center, Tampa, Fl), Mr. Reginald Claypool, Ms. Tanya Glaser, and Ms. Meghan O'Connell (National Institute of Allergy and Infectious Diseases, Bethesda, MD), Dr. Jeffrey Atkinson and Ms. Jane Quante (Washington University in St. Louis, Mo), Ms. Shelley Mann (The Children's Hospital, Denver, CO), Drs. Ronald Gibson and Moira Aitken and Ms. Sharon McNamara (Children's Hospital and Regional Medical Center, Seattle, WA), Dr. Carlos Milla and Ms. Jacquelyn Zirbes (Stanford University Medical Center, Palo Alto, CA), Ms. Donna Wilkes (The Hospital for Sick Children, Toronto, Ontario, Canada), and Ms. Caroline O'Connor (University of North Carolina at Chapel Hill). The authors also thank Drs. Larry Ostrowski, Peadar Noone, Hilda Metjian, Deepika Polineni, Adam Shapiro, Jessica Pittman, and Mr. Kunal Chawla, for thoughtful discussion; Ms. Lu Huang and Ms. Rhonda Pace, for technical assistance; and Ms. Elizabeth Godwin and Ms. Cindy Sell, for administrative support. Authors would like to acknowledge people for providing DNA from PCD patients and their families: Dr. Eitan Kerem from Hadassah University Hospital, Israel; Dr. H. Blau from Schneider Medical Center of Israel, Israel, Dr. Israel Amirav from Ziv Medical Center, Israel, Dr. Lucy Morgan from Concord Hospital, Australia, Dr. Robbert de Iongh from University of Melbourne, Australia, Dr. Scott Bell from The Prince Charles Hospital, Australia, Dr. Hannah Mitchison from University College London, England, Dr. Ugo Pradal from Cystic Fibrosis Center Verona, Italy.
Funding Research Support: MRK, MWL, JLC, MJH, SLM, SDD, TWF, KNO, SDS, MR, KEB, MCA, AL, and MAZ are supported by National Institute of Health research grant 5 U54 HL096458-06, funded by the Office of the Director, and supported by ORDR and NHLBI, NIH.
MRK and MAZ are supported by National Institutes of Health grant 5 R01HL071798.
TWF is supported by R01 HL08265 and Children's Discovery Institute.
KNO is supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
JLC is supported by Clinical Innovator Award by Flight Attendant Medical Research Institute.
HO is supported by a grant from the Deutsche Forschungsgemeinschaft (DFG Om 6/4, GRK1104, SFB592).
Resequencing was provided by the University of Washington, Department of Genome Sciences, under U.S. Federal Government contract number N01-HV-48194 from National Heart, Lung, and Blood Institute.
This work was supported in part by grants RR00046, UL1 RR025747 and UL1 RR025780 from the National Center of Research Resources, NHLBI P01 HL034322, NIH and CFF R026-CR07.
This consortium, Genetic Disorders of Mucociliary Clearance is part of NIH Rare Diseases Clinical Research Network (RDCRN). Funding and/or programmatic support for this project was provided by grant 5 U54 HL096458-06 from the NHLBI and the NIH Office of Rare Diseases Research (ORDR). The views expressed do not necessarily reflect the official policies of the Department of Health and Human Services; nor does mention by trade names, commercial practices, or organizations imply endorsement by the U. S. Government.
Footnotes
At a Glance Commentary: Primary ciliary dyskinesia (PCD) is an autosomal recessive, genetically heterogeneous disorder with oto-sino-pulmonary disease. Most patients are diagnosed on the basis of ciliary ultrastructural defects. This study identified biallelic mutations in DNAH11 in 22% of 58 unrelated patients with normal ciliary ultrastructure, which validates the concepts of 1) ciliary dysfunction in the presence of normal ultrastructure, and 2) the use of genetic analysis to facilitate the diagnosis of PCD.
REFERENCES
- 1.Zariwala M, Knowles M, Leigh M. Primary Ciliary Dyskinesia. GeneReviews at GeneTests: Medical Genetics Information Resource [database online] 2007 Available from: URL: http://www.genetesls.org.
- 2.Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423–450. doi: 10.1146/annurev.physiol.69.040705.141301. [DOI] [PubMed] [Google Scholar]
- 3.Leigh MW, Pittman JE, Carson JL, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy MP, Omran H, Leigh MW, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 5.Pennarun G, Escudier E, Chapelin C, et al. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazor M, Alkrinawi S, Chalifa-Caspi V, et al. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guichard C, Harricane MC, Lafitte JJ, et al. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome) Am J Hum Genet. 2001;68:1030–1035. doi: 10.1086/319511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zariwala M, Noone PG, Sannuti A, et al. Germline mutations in an intermediate chain dynein cause primary ciliary dyskinesia. Am J Respir Cell Mol Biol. 2001;25:577–583. doi: 10.1165/ajrcmb.25.5.4619. [DOI] [PubMed] [Google Scholar]
- 9.Zariwala MA, Leigh MW, Ceppa F, et al. Mutations of DNAI1 in primary ciliary dyskinesia: Evidence of founder effect in a common mutation. Am J Respir Crit Care Med. 2006;174:858–866. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Failly M, Saitta A, Munoz A, et al. DNAI1 mutations explain only 2% of primary ciliary dykinesia. Respiration. 2008;76:198–204. doi: 10.1159/000128567. [DOI] [PubMed] [Google Scholar]
- 11.Loges NT, Olbrich H, Fenske L, et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olbrich H, Haffner K, Kispert A, et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 13.Hornef N, Olbrich H, Horvath J, et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Failly M, Bartoloni L, Letourneau A, et al. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J Med Genet. 2009;46:281–286. doi: 10.1136/jmg.2008.061176. [DOI] [PubMed] [Google Scholar]
- 15.Loges NT, Olbrich H, Becker-Heck A, et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am J Hum Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omran H, Kobayashi D, Olbrich H, et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duriez B, Duquesnoy P, Escudier E, et al. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc Natl Acad Sci USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castleman VH, Romio L, Chodhari R, et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartoloni L, Blouin JL, Pan Y, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwabe GC, Hoffmann K, Loges NT, et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat. 2008;29:289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- 21.Pifferi M, Michelucci A, Conidi ME, et al. New DNAH11 mutations in primary ciliary dyskinesia with normal axonemal ultrastructure. Eur Respir J. 2010;35:1413–1416. doi: 10.1183/09031936.00186209. [DOI] [PubMed] [Google Scholar]
- 22.Becker-Heck A, Zohn IE, Okabe N, et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merveille AC, Davis EE, Becker-Heck A, et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbato A, Frischer T, Kuehni CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009;34:1264–1276. doi: 10.1183/09031936.00176608. [DOI] [PubMed] [Google Scholar]
- 25.Noone PG, Leigh MW, Sannuti A, et al. Primary ciliary dyskinesia: Diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 26.ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide. Am J Respir Crit Care Med. 2005;171:912–930. doi: 10.1164/rccm.200406-710ST. [DOI] [PubMed] [Google Scholar]
- 27.Carson JL, Collier AM. Ciliary defects: cell biology and clinical perspectives. Adv Pediatr. 1988;35:139–165. [PubMed] [Google Scholar]
- 28.Olin JT, Burns K, Carson JL, et al. Diagnostic yield of nasal scrape biopsies in primary ciliary dyskinesia: A multicenter experience. Pediatr Pulmonol. 2011;46:483–488. doi: 10.1002/ppul.21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fliegauf M, Olbrich H, Horvath J, et al. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med. 2005;171:1343–1349. doi: 10.1164/rccm.200411-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Wang X, Brighton L, et al. Increased nasal epithelial ciliary beat frequency associated with lifestyle tobacco smoke exposure. Inhal Toxicol. 2009;21:875–881. doi: 10.1080/08958370802555898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zariwala M, O'Neal WK, Noone PG, et al. Investigation of the possible role of a novel gene, DPCD, in primary ciliary dyskinesia. Am J Respir Cell Mol Biol. 2004;30:428–434. doi: 10.1165/rcmb.2003-0338RC. [DOI] [PubMed] [Google Scholar]
- 32.Supp DM, Witte DP, Potter SS, et al. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Layton WM., Jr Random determination of a developmental process: reversal of normal visceral asymmetry in the mouse. J Hered. 1976;67:336–338. doi: 10.1093/oxfordjournals.jhered.a108749. [DOI] [PubMed] [Google Scholar]
- 34.Supp DM, Brueckner M, Kuehn MR, et al. Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development. 1999;126:5495–5504. doi: 10.1242/dev.126.23.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papon JF, Coste A, Roudot-Thoraval F, et al. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2010;35:1057–1063. doi: 10.1183/09031936.00046209. [DOI] [PubMed] [Google Scholar]
- 36.Escudier E, Duquesnoy P, Papon JF, et al. Ciliary defects and genetics of primary ciliary dyskinesia. Paediatr Respir Rev. 2009;10:51–54. doi: 10.1016/j.prrv.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Sakakibara H, Takada S, King SM, et al. A Chlamydomonas outer arm dynein mutant with a truncated β-heavy chain. J Cell Biol. 1993;122:653–661. doi: 10.1083/jcb.122.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brokaw CJ, Luck DJ, Huang B. Analysis of the movement of Chlamydomonas flagella:” the function of the radial-spoke system is revealed by comparison of wild-type and mutant flagella. J Cell Biol. 1982;92:722–732. doi: 10.1083/jcb.92.3.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brokaw CJ, Kamiya R. Bending patterns of Chlamydomonas flagella: IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskeleton. 1987;8:68–75. doi: 10.1002/cm.970080110. [DOI] [PubMed] [Google Scholar]
- 40.Porter ME, Knott JA, Gardner LC, et al. Mutations in the SUP-PF-1 locus of Chlamydomonas reinhardtii identify a regulatory domain in the β-dynein heavy chain. J Cell Biol. 1994;126:1495–1507. doi: 10.1083/jcb.126.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucas JS, Adam EC, Goggin P, et al. Static respiratory cilia with normal ultrastructure in inversus viscerum (iv) mouse - a potential model of primary ciliary dyskinesia? [Abstract] Am J Respir Crit Care Med. 2010;181:A6724. [Google Scholar]
- 42.Smith CM, Hirst RA, Bankart MJ, et al. Cooling of cilia allows functional analysis of beat pattern for diagnostic testing. Chest. 2010;140:186–190. doi: 10.1378/chest.10-1920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.