

Abstract
Objective
Cocaine use is associated with arterial thrombosis, including myocardial infarction and stroke. Cocaine use results in increased plasma von Willebrand Factor (VWF), accelerated atherosclerosis, and platelet-rich arterial thrombi, suggesting that cocaine activates the endothelium, promoting platelet-VWF interactions.
Approach and Results
Human umbilical vein (HUVEC), brain microvasculature (BMVEC), or coronary artery (CAEC) endothelial cells were treated with cocaine or metabolites benzoylecgonine, cocaethylene, norcocaine, or ecgonine methylester. Supernatant VWF concentration and multimer structure were measured, and platelet–VWF strings formed on the endothelial surface under flow were quantified. Cocaine, benzoylecgonine, and cocaethylene induced endothelial VWF release, with the two metabolites being more potent than the parent molecule. BMVEC were more sensitive to cocaine and metabolites than were HUVEC or CAEC. CAEC released VWF into the supernatant but did not form VWF–platelet strings. Intracellular cAMP concentration was not increased after treatment with cocaine or its metabolites.
Conclusions
Both cocaine and metabolites benzoylecgonine and cocaethylene induced endothelial VWF secretion, possibly explaining thrombotic risk after cocaine ingestion. VWF secretion is likely to vary between vascular beds, with brain endothelial cells being particularly sensitive. These results suggest that clinical management of cocaine-induced ischemia may benefit from therapies aimed at disrupting the VWF–platelet interaction.
Keywords: von Willebrand factor, thrombosis, cocaine, platelets, endothelium
INTRODUCTION
Cocaine use is associated with an increased risk of a variety of thrombotic events, including stroke and myocardial infarction (MI)1, 2 and, less commonly, venous thromboembolism3 and thrombotic thrombocytopenic purpura.4 The risk of myocardial infarction is estimated to increase 6-fold with cocaine use.5 Although cocaine is rapidly metabolized by liver and plasma enzymes, the risk of thrombosis remains elevated for many days after its ingestion,6 possibly an effect of cocaine metabolites, some of which retain adrenergic activity and can persist in blood for 1–2 weeks.7 It is unknown how these metabolites, which include benzoylecgonine, ecgonine methylester, and norcocaine, affect platelet or endothelial functions. Another metabolite, cocaethylene, which is produced when cocaine is consumed with ethanol, is known to retain potent neuronal stimulation ability8 and is associated with an extremely high risk of sudden death.9 Its relationship to thrombotic risk is also unknown.
Although one likely cause of tissue ischemia is cocaine-induced vasospasm in the setting of increased tissue oxygen demand,10, 11 platelet-rich arterial thrombi have also been observed in coronary and cerebral vessels of up to 14% of cocaine users who died suddenly after ingesting cocaine.12, 13 Furthermore, a 10-fold increased incidence of acute coronary stent thrombosis, a platelet-dependent process, was observed in cocaine users14 which was not explained by non-compliance with antiplatelet therapy. Thus, in addition to cocaine’s sympathomimetic effects leading to increased tissue oxygen demand (increased heart rate, blood pressure, contractility, and metabolism) and decreased tissue oxygen delivery (vasoconstriction), cocaine likely contributes to ischemic events by promoting platelet aggregation on endovascular surfaces.
The primary endothelial determinant of platelet–endothelial attachment is von Willebrand Factor (VWF), a multimeric protein secreted from endothelial cells. Upon endothelial cell activation, VWF is rapidly secreted into plasma from storage granules called Weibel-Palade bodies. VWF is secreted as a very high molecular weight form referred to as ultra-large VWF (ULVWF). ULVWF can be retained on the vascular surface via P-selectin15 and/or integrin αvβ3,16 with long ULVWF strands extending up to several millimeters into the bulk flow.17 VWF binds platelets via the platelet glycoprotein Ib-IX-V complex, with ULVWF, unlike normal plasma forms of VWF, being able to spontaneously bind this receptor.18 The binding of platelets to VWF is the first step in thrombus formation, promoting subsequent platelet activation and aggregation. Under normal circumstances, ULVWF is processed proteolytically by the plasma metalloproteinase ADAMTS13 into the smaller, less-reactive VWF forms normally found in plasma.
Cocaine use is associated with a 40% increase in plasma VWF concentration,19 suggesting that cocaine induces endothelial secretion of VWF and supporting a role for platelet adhesion to the endothelium in cocaine-associated arterial thrombosis. In addition, because platelet binding to processed plasma VWF is increased under high shear stress,18 cocaine-induced vasoconstriction would be expected to promote thrombus formation. Histopathologic studies support this scenario, demonstrating numerous endovascular abnormalities in cocaine abusers, who tend to be young (less than age 35) with few, if any, additional cardiovascular risk factors. These findings include accelerated atherosclerosis,13, 20 intimal hyperplasia,21 and increased adventitial inflammation.22 Endothelial dysfunction induced by cocaine is also suggested by the findings of upregulated endothelial adhesion molecules,23, 24 increased vascular permeability,25 and increased circulating endothelial cells24 following cocaine exposure.
We hypothesize that cocaine acts as an endothelial agonist and VWF secretogogue, promoting platelet-mediated thrombosis by inducing exposure of ULVWF on endothelial surfaces. Given the prolonged thrombotic risk following cocaine ingestion, we also hypothesize that cocaine metabolites induce ULVWF release from endothelial cells. We therefore assessed the ability of cocaine and its metabolites to induce VWF release from cultured human vascular endothelial cells from a variety of tissue beds including umbilical vein, brain, and coronary artery.
MATERIALS AND METHODS
Materials and Methods can be found in the online supplement.
RESULTS
VWF is secreted from endothelial cells in response to a variety of agonists including histamine, epinephrine, dDAVP, and hypoxia.26 After secretion, VWF can either remain tethered to the endothelial surface or be released into the supernatant. We assessed the ability of cocaine and cocaine metabolites to directly induce VWF secretion from human endothelial cells of various tissues.
Cocaine induces von Willebrand Factor release from endothelial cells
We treated endothelial cells from three different sources (HUVEC, BMVEC and CAEC) with cocaine at several concentrations and assessed their secretion of VWF. Cocaine induced VWF secretion in a concentration-dependent manner from the three endothelial types (Figure 1A). In all three cell types, cocaine at a concentration of 1 μg/ml induced a similar extent of VWF secretion as 50 ☐M histamine or 4 μg/ml dDAVP, both known potent VWF secretogogues. This cocaine concentration, which is within the range detected in the plasma of cocaine users,7 increased VWF secretion more than 300% above the unstimulated baseline. Lower concentrations of cocaine (0.5 ☐g/ml) also induced endothelial VWF release, to twice that of unstimulated HUVEC and BMVEC (Figure 1A, 1B, and 1C). In contrast, VWF release from CAEC treated with 0.5 ☐g/ml cocaine was not different than from untreated cells (p = 0.99, Figure 1D). For all concentrations of cocaine tested, secretion from BMVEC was greater than from HUVEC and CAEC.
Figure 1. Cocaine induces VWF release from HUVEC, BMVEC, and CAEC.
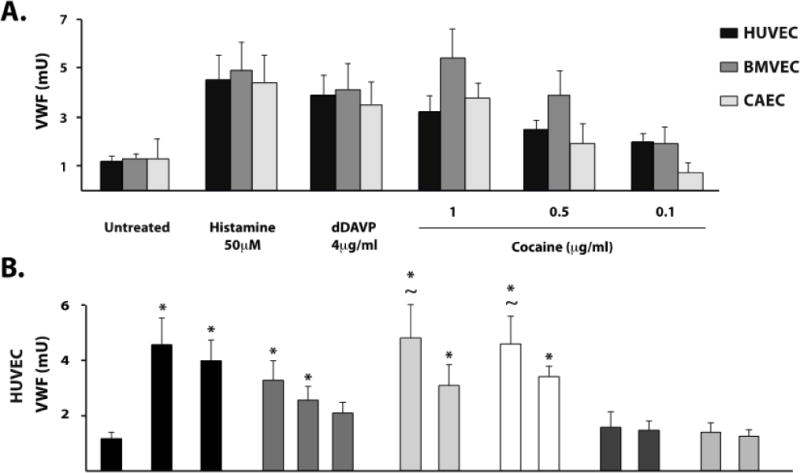
Cultured endothelial cells at passage 2–4 were incubated with the indicated agonists or serum-free medium (untreated) at 37°C. VWF abundance was determined in cell supernatants by dot blot analysis using an HRP-conjugated detection antibody and expressed in milliunits (mU) calibrated to a standard curve of normal human reference plasma. A) VWF release following incubation with cocaine at the indicated concentrations for each of the three endothelial cell types. VWF release following incubation with cocaine metabolites was also assessed from HUVEC (B), BMVEC (C), and CAEC (D) as compared to histamine 50μM or dDAVP 4μg.ml. The results are expressed as means +/− SEM. * Tukey’s HSD test, as compared to untreated cells (p < 0.05). ~ Tukey’s HSD test, not different than histamine treated cells (p > 0.05).
The cocaine metabolites benzoylecgonine and cocaethylene induce VWF release from human endothelial cells
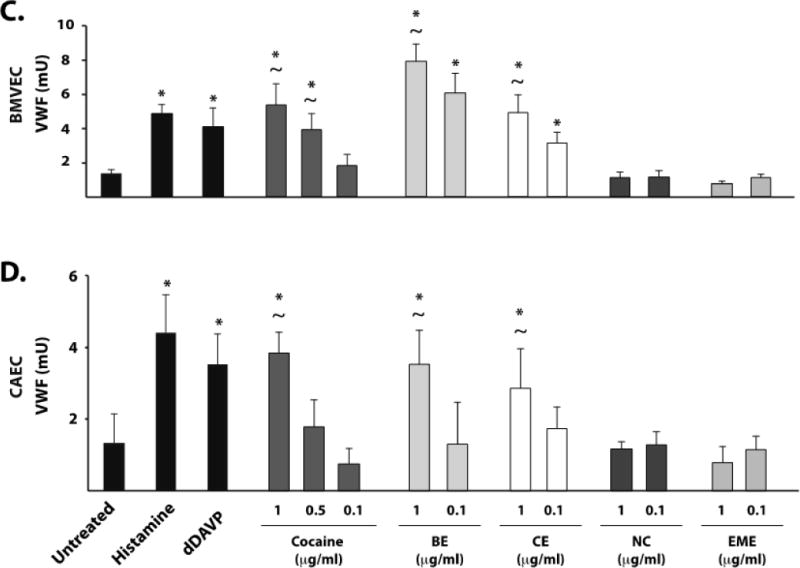
The risk of cardiovascular complications following cocaine ingestion persist long after cocaine levels fall below the minimum cocaine concentration able to stimulate endothelial VWF in our assay.6 Therefore, we also studied four of the major cocaine metabolites for their ability to induce VWF release from endothelial cells. Only benzoylecgonine and cocaethylene induced VWF release and induced it in all three cell types (Figure 1B–D). The concentrations of cocaine metabolites used here have been reported in the plasma of cocaine users.7 In general, BMVEC released larger quantities of VWF than HUVEC or CAEC in response to the two metabolites. Benzoylecgonine and cocaethylene at both 1 μg/ml and 0.1 μg/ml induced significant increases in VWF secretion in both HUVEC (Figure 1B) and BMVEC (Figure 1C) compared to untreated cells (p < 0.05). However, CAEC responded robustly only at the higher concentration (Figure 1D). Both benzoylecognine and cocaethylene were more potent than cocaine in inducing endothelial VWF release, as cocaine at 0.1 μg/ml failed to induce VWF release from any of the cell types.
ULVWF released from HUVEC and BMVEC remains tethered to the endothelial surface and binds platelets under flow
Upon initial secretion, a subset of ULVWF molecules remains tethered to the endothelial surface, forming fibers under flow capable of reaching several millimeters in length.17 Such incredibly long ULVWF strings can spontaneously bind hundreds of platelets and can be easily visualized as “beads on a string” by phase-contrast microscopy (Figure 2A). This platelet–VWF interaction is dependent on the platelet VWF receptor, GPIb.17 Using a parallel-plate flow chamber, we assessed VWF–platelet string formation in cocaine-treated HUVECs (Figure 2B) and BMVECs (Figure 2C) exposed to a constant shear stress of 2.5 dyne/cm2. Mock-treated HUVECs and BMVECs failed to form strings (Figure 2B), whereas cocaine (1μg/ml) induced numerous VWF–platelet strings in both HUVEC and BMVEC (Figures 2B and 2C). Benzoylecgonine and cocaethylene also induced VWF–platelet string formation on HUVEC and BMVEC at concentrations reported to occur in plasma,7 but norcocaine and ecgonine methylester did not (Figures 2B and 2C). In HUVEC, the lowest concentration of cocaethylene (0.1 μg/ml) induced a similar number of VWF–platelet strings as 50 μM histamine, whereas 1 μg/ml cocaine produced fewer platelet–VWF strings than histamine. In BMVEC, benzoylecgonine and cocaethylene at both concentrations and cocaine at 1 μg/ml induced similar numbers of VWF–platelet strings as 50 μM histamine. This indicates that BMVEC are more sensitive than HUVEC to cocaine and the longer-lived cocaine metabolites. As has been reported, CAEC did not produce VWF–platelet strings after treatment with histamine.17 We obtained a similar result in CAEC treated with cocaine or cocaine metabolites (data not shown). As expected, formation of VWF–platelet strings in cocaine- and cocaine metabolite–treated HUVEC was prevented by pretreating the platelets with a GPIb-blocking antibody (Figure 2D).
Figure 2. Cocaine, benzoylecgonine, and cocaethylene induce platelet–VWF string formation on the surface of stimulated HUVEC and BMVEC.
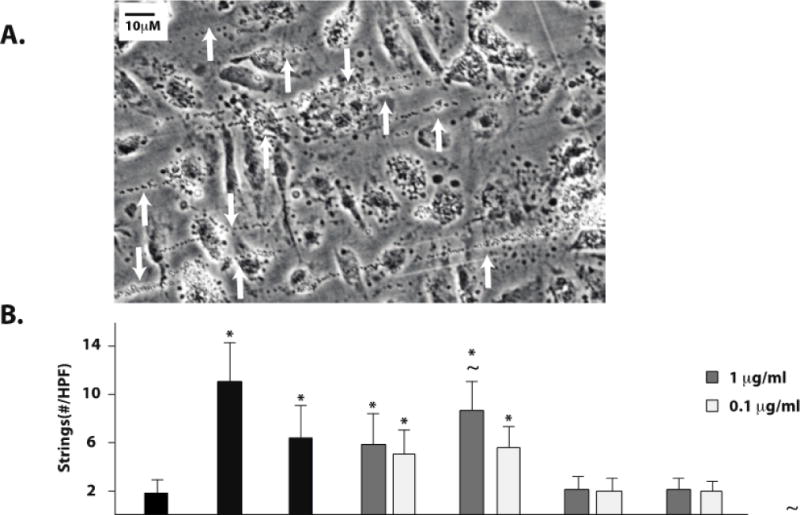
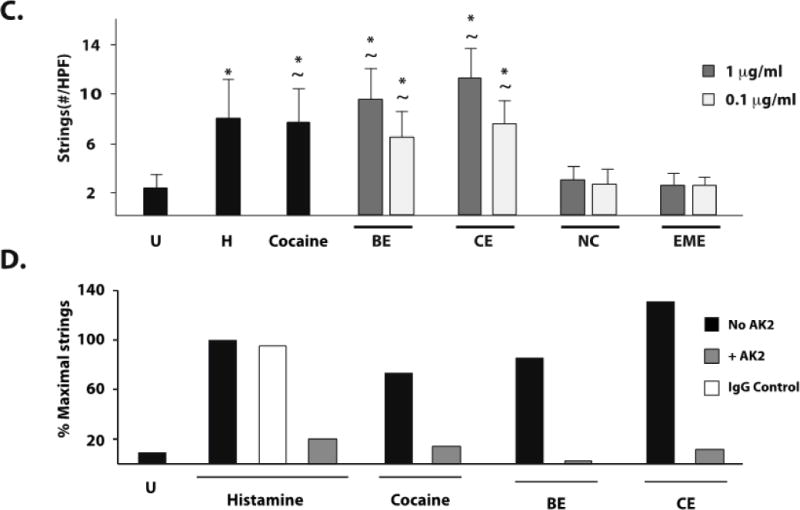
HUVEC or BMVEC were grown to confluence on glass coverslips and stimulated with histamine 50 μM (H), cocaine 1μg/ml, serum free media (untreated, U), or cocaine metabolites. The coverslips were then placed in a parallel-plate flow chamber and perfused with platelets at 2.5dyne/cm2. Platelet–VWF strings were visualized by phase-contrast videomicroscopy. Arrows indicate platelet-VWF strings. A) Video capture of platelet-VWF strings in a representative 40X field of histamine-stimulated HUVEC cells. Strings per 40X field were counted for HUVEC (B) and BMVEC (C). The results are expressed as means +/− SEM. * Tukey’s HSD test, as compared to untreated cells (p < 0.05). ~ Tukey’s HSD test, indicating no difference as compared to histamine treated cells (p > 0.05). D) GPIb dependence of string formation was assessed in the presence or absence of the GPIbα antibody AK2 after stimulation with no agonist (Untreated, U), cocaine, benzoylecgonine, or cocaethylene at 1μg/ml. The results are expressed as the percent maximal strings formed after histamine stimulation from a representative experiment.
Endothelial cell VWF content varies between tissue sources
We investigated whether BMVEC secreted more VWF than CAEC when treated with cocaine or its active metabolites because the former cell line contained more intracellular VWF. Quantification of the immunofluorescence signals from anti-VWF labeled permeabilized cells indeed revealed that BMVEC contained more intracellular VWF than either HUVEC or CAEC (Figure 3).
Figure 3. HUVEC, BMVEC, and CAEC VWF and P-selectin content.
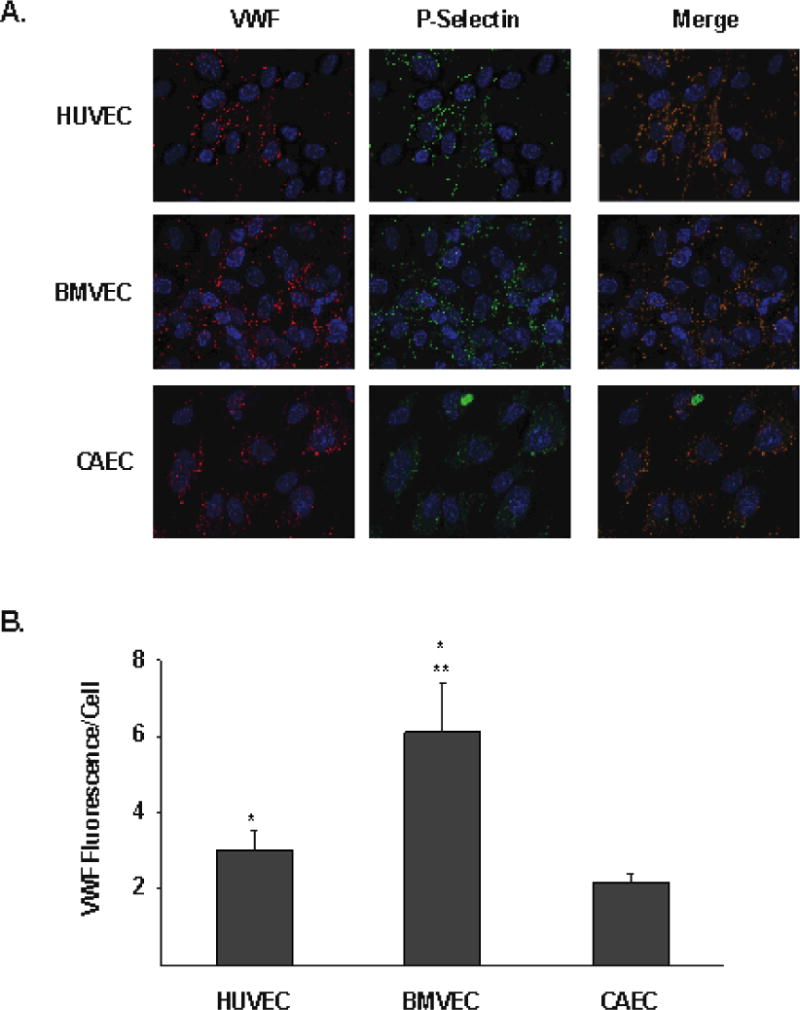
A) Cultured endothelial cells were fixed and permeabilized. VWF (red) and P-Selectin (green) was then assessed by immunofluorescence. Cell nuclei were stained with DAPI (blue). The merged image shows colocalization of VWF and P-Selectin in each cell type. B) VWF immunflourescence per DAPI-positive cell was calculated and is shown for each indicated endothelial cell type (arbitrary units). The results are expressed as means +/− SEM. * Tukey’s HSD test, as compared to CAEC (p = 0.0002). ** Tukey’s HSD test, as compared to HUVEC (p < 0.0001).
CAEC secrete ULVWF in response to cocaine and cocaine metabolites and express P-selectin, αvβ3, and ADAMTS13
One potential explanation for the inability of CAEC to produce platelet–VWF strings after stimulation by agonists is that they do not secrete VWF in the ULVWF form. We therefore assessed the multimer pattern in VWF secreted from CAECs treated with histamine, cocaine, benzoylecgonine, or cocaethylene (Figure 4). As compared to VWF in normal human plasma, stimulated CAEC supernatants contained abundant ULVWF but apparently less than HUVEC or BMVEC supernatants and of lower maximum size (Figure 4).
Figure 4. Cocaine, benzoylecgonine, and cocaethylene induce secretion of ULVWF.
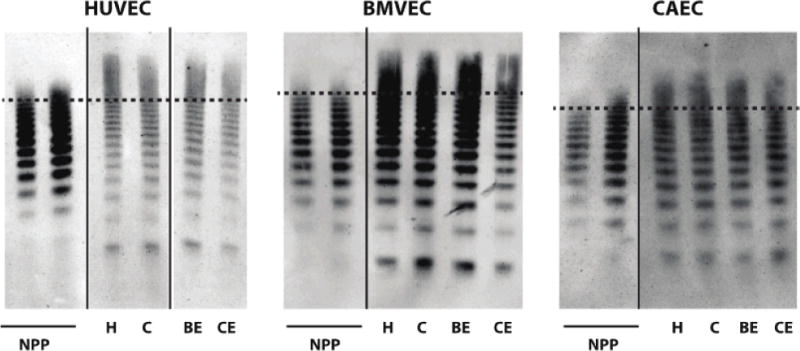
Endothelial cells exposed to the indicated agonsists (H = histamine 100μM, C = cocaine1μg/ml, BE = benzoyecgonine 10μg/ml, CE = cocaethylene 10μg/ml). Supernatants were then subjected to non-reducing gel electrophoresis, and VWF multimer patterns were assessed by Western blot. Normal pooled human plasma (NPP) was used as a control with 0.0125 μl loaded (left lane) and 0.025 μl loaded (right lane). For each panel, lanes are derived from a single gel. The dashed line represents the same band number (14) counted from the bottom applied to each cell type. Higher molecular weight forms (ULVWF) are present above the dotted line.
We also investigated whether the inability of CAECs to form platelet strings was because these cells are deficient in P-selectin or the integrin αvβ3, loss of either of which could prevent tethering of secreted ULVWF to the endothelial surface. By immunofluorescence of unstimulated endothelial cells, P-selectin was present in endothelial cells from each tissue type, colocalizing with VWF in a distribution consistent with Weibel-Palade bodies (Figure 3). Following agonist stimulation of CAEC, P-selectin appeared on the cell surface as it did in HUVEC (Figure 5A). In addition, integrin αvβ3 was present on the surfaces of the three types of endothelial cells (Figure 5B). This indicates that CEAC express the appropriate cell surface proteins to retain secreted ULVWF on the cell surface.
Figure 5. CAEC translocate P-selectin to the cell surface and express αvβ3 and ADAMTS13.
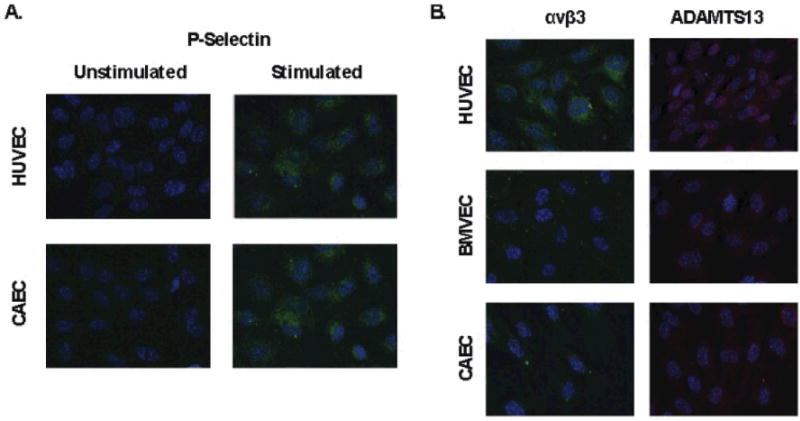
A) Endothelial cells were incubated with serum free media (unstimulated) or cocaine 1μg/ml (stimulated). Cells were then fixed without permeabilization and stained with an anti-P-selectin antibody (green). B) Endothelial cells fixed without permeabilization (αvβ3) or with permeabilization (ADAMTS13). Cell surface αvβ3 (green) and intracellular ADAMTS13 (red) was detected for each indicated endothelial cell type. In all panels, nuclei are stained with DAPI (blue).
Because ADAMTS13 normally cleaves ULVWF, and could remove VWF from the endothelial surface after secretion from endothelial cells, we evaluated the possibility that CAEC express more ADAMTS13 than do BMVEC. ADAMTS13 distribution within each endothelial cell type was similar (Figure 5B).
VWF secretion induced by cocaine and its metabolites does not involve increased intracellular cAMP
The mechanism by which cocaine and its metabolites induce endothelial VWF release is unknown. Increased intracellular cAMP precedes Weibel-Palade body exocytosis and VWF secretion in response to several agonists, including dDAVP but not histamine. We therefore examined intracellular cAMP concentrations in lysates of endothelial cells treated with cocaine or its metabolites. The intracellular cAMP concentration did not change relative to untreated cells after incubation with cocaine and cocaine metabolites at concentrations shown to induce VWF secretion in HUVEC (Figure 6). Similar results were obtained in BMVEC and CAEC (data not shown).
Figure 6. Cocaine and cocaine metabolites do not increase intracellular cAMP levels.
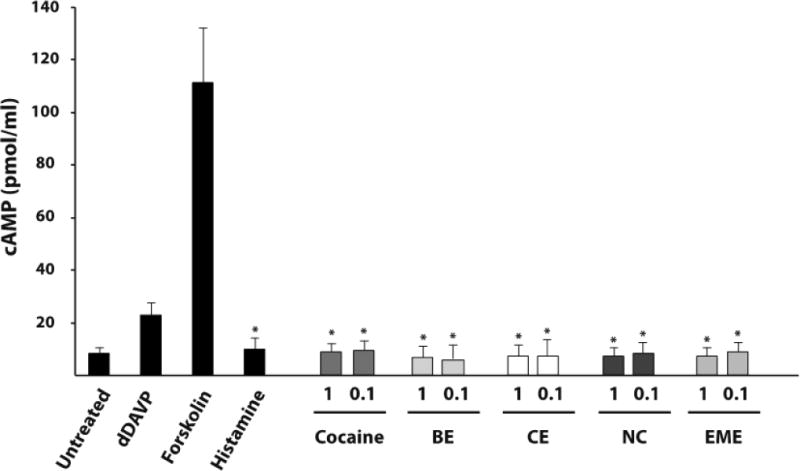
HUVEC were grown to confluence and incubated with the indicated agonists (dDAVP 4μg/ml, forskolin 2μM, and histamine 50μM) or serum free media (untreated). Cocaine and metabolite concentrations indicated are in μg/ml. Intracellular cAMP concentration was determined in cell lysates for each condition as indicated. The results are expressed as means +/− SEM. * Tukey’s HSD test, as compared to untreated cells (p < 0.05).
DISCUSSION
Cocaine abuse remains a major public health problem, contributing to over 400,000 emergency room visits per year.27 In addition to the problem of drug addiction and its attendant social and medical issues, cocaine ingestion is associated with both acute and chronic cardiovascular disease, including myocardial infarction28 and stroke.29 The thrombotic effects of cocaine include increased rates of both arterial and venous thrombosis.30 These thrombotic events can be delayed after cocaine use, sometimes occurring when cocaine is no longer detectable in plasma. Our results indicate that this prolonged effect of cocaine on thrombotic risk is related to the continued pro-thrombotic activity of the long-lived cocaine metabolites benzoylecgonine and cocaethylene.
Our results indicate that endothelial cells in culture release VWF in response to agonism by cocaine, benzoylecgonine, and cocaethylene, but not by cocaine metabolites norcocaine nor ecgonine methyl ester. Cocaine, benzoylecgonine, and cocaethylene are extremely potent VWF secretagogues, with similar ability to induce VWF release as 50μM histamine or 4μg/ml dDAVP, both known strong VWF secretagogues. Our results also indicate that the metabolites benzoylecgonine and cocaethylene are at least as potent at inducing endothelial VWF as cocaine, and in some endothelial cell types are more potent agonists than cocaine and able to induce significant VWF release from endothelial cells at very low concentrations. Particularly significant is the effect of cocaethylene, which is only formed when cocaine is ingested simultaneously with ethanol. This finding may explain the particularly elevated risk for cardiovascular events seen accompanying combined cocaine and ethanol use.9 Brain microvascular endothelial cells are especially sensitive to cocaine, benzoylecgonine, and cocaethylene as compared to umbilical vein or coronary artery derived endothelial cells. This was true both for VWF released into the culture supernatant in ULVWF form as well as for secreted VWF that remains tethered to the endothelial surface. Platelet binding tp endothelial surface bound VWF was found to be dependent on the platelet receptor, GPIb. This supports a model in which cocaine associated thrombosis involves direct action of cocaine and cocaine metabolites on endothelial cells to secrete VWF, followed by platelet binding and subsequent activation.
Several years ago, it became clear that platelets were a target of cocaine;31–33 the mechanisms of this effect have yet to be fully elucidated. In response to cocaine, platelets become activated and release constituents of their alpha granules, promoting thrombosis.32, 34 Cocaine also decreases nitric oxide levels, which in addition to inhibiting vasodilatation, can lower the threshold for platelet activation.35 Further evidence that platelets are involved in the cardiovascular pathophysiology associated with cocaine is the high incidence of acute coronary stent thrombosis in cocaine users, even in those compliant with anti-platelet therapy.14, 36 It is not known whether benzoylecgonine, or cocaethylene, which we found induce endothelial VWF release, can also activate platelets.
Platelets bind ULVWF spontaneously via an interaction of the VWF A1 domain with the platelet receptor GPIb,37 initiating platelet aggregation and thrombosis. When VWF is cleaved to lower–molecular-weight VWF by ADAMTS13, the A1 domain is no longer able to spontaneously bind platelets in the absence of high shear stress, collagen binding, pharmacologic reactivation as by ristocetin, or oxidative modification.18, 37–39 Accumulation of ULVWF is associated with the devastating thrombotic disease, thrombotic thrombocytopenic purpura (TTP).40 Consistent with a similar mechanism for cocaine-associated thrombosis, a TTP-like syndrome has been reported in cocaine users4 suggesting that in at least a subset of this group there is, in fact, an accumulation of ULVWF. Whether increased ULVWF can be detected in cocaine abusers is unknown.
Previous studies have demonstrated probable endothelial activation following cocaine use, as documented by increased endothelial release of platelet-derived growth factor,41 endothelin-1,35 stromal cell derived factor-1,24 monocyte chemotactic protein-1,24 and soluble intercellular adhesion molecule-1.24 In addition, cocaine is associated with decreased endothelial nitric oxide synthase (eNOS) activity and nitric oxide production,35 activation of endothelial cell NFκB activity,42 and increased numbers of circulating endothelial cells.24 Signals that induce such endothelial activation would also be expected to induce ULVWF secretion. Indeed, increased circulating VWF has been detected in chronic cocaine abusers.19 Whether the increased VWF concentration is due to increased production or delayed clearance cannot be discerned from measurements of the plasma concentration. Our studies suggest that the elevated VWF concentration in the blood of cocaine users is largely due to increased endothelial secretion in response to cocaine and two of its major metabolites.
The cardiovascular complications of cocaine use had been presumed to be primarily caused by the effects of cocaine on the sympathetic nervous system, causing vasoconstriction of coronary vessels while simultaneously increasing myocardial oxygen demand by increasing heart rate, contractility, blood pressure, and cellular metabolism. Cocaine metabolites have also been shown to induce cerebral and coronary vasoconstriction, particularly benzoylecgonine and cocaethylene.43, 44 While these mechanisms likely contribute to cocaine-induced myocardial ischemia, our results demonstrate that the effects of cocaine and its metabolites on the vascular endothelium are also mechanistically important and likely additive. Vasoconstriction induced by cocaine and its metabolites would be expected to potentiate the prothrombotic effect of increased VWF concentrations. The ability of all forms of VWF to bind and activate platelets is enhanced by high fluid shear stress.45 This physical force is increased in proportion to the velocity of blood flow and in an inverse relationship to the radius of the blood vessel. Flow velocity increases and vessel diameter decreases during vasoconstriction, increasing the likelihood of VWF-dependent shear-induced platelet aggregation. The threshold for aggregation will be further decreased and the extent increased in the presence of elevated VWF concentrations. Furthermore, the propensity to develop a flow-restricting thrombus would be further increased by additional effects of cocaine and cocaine metabolites on platelet activation.
Cocaine’s anesthetic properties relate to its ability to slow nerve conduction by blocking voltage-gated sodium channels.46 It produces addiction by blocking presynaptic dopamine and serotonin reuptake receptors, potentiating the effect of these neurotransmitters in the synaptic cleft in reward centers of the brain.7 Similarly, the sympathomimetic effects of cocaine are a consequence of its ability to block presynaptic reuptake of catecholamines at sympathetic nerve terminals and stimulate central sympathetic outflow.47, 48 It is unknown how cocaine and its metabolites activate endothelial cells to secrete VWF, whether by engaging a cell surface receptor or an intracellular target. Also unknown are the signaling pathways involved. We found that endothelial cAMP concentrations do not increase in endothelial cells treated with cocaine or its metabolites.
It is noteworthy that myocardial infarction is a more common thrombotic consequence of cocaine abuse than is stroke. Our studies in a cell culture system suggest that brain-derived endothelial cells contain more VWF and are more sensitive to cocaine and cocaine metabolites than coronary artery-derived endothelial cells. The inability of CAEC to produce platelet–VWF strings is of unclear significance in light of the clinical and pathologic data supporting a role for platelet–VWF interaction in cocaine-associated myocardial infarction. Although we have shown that CAEC contain similar intracellular ADAMTS13 quantities as the two other endothelial types studied, it is possible that ADAMTS13 on the surface of CAEC is more active than in other cell types, leading to increased clearance of VWF from the surface of CAEC in the static assays used in our studies. In addition, unlike other tissue beds, coronary arterial flow is subjected to compressive forces exerted by cardiac muscle and differs from flow in other arteries in being highest during diastole instead of systole. It remains possible as well that cocaine- and cocaine metabolite–activated coronary artery endothelium is able to capture circulating VWF and/or platelet–VWF aggregates, a process that would be promoted by increased VWF and/or ULVWF abundance in the blood, platelet activation, and vaso-constriction-induced shear stress. It is possible that in vivo models may be more revealing than in vitro studies.
Interestingly, evidence for cocaine-induced thrombosis has been reported after low-dose intranasal cocaine delivery to treat epistaxis.49 It is interesting to speculate that cocaine’s ability to arrest epistaxis and other bleeding events is not due simply to local vasospasm, as has been postulated, but rather to platelet-mediated thrombosis within bleeding vessels. The septal necrosis reported in chronic cocaine abusers whose preferred route of administration is “snorting” may thus represent thrombosis as opposed to sequelae of vasoconstriction.
In summary, we report here that cocaine and two of its metabolites, benzoylecgonine and cocaethylene, potently stimulate release of hyperadhesive forms of VWF from endothelium, providing at least one mechanism by which this commonly used recreational drug can cause potentially catastrophic thrombotic events. The likelihood of developing occlusive coronary thrombi is increased even further by the vasoconstrictive effect of these drugs. These data also suggest that therapy for these events should be tailored toward disrupting this mechanism of thrombosis.
Supplementary Material
SIGNIFICANCE.
Individuals who abuse cocaine are at high risk of thrombosis, particularly stroke and myocardial infarction. Over 240,000 individuals are seen each year in US Emergency Rooms with cocaine induced chest pain, of which an estimated 15,000 suffer myocardial infarction. The risk period for cocaine-associated thrombosis extends days after cocaine is no longer detectable in plasma, including a 10-fold increased risk of acute coronary stent thrombosis following angioplasty. We hypothesized that longer-lived cocaine metabolites participate in increased thrombotic risk by directly activating endothelial cells to release von Willebrand Factor (VWF). We found that two cocaine metabolites, benzoylecgonine and cocaethylene, are potent inducers of VWF release from brain- and coronary-artery derived endothelial cells, and in some cases more potent than cocaine itself. This suggests that a common mechanism of cocaine-induced thrombosis is VWF dependent and could be treated by disrupting the platelet-VWF interaction.
Acknowledgments
The authors thank Dr. Jing-fei Dong for providing the anti-ADAMTS13 antibody, and Dr. Adam D. Munday for helpful suggestions and technical assistance with microscopy.
SOURCES OF FUNDING:
This work was supported by NIH R01 HL091153 (López), NIH K12 HL087165 (Hobbs), and institutional funds from the Puget Sound Blood Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES:
The authors report no disclosures.
References
- 1.Bhattacharya P, Taraman S, Shankar L, Chaturvedi S, Madhavan R. Clinical profiles, complications, and disability in cocaine-related ischemic stroke. J Stroke Cerebrovasc Dis. 2011;20:443–449. doi: 10.1016/j.jstrokecerebrovasdis.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 2.Phillips K, Luk A, Soor GS, Abraham JR, Leong S, Butany J. Cocaine cardiotoxicity: A review of the pathophysiology, pathology, and treatment options. Am J Cardiovasc Drugs. 2009;9:177–196. doi: 10.2165/00129784-200909030-00005. [DOI] [PubMed] [Google Scholar]
- 3.Lisse JR, Davis CP, Thurmond-Anderle ME. Upper extremity deep venous thrombosis: Increased prevalence due to cocaine abuse. Am J Med. 1989;87:457–458. doi: 10.1016/s0002-9343(89)80832-0. [DOI] [PubMed] [Google Scholar]
- 4.Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis. 2000;35:1–5. doi: 10.1016/S0272-6386(00)70321-0. [DOI] [PubMed] [Google Scholar]
- 5.Qureshi AI, Suri MF, Guterman LR, Hopkins LN. Cocaine use and the likelihood of nonfatal myocardial infarction and stroke: Data from the third national health and nutrition examination survey. Circulation. 2001;103:502–506. doi: 10.1161/01.cir.103.4.502. [DOI] [PubMed] [Google Scholar]
- 6.Mittleman MA, Mintzer D, Maclure M, Tofler GH, Sherwood JB, Muller JE. Triggering of myocardial infarction by cocaine. Circulation. 1999;99:2737–2741. doi: 10.1161/01.cir.99.21.2737. [DOI] [PubMed] [Google Scholar]
- 7.Johanson CE, Fischman MW. The pharmacology of cocaine related to its abuse. Pharmacol Rev. 1989;41:3–52. [PubMed] [Google Scholar]
- 8.Rose JS. Cocaethylene: A current understanding of the active metabolite of cocaine and ethanol. Am J Emerg Med. 1994;12:489–490. doi: 10.1016/0735-6757(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 9.Andrews P. Cocaethylene toxicity. J Addict Dis. 1997;16:75–84. doi: 10.1300/J069v16n03_08. [DOI] [PubMed] [Google Scholar]
- 10.Benzaquen BS, Cohen V, Eisenberg MJ. Effects of cocaine on the coronary arteries. Am Heart J. 2001;142:402–410. doi: 10.1067/mhj.2001.117607. [DOI] [PubMed] [Google Scholar]
- 11.Treadwell SD, Robinson TG. Cocaine use and stroke. Postgrad Med J. 2007;83:389–394. doi: 10.1136/pgmj.2006.055970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konzen JP, Levine SR, Garcia JH. Vasospasm and thrombus formation as possible mechanisms of stroke related to alkaloidal cocaine. Stroke. 1995;26:1114–1118. doi: 10.1161/01.str.26.6.1114. [DOI] [PubMed] [Google Scholar]
- 13.Lucena J, Blanco M, Jurado C, Rico A, Salguero M, Vazquez R, Thiene G, Basso C. Cocaine-related sudden death: A prospective investigation in south-west spain. Euro Heart J. 2010;31:318–329. doi: 10.1093/eurheartj/ehp557. [DOI] [PubMed] [Google Scholar]
- 14.Karlsson G, Rehman J, Kalaria V, Breall JA. Increased incidence of stent thrombosis in patients with cocaine use. Cath Cardiovasc Interv. 2007;69:955–958. doi: 10.1002/ccd.21151. [DOI] [PubMed] [Google Scholar]
- 15.Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, López JA, Dong JF. P-selectin anchors newly released ultralarge von Willebrand Factor multimers to the endothelial cell surface. Blood. 2004;103:2150–2156. doi: 10.1182/blood-2003-08-2956. [DOI] [PubMed] [Google Scholar]
- 16.Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand Factor strings under fluid shear stress. Blood. 2009;113:1589–1597. doi: 10.1182/blood-2008-05-158584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, López JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand Factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, López JA. Interactions of platelets with subendothelium and endothelium. Microcirculation. 2005;12:235–246. doi: 10.1080/10739680590925484. [DOI] [PubMed] [Google Scholar]
- 19.Siegel AJ, Sholar MB, Mendelson JH, Lukas SE, Kaufman MJ, Renshaw PF, McDonald JC, Lewandrowski KB, Apple FS, Stec JJ, Lipinska I, Tofler GH, Ridker PM. Cocaine-induced erythrocytosis and increase in von willebrand factor: Evidence for drug-related blood doping and prothrombotic effects. Arch Intern Med. 1999;159:1925–1929. doi: 10.1001/archinte.159.16.1925. [DOI] [PubMed] [Google Scholar]
- 20.Dressler FA, Malekzadeh S, Roberts WC. Quantitative analysis of amounts of coronary arterial narrowing in cocaine addicts. Am J Cardiol. 1990;65:303–308. doi: 10.1016/0002-9149(90)90292-9. [DOI] [PubMed] [Google Scholar]
- 21.Ramondo AB, Mistrorigo F, Angelini A. Intimal hyperplasia and cystic medial necrosis as substrate of acute coronary syndrome in a cocaine abuser: An in vivo/ex vivo pathological correlation. Heart. 2009;95:82. doi: 10.1136/hrt.2007.140509. [DOI] [PubMed] [Google Scholar]
- 22.Kolodgie FD, Virmani R, Cornhill JF, Herderick EE, Smialek J. Increase in atherosclerosis and adventitial mast cells in cocaine abusers: An alternative mechanism of cocaine-associated coronary vasospasm and thrombosis. J Am Coll Cardiol. 1991;17:1553–1560. doi: 10.1016/0735-1097(91)90646-q. [DOI] [PubMed] [Google Scholar]
- 23.Gan X, Zhang L, Berger O, Stins MF, Way D, Taub DD, Chang SL, Kim KS, House SD, Weinand M, Witte M, Graves MC, Fiala M. Cocaine enhances brain endothelial adhesion molecules and leukocyte migration. Clinical Immunol. 1999;91:68–76. doi: 10.1006/clim.1998.4683. [DOI] [PubMed] [Google Scholar]
- 24.Sáez CG, Olivares P, Pallavicini J, Panes O, Moreno N, Massardo T, Mezzano D, Pereira J. Increased number of circulating endothelial cells and plasma markers of endothelial damage in chronic cocaine users. Thrombosis Research. 2011;128(4):e18–23. doi: 10.1016/j.thromres.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Kolodgie FD, Wilson PS, Mergner WJ, Virmani R. Cocaine-induced increase in the permeability function of human vascular endothelial cell monolayers. Exp Mol Path. 1999;66:109–122. doi: 10.1006/exmp.1999.2253. [DOI] [PubMed] [Google Scholar]
- 26.Blann AD. Plasma von Willebrand Factor, thrombosis, and the endothelium: The first 30 years. Thromb Haemost. 2006;95:49–55. [PubMed] [Google Scholar]
- 27.Substance Abuse and Mental Health Services Administration. The DAWN report: Highlights of the 2009 Drug Abuse Warning Network (DAWN) findings on drug-related emergency department visits. 2010. [PubMed] [Google Scholar]
- 28.Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558–2569. doi: 10.1161/CIRCULATIONAHA.110.940569. [DOI] [PubMed] [Google Scholar]
- 29.Esse K, Fossati-Bellani M, Traylor A, Martin-Schild S. Epidemic of illicit drug use, mechanisms of action/addiction and stroke as a health hazard. Brain and Behavior. 2011;1:44–54. doi: 10.1002/brb3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Killam AL. Cardiovascular and thrombosis pathology associated with cocaine use. Hematol Oncol Clinics of North Am. 1993;7:1143–1151. [PubMed] [Google Scholar]
- 31.Heesch CM. Cocaine activates platelets and increases the formation of circulating platelet containing microaggregates in humans. Heart. 2000;83:688–695. doi: 10.1136/heart.83.6.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kugelmass AD, Oda A, Monahan K, Cabral C, Ware JA. Activation of human platelets by cocaine. Circulation. 1993;88:876–883. doi: 10.1161/01.cir.88.3.876. [DOI] [PubMed] [Google Scholar]
- 33.Kugelmass AD, Shannon RP, Yeo EL, Ware JA. Intravenous cocaine induces platelet activation in the conscious dog. Circulation. 1995;91:1336–1340. doi: 10.1161/01.cir.91.5.1336. [DOI] [PubMed] [Google Scholar]
- 34.Rinder HM, Ault KA, Jatlow PI, Kosten TR, Smith BR. Platelet alpha-granule release in cocaine users. Circulation. 1994;90:1162–1167. doi: 10.1161/01.cir.90.3.1162. [DOI] [PubMed] [Google Scholar]
- 35.Pradhan L, Mondal D, Chandra S, Ali M, Agrawal KC. Molecular analysis of cocaine-induced endothelial dysfunction: Role of endothelin-1 and nitric oxide. Cardiovasc Toxicology. 2008;8:161–171. doi: 10.1007/s12012-008-9025-z. [DOI] [PubMed] [Google Scholar]
- 36.McKee SA, Applegate RJ, Hoyle JR, Sacrinty MT, Kutcher MA, Sane DC. Cocaine use is associated with an increased risk of stent thrombosis after percutaneous coronary intervention. Am Heart J. 2007;154:159–164. doi: 10.1016/j.ahj.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, López JA. Ultralarge multimers of von Willebrand Factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: Studies using optical tweezers. Blood. 2002;99:3971–3977. doi: 10.1182/blood-2001-11-0060. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, Chung DW, López JA. Oxidative modification of von Willebrand Factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115:706–712. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson DM, Stathopoulos NA, Giorgio TD, Hellums JD, Moake JL. Shear-induced platelet aggregation requires von Willebrand Factor and platelet membrane glycoproteins Ib and IIb-IIIa. Blood. 1987;69:625–628. [PubMed] [Google Scholar]
- 40.Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, Deykin D. Unusually large plasma factor VIII:von Willebrand Factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307:1432–1435. doi: 10.1056/NEJM198212023072306. [DOI] [PubMed] [Google Scholar]
- 41.Yao H, Duan M, Buch S. Cocaine-mediated induction of platelet-derived growth factor: Implication for increased vascular permeability. Blood. 2011;117:2538–2547. doi: 10.1182/blood-2010-10-313593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YW, Hennig B, Fiala M, Kim KS, Toborek M. Cocaine activates redox-regulated transcription factors and induces TNF-alpha expression in human brain endothelial cells. Brain Res. 2001;920:125–133. doi: 10.1016/s0006-8993(01)03047-5. [DOI] [PubMed] [Google Scholar]
- 43.Brogan WC, 3rd, Lange RA, Glamann DB, Hillis LD. Recurrent coronary vasoconstriction caused by intranasal cocaine: Possible role for metabolites. Ann Intern Med. 1992;116:556–561. doi: 10.7326/0003-4819-116-7-556. [DOI] [PubMed] [Google Scholar]
- 44.Kurth CD, Monitto C, Albuquerque ML, Feuer P, Anday E, Shaw L. Cocaine and its metabolites constrict cerebral arterioles in newborn pigs. J Pharmacol Exp Ther. 1993;265:587–591. [PubMed] [Google Scholar]
- 45.López JA, Dong JF. Shear stress and the role of high molecular weight von wWillebrand Factor multimers in thrombus formation. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S11–16. doi: 10.1097/01.mbc.0000167657.85143.ad. [DOI] [PubMed] [Google Scholar]
- 46.Benowitz NL. Clinical pharmacology and toxicology of cocaine. Pharmacol Toxicol. 1993;72:3–12. doi: 10.1111/j.1600-0773.1993.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 47.Lange RA, Hillis LD. Cardiovascular complications of cocaine use. New Eng J Med. 2001;345:351–358. doi: 10.1056/NEJM200108023450507. [DOI] [PubMed] [Google Scholar]
- 48.Vongpatanasin W, Mansour Y, Chavoshan B, Arbique D, Victor RG. Cocaine stimulates the human cardiovascular system via a central mechanism of action. Circulation. 1999;100:497–502. doi: 10.1161/01.cir.100.5.497. [DOI] [PubMed] [Google Scholar]
- 49.Patel R, Shah R, Baredes S, Spillert CR, Lazaro EJ. Nasal toxicity of cocaine: A hypercoagulable effect? J Natl Med Assoc. 2000;92:39–41. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.