

Abstract
Extracorporeal membrane oxygenation (ECMO) supports infants and children with severe cardiopulmonary compromise. Nutritional support for these children includes provision of medium- and long-chain fatty acids (FAs). However, ECMO induces a stress response, which could limit the capacity for FA oxidation. Metabolic impairment could induce new or exacerbate existing myocardial dysfunction. Using a clinically relevant piglet model, we tested the hypothesis that ECMO maintains the myocardial capacity for FA oxidation and preserves myocardial energy state. Provision of 13-Carbon labeled medium-chain FA (octanoate), long-chain free FAs (LCFAs), and lactate into systemic circulation showed that ECMO promoted relative increases in myocardial LCFA oxidation while inhibiting lactate oxidation. Loading of these labeled substrates at high dose into the left coronary artery demonstrated metabolic flexibility as the heart preferentially oxidized octanoate. ECMO preserved this octanoate metabolic response, but also promoted LCFA oxidation and inhibited lactate utilization. Rapid upregulation of pyruvate dehydrogenase kinase-4 (PDK4) protein appeared to participate in this metabolic shift during ECMO. ECMO also increased relative flux from lactate to alanine further supporting the role for pyruvate dehydrogenase inhibition by PDK4. High dose substrate loading during ECMO also elevated the myocardial energy state indexed by phosphocreatine to ATP ratio. ECMO promotes LCFA oxidation in immature hearts, while maintaining myocardial energy state. These data support the appropriateness of FA provision during ECMO support for the immature heart.
Keywords: Extracorporeal membrane oxygenation, immature heart, fatty acid oxidation, Nuclear magnetic resonance, substrate metabolism
1. Introduction
Extracorporeal membrane oxygenation (ECMO) provides a form of rescue for infants and children with severe pulmonary or cardiac disease [1, 2]. Respiratory failure and/or pulmonary hypertension such as occur with diaphragmatic hernia are the primary indications in neonates, although ECMO is used predominantly for severe acute cardiac decompensation in older infants and children [2]. Veno-arterial (V-A) ECMO redirects systemic venous return into a mechanical circuit containing an oxygenator. A pump within the circuit returns oxygenated blood to a major artery and maintains mean systemic blood flow pressure, while markedly reducing aortic pulse pressure. Thus, V-A ECMO provides a form of biventricular pressure and volume unloading, which theoretically allows the heart to rest and recover from injury. However, ECMO can induce a cardiac stun syndrome starting within a few hours of instituting support [3-6]. Stunning evidenced by severe cardiac dysfunction occurs in infants and children even without prior cardiac injury [4, 5], and similarly occurs in ECMO animal models [7]. Occurrence of stun impedes weaning or removal from the circuit which is the overall goal of therapy.
The mechanisms responsible for stunning still require elucidation. Some investigators have suggested that a surge in proinflammatory cytokines induced by the circuit is at least partially responsible [8-10]. In particular, we have previously noted that ECMO performed in immature swine promotes a tremendous increase in plasma interleukin-6 levels [11]. This particular proinflammatory cytokine decreases insulin sensitivity, possibly explaining skeletal muscle wasting noted during infant ECMO despite relatively high caloric supply [12]. However, the impact of ECMO on cardiac substrate metabolism has not been previously elucidated. The heart for many species undergoes an early postnatal metabolic shift towards preference for fatty acid (FA) oxidation [13, 14]. This shift is followed by further marked inhibition of carbohydrate oxidation during the period of rapid cardiac growth. Accordingly, the immature heart depends primarily on fats for provision of oxidative substrate to the citric acid cycle (CAC). Both medium- and long-chain FAs within emulsions are typically infused intravenously as fuel for infants and children undergoing ECMO procedures [15, 16]. However, it is unclear if the heart can sustain a further shift towards FA oxidation under ECMO conditions which may inhibit glucose oxidation. ECMO impairment of cardiac substrate oxidation could lead to reductions in mitochondrial ATP production or to a lower myocardial energy state, which could both explain the contractile dysfunction associated with the stun phenomenon. We tested the hypothesis that the ECMO support for the immature heart maintains the myocardial capacity for FA oxidation and thereby preserves the myocardial energy state. Using an immature piglet model emulating V-A ECMO in infants and children, we first determined if ECMO induced shifts in utilization of the three principal substrates oxidized by the pig myocardium: lactate, medium-chain FA (MCFA), exemplified by octanoate, and long-chain mixed FAs (LCFAs). Then we determined if the heart was capable of increasing relative acetyl-CoA contribution to the CAC from these substrates after direct perfusion infusion into the coronary arteries. Nuclear magnetic resonance (NMR) methods were used to determine substrate fractional contributions and metabolomic profiles in these hearts. Myocardial energy state was indexed by phosphocreatine (PCr) to ATP ratio, a typical surrogate for phosphorylation potential.
2. Methods
2.1. Animals
All experimental procedures were approved by Seattle Children’s Institutional Animal Use and Care Committee. Twenty-two male Yorkshire piglets (body weight 10.6-15.6 kg, age 25-38 days) were used. They were fasted overnight with free access to water. They were premedicated with an intramuscular injection of ketamine (33 mg/kg) and xylazine (2 mg/kg). After intubation through surgical tracheostomy, the piglets were mechanically ventilated (FiO2 40-60%, volume control 15ml/kg, PEEP 3cmH2O, respiratory rate 14-18/min), and isoflurane (1-2%) to maintain general anesthesia. An arterial pCO2 of 35-45 mmHg was maintained by adjusting minute ventilation.
2.2. Protocol
Animals were separated into experimental groups first by mode of circulation either normal without extracorporeal support (CON) or ECMO which provides mechanical unloading. The ECMO circuit is described below. Sham animals (CON) received anesthesia, assisted ventilation, and heparinization similar to ECMO animals, but were not connected to the ECMO circuit. Within each of these groups the animals were further separated by mode of 13-Carbon (13C) substrate delivery either in tracer amounts through systemic venous route (CON-S; ECMO-S) or delivery at high concentration to the heart via intracoronary (CON-IC; ECMO-IC) infusion into the left anterior descending coronary artery (LAD). Animals were maintained for 8 hours and received steady-state 13C substrate infusions over the final hour. Immediately upon completion of the labeled infusion, the portions of left ventricular (LV) myocardium perfused by the LAD were rapidly freeze-clamped and stored under liquid nitrogen for later extraction. Arterial and coronary venous blood samples were collected at multiple time points: after anesthesia induction and before ECMO as a baseline, 1, 2, 4 and 7 hours after starting ECMO, and just before completion of the labeled infusion as an endpoint. Baseline data was obtained after administration of heparin. Blood samples were immediately centrifuged and aliquots of plasma were stored at −80 °C. Plasma lactate and free FA concentrations were measured using commercial kits (BioVision, Mountain View, CA and Cayman, Ann Arbor, MI). Blood glucose was measured using a Bayer Contour point-of-care glucometer (Bayer HealthCare, Tarrytown, NY). Blood pH, pCO2, pO2, and hemoglobin were measured at regular intervals by a Radiometer ABL 800 (Radiometer America, Westlake, OH). Myocardial oxygen consumption (MVO2) was calculated from coronary venous flow and blood gas analysis. Ventilator settings as described above were maintained during ECMO.
2.3. Hemodynamic monitoring
An arterial line for systemic blood pressure monitoring and blood sampling was placed in the femoral artery. A saline-filled catheter was inserted into the internal jugular vein for continuous heparin infusion and connected to a pressure-membrane transducer for recording of the central venous pressure. A flow probe was placed around the ascending aorta to measure cardiac output (TS420, Transonic Systems Inc, Ithaca, NY). A 5-French high-fidelity micromanometer (Millar Instruments, Houston, TX) was inserted via the apex to measure LV pressure. To measure coronary venous flow, a cannula with an inflatable balloon cuff was placed into the coronary sinus via the right atrium; blood was returned to the superior vena cava by a shunt loop. A Transonic flow probe was placed around this shunt for continuous flow monitoring. The hemiazygous vein, which drains systemic venous blood to the coronary sinus in swine, was ligated to avoid systemic contamination of the coronary venous blood. A PowerLab 16/30 recorder (AD Instruments Inc., Colorado Springs, CO) continuously recorded hemodynamic data in all cases.
2.4. ECMO circuit and management
We used a miniaturized extracorporeal circuit to minimize hemodilution and avoid the need for blood transfusions. The circuit consisted of the following: a roller peristaltic pump console (Sarn8000 Terumo, Tokyo, Japan); a hollow fiber membrane oxygenator (CX-RX05RW, Terumo, Tokyo, Japan). The circuit was primed with dextran 40 in 0.9% sodium chloride, 5% dextrose and 2,000 units of heparin. The total prime volume was 80 mL.
After median sternotomy, the ascending aorta and right atrium were cannulated to create a V-A ECMO circuit. Management during ECMO maintained the pump flow rates of 80-100 mL/kg/min. We maintained a pH of 7.35 to 7.45, an arterial pCO2 of 35 to 45 mmHg, pO2 of >100 mmHg and a rectal temperature of 36 to 37.5 °C. ECMO duration time was 8 hours.
2.5. Infusion of labeled substrates
As noted previously, two separate substrate delivery methods were used. [2-13C]lactate and [2,4,6,8-13C]octanoate, MCFA, were obtained from Sigma (St. Louis, MO), and [U-13C]LCFAs was obtained from Cambridge Isotope Laboratories (Andover, MA). LCFAs consist of palmitic acid (45-55%), palmitoleic acid (10-15%), oleic acid (20-30%) and linoleic acid (10-15%). Labeled substrates in all protocols were infused for the final 60 minutes of the protocol. For the systemic delivered dose, [2-13C]lactate [2,4,6,8-13C]octanoate and [U-13C]LCFAs were used at 2.6, 0.8 and 0.8 μmol/kg body weight/min respectively, and were delivered into the left atrium (CON-S) or the aortic return cannula (ECMO-S). Intracoronary final concentrations of metabolites are equal to systemic arterial plasma levels at endpoint (Table 2) due to dilution with circulating blood in these 2 groups.
Table 2.
Systemic arterial substrate concentration.
| CON-S | ECMO-S | |||||
|---|---|---|---|---|---|---|
| Baseline | Endpoint | p | Baseline | Endpoint | p | |
| Lactate | 1.19 ± 0.27 | 1.38 ± 0.14 | 0.36 | 1.26 ± 0.16 | 1.43 ± 0.27 | 0.24 |
| Nonesterified FA | 0.20 ± 0.02 | 0.21 ± 0.03 | 0.33 | 0.24 ± 0.02 | 0.20 ± 0.02 | 0.94 |
| Glucose | 104 ± 11 | 108 ± 11 | 0.68 | 101 ± 10 | 116 ± 16 | 0.55 |
FA, fatty acid. Values are means ± SE; n = 6 per group.
For substrate loading by intracoronary infusion, the stable isotopes were infused directly into the LAD via a 24-gauge BD Saf-T-catheter (Becton Dickinson, Sandy, UT) inserted just distal to the origin of the first branch. The intracoronary doses were adjusted to achieve 1.2 mM [2-13C]lactate, 0.4 mM [2,4,6,8-13C]octanoate and 0.4 mM [U-13C]LCFAs concentrations and were based upon the mean LV coronary artery flow per body weight calculated in preliminary immature pig experiments [17, 18].
2.6. Metabolite analyses by NMR
Briefly, freeze-clamped hearts were ground into fine powder under liquid nitrogen and 0.5 mg further homogenized in 2.5 ml of a methanol/ddH2O (1:0.25) mix. A 2:1 chloroform/ddH2O mix was added to the homogenate, vortexed, and placed on ice for 10 minutes. The samples were next centrifuged for 10 minutes at 2000g for 15 minutes. The top layer was removed to a fresh tube and subjected to vacuum lyophilization. The resulting precipitate was dissolved in deuterium oxide (DLM11-100, Cambridge Isotopes, Andover, MA) and Chenomx ISTD (DSS) (IS-1, Chenomx, Alberta Canada) at a 9:1 ratio, filtered through a 0.22 μM syringe filter into NMR sample tubes (WG-1241-8, Wilmad LabGlass, Vineland, NJ).
13C-NMR spectra were acquired on a Varian Direct Drive (VNMRS) 600 MHz spectrometer (Varian Inc., Palo Alto, CA) equipped with a Dell Precision 390 Linux workstation running VNMRJ 2.2C. The spectrometer system was fitted with a Varian triple resonance salt-tolerant cold probe with a cold carbon preamplifier. Protons were decoupled with a Waltz decoupling scheme. Final spectra were obtained using a 45° excitation pulse (7.05 μs at 58 dB), with an acquisition time of 1.3 seconds, a recycle delay of 3 seconds, and a spectral width of 224.1 ppm. Fourier-transformed spectra were fitted with commercial software (NUTS, Acorn NMR Inc., Livermore, CA). All of the labeled carbon resonances (C1–C5) of glutamate were integrated using the Lorentzian peak-fitting subroutine in the acquisition program, NUTS as previously described [17-19]. The individual integral values were used as starting parameters for the CAC analysis-fitting algorithm tcaCALC (kindly provided by the Advanced Imaging Research Center at the University of Texas, Southwestern). This algorithm provided the fractional contribution (Fc) of each substrate to the acetyl-CoA pool entering the CAC. Label from exogenous [2-13C]lactate gives rise to label in the Carbon 1 of acetyl-CoA (Fc1), while [2,4,6,8-13C]octanoate labels Carbon 2 (Fc2) and [U-13C]LCFAs labels both Carbons 1 and 2 (Fc12), allowing for determination of each substrate’s Fc to the CAC (Figure 1).
Figure 1. Schematic demonstrating the 13C-labeling of acetyl-CoA and glutamate with [2-13C]lactate, [U-13C]LFCAs and [2,4,6,8-13C4]octanoate as substrates at the end of first turn of the CAC.

Double circles=13C from [2-13C]lactate; Filled gray circles=13C from [2,4,6,8-13C4]octanoate; Filled black circles=13C from [U-13C]LFCAs; Open circles=12C. Fc, fraction of acetyl-CoA enriched in 13C. The arrow with a dotted line comes from endogenous substrates.
Methanol extracted metabolites from tissue samples were analyzed using 1H-NMR, also performed on the Varian system to determine myocardial concentration of various metabolites. A standard one dimensional proton NOESY with presaturation (TMNNOESY) was collected on each sample, using the Chenomx standard data collection protocol [20]: a nonselective 90° excitation pulse (approximately 7 μs at 53 dB), a mixing time of 100 millisecond, acquisition time of 4 seconds, a recycle delay of 1 second, a sweep width of 12 ppm, and temperature control set to 25 °C. Collected spectra were analyzed using Chenomx software version 7.1 (Chenomx Inc., Edmonton, Canada), with quantifications based on spectral intensities relative to 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonate, which was added as a spike to each sample.
2.7. Immunoblot analyses
Immunoblot analyses were used to evaluate expression of GLUT4 and CD36, the predominant glucose and FA transporters in the heart, as well as for phosphorylation of key proteins regulating substrate oxidation.
Fifty micrograms of total protein extract from heart tissue were electrophoresed through 4.5% stacking and 10% running SDS-polyacrylamide gels and electroblotted onto PDVF-plus membranes. Membranes were blocked for 1 hour at room temperature with 5% nonfat milk in Tris-buffered saline plus Tween-20 (TBST: 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween-20). Equal protein loading of samples was determined by a protein assay (BioRad, Hercules, CA) and confirmed by Ponceau S staining (Sigma, St. Louis, MO) and probing with antibodies against GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were probed overnight at 4 °C with primary antibodies dissolved in PBS-T containing 5% milk or bovine serum albumin. The primary antibodies used in this study were 5′ adenosine monophosphate-activated protein kinase α (AMPKα), acetyl-CoA carboxylase (ACC) and GLUT4 obtained from Cell Signaling Technology (Danvers, MA). The CD36 antibody was purchased from Novus biologicals (Littleton, CO). The pyruvate dehydrogenase kinase (PDK)-2 and PDK4 antibodies were obtained as personal gifts from Dr. Robert Harris [21]. After two 5-minute washes with TBST and one 5-minute wash with TBS, blots were incubated at room temperature for 1 hour with the appropriate secondary antibody conjugated to horseradish peroxidase. The blots were washed twice for 10 minutes with TBST and visualized with enhanced chemiluminescence after exposure to Kodak Biomax light ML-2 film. The densitometric intensities were determined using Image J analysis software (National Institutes of Health, Bethesda, MD). Western blots were repeated in triplicate to confirm the findings.
2.8. Statistical analyses
Reported values are means ± standard error (SE) in figures, text, and tables. Results of Fc to acetyl-CoA by 13C-NMR were compared and analyzed with Mann-Whitney U test. Results of arterial substrate concentrations were analyzed with paired t-tests. ANOVA for multiple time points were used where appropriate e.g. time related changes during ECMO. Other statistical analyses were performed by one-way ANOVA with Tukey’s post hoc test. Criterion for significance was P < 0.05 for all comparisons.
3. Results
3.1. Cardiac function and myocardial oxygen consumption during ECMO
The animals did not receive any blood transfusions or inotropic or vasoactive drugs. The miniaturized extracorporeal circuit minimized hemodilution, and maintained hemoglobin within an expected range (9.5 ± 0.4 g/dl to 6.9 ± 0.3) in ECMO. Table 1 shows the parameters of cardiac function measured at a baseline and during ECMO in each group. We report LV functional parameters for the ECMO groups with the understanding that the accuracy of these measures acquired via a pressure-volume catheter is diminished in a ventricle with minimal volume. The LV functional data is shown in ECMO hearts more to demonstrate consistency in parameters between ECMO-S and ECMO-IC groups than to show alterations in function caused by ECMO. However, pressure hemodynamics obtained in the aorta show that ECMO markedly reduced the developed pressure (5.8 ± 1 mmHg), but maintained mean systemic blood pressure (53 ± 2 mmHg). In context of reduced accuracy in volume depleted ventricle, ECMO did significantly decrease the LV end-diastolic pressure when compared to baseline (P < 0.05). The calculate data from ECMO flow and aortic flow showed that about 90% of total cardiac output was supplied from ECMO flow.
Table 1.
Parameters of cardiac function at beginning and endpoints for each group.
| CON-S (n = 6) | ECMO-S (n = 5) | CON-IC (n = 6) | ECMO-IC (n = 5) | |||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Endpoint | Baseline | Endpoint | Baseline | Endpoint | Baseline | Endpoint | |
| Hemoglobin, g/dL | 9.1 ± 0.2 | 8.6 ± 0.3 | 9.1 ± 0.7 | 6.7 ± 0.5*† | 9.5 ± 0.2 | 9.1 ± 0.2 | 9.9 ± 0.2 | 7.1 ± 0.3*† |
| HR, BPM | 87 ± 2 | 91 ± 3 | 97 ± 6 | 110 ± 9 | 96 ± 6 | 103 ± 9 | 96 ± 3 | 117 ± 7 |
| CO, L/min | 1.2 ± 0.1 | 1.3 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.2 | 1.2 ± 0.1 | 1.2 ± 0.1 |
| Mean SBP, mmHg | 66 ± 3 | 58 ± 2 | 66 ± 4 | 52 ± 2* | 59 ± 5 | 57 ± 1 | 58 ± 3 | 54 ± 3 |
| DP, mmHg | 24 ± 2 | 24 ± 2 | 22 ± 1 | 5 ± 1*† | 25 ± 2 | 24 ± 3 | 27 ± 4.0 | 7 ± 2*† |
| LVEDP, mmHg | 10 ± 1 | 9 ± 1 | 11 ± 1 | 7 ± 1*† | 7 ± 1 | 7± 1 | 7 ± 1 | 5 ± 1*† |
| dP/dt max, mmHg/s | 906 ± 76 | 1023 ± 82 | 905 ± 74 | 611 ± 141† | 810 ± 50 | 892 ± 54 | 887 ± 38 | 754 ± 127 |
| dP/dtmin, mmHg/s | −1442 ± 72 | −1147 ± 85 | −1622 ± 126 | −657 ± 67*† | −1442 ± 240 | −1326 ± 100 | −1320 ± 35 | −662 ± 97*† |
| MVO2, μmol/min/g | 1.9 ± 0.1 | 1.8 ± 0.1 | 1.9 ± 0.2 | 1.4 ± 0.1* | 2.2 ± 0.3 | 1.9 ± 0.2 | 2.3 ± 0.2 | 1.7 ± 0.2* |
CO, mean value of cardiac output; Mean SBP, mean systemic blood pressure; DP, developed pressure (aortic systolic pressure – diastolic pressure); LVEDP, left ventricular end diastolic pressure; dP/dt, first derivative of LV pressure; MVO2, myocardial oxygen consumption. Values are means ± SE; n = 5-6 per group.
P < 0.05 vs baseline.
P < 0.05 vs Control.
ECMO reduced MVO2 approximately 30% from the pre-ECMO baseline, while substrate infusion itself did not change the calculated MVO2 (Figure 2). This change was due in large part to increased coronary venous oxygen content and decreased extraction, rather than to changes in hemoglobin concentration or coronary flow.
Figure 2. Myocardial oxygen consumption rate (MVO2) during ECMO.

MVO2 (A) was determined through arteriovenous oxygen content difference (B) and coronary sinus return flow (C). MVO2 was significantly decreased with ventricular unloading. Substrate infusion itself did not lead to change MVO2. Coronary venous oxygen content was increased after starting ECMO, and then it was gradually decreased parallel to coronary arterial oxygen content during ECMO. Thus difference between arterial and venous oxygen content was significantly decreased during ECMO, whereas coronary flow was not changed. A-V, arteriovenous oxygen content difference (circle with dotted line). Values are means ± SE; n = 10. *: P < 0.05 vs pre (baseline prior to ECMO).
Substrate infusion did not affect systemic hemodynamics (data not shown).
3.2. Metabolism in the immature swine heart on ECMO
We delivered relatively low amounts of stable isotopes into the systemic circulation in order to determine if ECMO alters acetyl-CoA fractional contribution for multiple substrates. Table 2 shows that these isotopic labels supplied into the systemic circulation did not increase total systemic arterial plasma metabolite levels, and thus did not alter coronary arterial supply of these substrates. Blood glucose levels were also stable during this protocol (Table 2).
Typical 13C spectra obtained from LV extracts are shown in Supplemental Figure 1. Spectral peak areas were entered into the tcaCALC program to determine the Fc of individual substrates to the CAC. The unlabeled component consists of circulating substrates taken up by the heart, or endogenous substrates within the heart undergoing oxidation. Figure 3A shows the absolute Fc for individual labeled substrates and the unlabeled substrates. As expected for delivery of tracer amounts the labeled substrates account for only a small portion of the acetyl-CoA (~15%) delivered to the CAC. However, ECMO-S increased overall Fc from labeled substrates compared to CON-S. ECMO-S increased LCFAs-Fc more than 3-fold (p = 0.006). When considering contributions from labeled substrates only, ECMO-S significantly increased the relative LCFAs-Fc while decreasing lactate-Fc. Octanoate contribution was not altered by ECMO-S.
Figure 3. Fractional contribution (Fc) to acetyl-CoA by 13C-NMR.

(A) Fc to acetyl-CoA with systemic infusion (S), including large unlabeled endogenous contribution. (B) Fc to acetyl-CoA with intracoronary infusion (IC). In comparison with S, Fc of [2,4,6,8-13C4]octanoate markedly increased with CON and ECMO. In both the systemic and intracoronary infusion groups, LCFA oxidation significantly increased with ECMO when compared to CON. White, Fc of unlabeled endogenous substrates; dark gray, Fc of [2-13C]lactate; light gray, Fc of [2,4,6,8-13C4]octanoate; black, Fc of [U-13C]LFCAs. (C) Anaplerotic component relative to CAC flux. Values are means ± SE; n = 5-6 per group. *: P < 0.05, †: P < 0.01 vs CON, ‡: P < 0.01 vs S.
Labeled substrate loading directly into the coronary artery markedly decreased the Fc from unlabeled substrate, and demonstrates the metabolic flexibility inherent in these immature hearts. Under these conditions the MCFA, octanoate, provides more than 60% of CAC oxidation via acetyl-CoA. However, just as occurred with systemic low dose labeled substrate loading, ECMO-IC increased in LCFAs-Fc relative to CON-IC. The ECMO group (Figure 3B) showed an approximate 2-fold greater LCFAs-Fc than observed in the Control group (7.4 ± 1.4 vs. 12.7 ± 1.8 %; P < 0.01). ECMO-IC also reduced the lactate-Fc (9.7 ± 0.7 vs. 3.4 ± 0.3 %; P < 0.05) in agreement with the results of systemic infusion, whereas MCFA oxidation was not altered by ECMO. Anaplerotic component relative to CAC flux demonstrated 15-20% in each group and were not significantly different among the 4 groups (Figure 3C).
Preliminary data from our samples showed that methanol extraction improved signal to noise and resolution compared to standard perchloric acid extraction. 1H-NMR spectra of myocardial metabolites showed a significantly increased alanine-to-lactate ratio with ECMO compared to Control group in both intracoronary and systemic infusions (Figure 4A, Supplemental Figure 2A). 13C-NMR also showed that the ratio of [2-13C]alanine to [2-13C]lactate increased with ECMO (Figure 4B). Absolute lactate concentration from extract LV tissue by 1H-NMR was similar between groups (Figure 4C). These results indicated that alanine labeling from lactate through pyruvate strongly increased, and pyruvate oxidation indexed by lactate-Fc decreased with ECMO.
Figure 4. Ratio of alanine to lactate as determined by (A) 1H-NMR and (B) 13C-NMR spectra and (C) absolute lactate concentration by 1H-NMR from left ventricular tissues at the end of the protocol.

1H-NMR shows absolute alanine and lactate ratios, whereas 13C-NMR shows tissue 13C labeled alanine and lactate. In both NMR, [alanine]/[lactate] significantly increased with ECMO compared to CON. Absolute lactate concentration was similar between groups. Values are means ± SE; n = 5-6 per group. *: P < 0.05, †: P < 0.01 vs systemic infusion (S).
Typical 1H spectra for the energy metabolites ares shown in Supplemental Figure 2B. Perchloric acid extracts typically produce low yield and unreliable data for energy metabolites. Methanol-chloroform extraction has shown optimal yields for these metabolites including NAD and NADH [22]. Tissue [Phosphocreatine (PCr)]/[ATP] from extracts in our study were comparable to those ratios previously obtained during 31phosphorous NMR in vivo in piglets of comparable ages [23, 24]. The ECMO induction of LCFAs-Fc did not modify [PCr]/[ATP] and [NADH]/[NAD+], demonstrating stability in the cellular energy state, despite the modest shift in substrate oxidation. However, high dose substrate infusion elevated [PCr]/[ATP] in ECMO-IC and [NADH]/[NAD+] in both CON-IC and ECMO-IC compared to CON-S and ECMO-S (Figure 5).
Figure 5. Energy metabolite ratios by 1H-NMR spectra from left ventricular tissues at the end of the protocol.
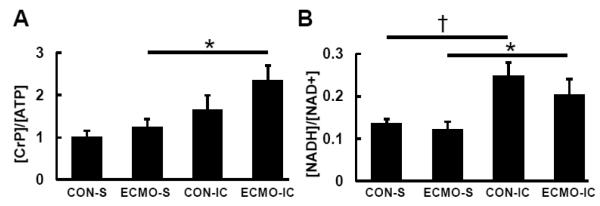
Values are means ± SE; n = 5-6 per group. *: P < 0.05, †: P < 0.01 vs systemic infusion (S).
3.3. Immunoblot analysis on carbohydrate and FA pathway under ECMO
To determine the mechanisms underlying these metabolic shifts, we assessed activation of carbohydrate and FA metabolic pathways by western blot analysis. Importantly, as shown in Figure 6A, the PDK4 expression level in ECMO animals was increased by 140% with systemic infusion and 70% with intracoronary infusion compared with CON. The PDK2 expression level did not differ between groups (Figure 6B). Moreover there were no differences in the expression of CD36 (Figure 6C) or GLUT4 (Figure 6D) between groups indicating that differences in FA oxidation were not due to alterations in expression of these membrane transporters. The phosphorylation of AMPKα (Thr172) and ACC (Ser79), regulators of one of the key pathways of FA oxidation, were also similar between groups (Figures 6E and F).
Figure 6. Representative immunoblots with densitometric analyses for proteins involved in regulation of fatty acid oxidation.

(A) The PDK4 expression levels shown normalized to mean CON-S intensity relative to GAPDH were increased in ECMO groups with systemic (S) and intracoronary infusion (IC) respectively compared with CON. No changes in protein expression between groups for PDK2 (B), CD36 (C) or Glut4 (D) were significant. Furthermore, no significant difference occurred between groups for phosphorylation state (blot intensity phosphorylation/total) for AMPK (E)) and ACC (F). Phosphorylation of each protein was detected on the same gel of each protein following re-probing of membranes. GAPDH was used as loading control and varied similarly to total protein by Ponceau S staining. All data are representative of at least three independent experiments. Values are means ± SE; n = 5-6 per group. *: P < 0.05 vs CON.
4. Discussion
This study represents the first evaluation of myocardial FA metabolism during ventricular unloading by ECMO. We showed that ECMO promotes myocardial FA oxidation relative to other substrates in the immature heart, while preserving the [PCr]/[ATP] and [NADH]/[NAD] ratios. Furthermore, ECMO does not limit the capacity for myocardial FA oxidation, as the heart accommodates to a robust free FA supply by elevating the mitochondrial acetyl-CoA contribution by those free FAs.
ECMO in infants promotes whole body protein turnover and skeletal muscle wasting [25]. This metabolic disadvantage is attributed to a persistent inflammatory state, which has been well documented in infants and animal models of ECMO [8-11]. Nutritional strategies have been formulated and attempted to prevent generalized protein wasting which can rapidly diminish total body mass in children [16]. Insulin resistance during ECMO caused in part by the inflammatory cytokines reduces glucose utilization and promotes a general catabolic state. Accordingly, researchers have attempted to modify the metabolic state and protein turnover by increasing glucose utilization with insulin clamp [26, 27]. Insulin doses maintained at steady-state levels as high as 20 times greater than normal minimally modified protein turnover. The effects of modifying energy balance and substrate utilization on the heart under these ECMO conditions has not been previously investigated. Adequate nutritional support of the heart is mandatory, as cardiac function serves as a primary obstacle to weaning from the circuit. Free FAs provide potential alternate substrates to carbohydrates, which efficiently sustain cardiac energy requirements during this period of ventricular unloading. Most nutritional strategies used for infants and children supported by ECMO include FA supplementation with both MCFA and LCFAs. The latter are transported into cytosol by sarcolemmal transporters, such as FA translocase (FAT/CD36), and into mitochondria through the carnitine shuttle consisting of three enzymes: carnitine palmitoyltransferase (CPT) -1, carnitine acylcarnitine translocase and CPT-2. In contrast, MCFAs do not rely on membrane transporters for their uptake into cells and mitochondria [28]. Despite the LCFA shuttling requirements, unloading by ECMO induces an increase in LCFA oxidation. Thus, unloading does not impair function of these shuttles and provision of LCFA under ECMO is appropriate for nutritional support of the heart.
Results from prior studies suggest that the heart subjected to unloading preferentially uses carbohydrates and exhibits impaired FA metabolism [29, 30]. Such impairments in FA handling can cause abnormal myocardial fat storage, lipotoxicity, and further damage the heart. Conditions for those studies as well as the developmental state of the hearts were markedly different than those prevailing for ECMO model, which contrasts substantially with regards to substrate utilization shifts promoted by unloading. Our observed shifts in FA oxidation by ECMO are accompanied by decreased relative lactate contribution and maintained MCFA contribution to the CAC via acetyl-CoA. Thus ECMO substantially increases LCFA-Fc under both substrate provisions and thereby decreases relative flux through pyruvate dehydrogenase (PDH).
We used [2-13C]lactate as the label to detect acetyl-CoA contribution through pyruvate decarboxylation. Under certain conditions such as cardiac hypertrophy or failure, pyruvate egress to the CAC substantially increases via malic enzyme or pyruvate carboxylase [31, 32] relative to pyruvate decarboxylation. Substantial CAC carbon entry along these anaplerotic pathways could conceivably confound modeling and the affect results provided by the tcaCALC algorithm. However, we have previously found that our immature porcine model is fairly resilient with respect to alterations in pyruvate anaplerosis relative to pyruvate decarboxylation after acute injury [17]. Under several different physiological conditions, including ischemia-reperfusion, pyruvate anaplerosis accounted for not more than 30% of total pyruvate entering the CAC. In the current study, 13C labeled lactate accounted for only a minor portion of acetyl-CoA entering the CAC, and thus pyruvate contribution through the carboxylase or malic enzyme would be substantially less. Thus, we feel that our tcaCALC modeling is appropriate. The model did provide estimates of overall anaplerosis compared to total CAC flux, which were comparable to previous studies using different methodology [17]. We showed minimal change in anaplerotic carbon contribution relative to CAC flux occurring after ECMO or after increased substrate loading. However, the total anaplerotic rate was maintained during the marked decrease in lactate oxidation caused by ECMO in the intracoronary substrate loaded group. This finding suggests that under these conditions anaplerosis does provide relatively higher contribution of carbons than through pyruvate decarboxylation. This study was not designed to specifically measure pyruvate anaplerosis, and other anaplerotic pathways could be contributing.
Reductions in relative flux via PDH could be caused by changes at either lactate dehydrogenase or at PDH itself. The 13C labeling patterns for alanine and lactate suggest that ECMO does not inhibit flux from lactate to pyruvate. The pools of intracellular pyruvate and alanine are in isotopic equilibrium, but lactate is not in isotopic equilibrium with pyruvate and hence alanine [33]. Thus, the shifts in the ratios for 13C labeling [alanine]/[lactate] caused by ECMO should reflect relative increases in flux through alanine transaminase versus lactate dehydrogenase. The increased alanine accumulation relative to lactate noted by 1H-NMR spectra also support this contention. However, due to compartmentalization of lactate we cannot be totally certain that the lactate detected by 1H-NMR represents the pool which exchanges with pyruvate and alanine.
One mechanism for these changes in substrate utilization appears to be PDK4 up regulation. PDK4 inhibits PDH through phosphorylation, thereby reciprocally increasing free FA oxidation via acyl-CoA dehydrogenases, as described by Randle [34]. This inhibition also accounts for the shuttling of pyruvate generated from lactate away from oxidative entry into the CAC and towards alanine. PDK protein responds rapidly to ECMO conditions, showing differences from control conditions within 8 hours. The rapidity of this response is consistent with prior multiple studies [35-37], which have shown that PDK4 transcription and mRNA levels in skeletal muscle respond within an hour to exercise. Surprisingly, relatively few studies have evaluated the time course for PDK4 protein accumulation after stimulation, although Holness et al showed that fasting caused 2-fold increases by 24 hours in murine anterior tibialis muscle [38]. Puthanveetil et al also demonstrated that dexamethasone treatment over 6 hours more than doubled PDK protein expression in cultured cardiomyocytes [39]. Although we could not eliminate all other potential mechanisms for increased FA oxidation, our results suggest that posttranslational regulation through phosphorylation of AMPK and ACC does not play an important role in our experimental model. Acetyl-CoA undergoes carboxylation by ACC to malonyl-CoA, an inhibitor of FA oxidation. Our findings are consistent with results from prior studies performed in porcine heart in vivo, which detected no alterations in AMPK and ACC phosphorylation during increases in FA oxidation associated with decreases in malonyl-CoA [40, 41]. However, our immunoblot data is limited by specificity and sensitivity of antibody-based detection of these proteins in pigs,
Our study also showed improvements in the myocardial energy state, indexed by [PCr]/[ATP] and [NADH]/[NAD] during relatively high FA oxidation rates promoted by the high free FA infusion dose. Some prior studies have shown that increasing FA oxidation relative to carbohydrates reduces LV mechanical efficiency by decreasing LV power for a given rate of MVO2 [42]. Compared with FAs, carbohydrates are traditionally presumed more oxygen efficient due to stoichiometric differences in ATP:oxygen ratios between these substrates. Additionally, high concentrations of FAs uncouple oxidative phosphorylation and increase MVO2 in isolated mitochondria and cardiomyocytes [43, 44]. One prior study suggested that lipid-enhancing interventions, such as Intralipid with heparin or glucose with insulin and potassium, increase work-independent MVO2 in adult swine hearts in vivo [45]. However, the basal or unloaded condition values were only assumed by those previous investigators. In the current study, we directly measured MVO2 under unloading conditions provide by ECMO. Consistent with prior studies in isolated perfused rat heart [46], high dose free FA oxidation produced a shift towards elevated cytosolic [NADH]/[NAD+] ratio as determined by 1H-NMR spectroscopy under both control and ECMO conditions. The cytosolic ratio [NADH]/[NAD+] operates in near equilibrium with mitochondrial [NADH]/[NAD+] ratio as well as with the cellular phosphorylation potential, which determines the free energy of ATP hydrolysis [47]. The high relative free FA oxidation in ECMO hearts supplied substrates at high doses also produced an elevated [PCr]/[ATP] ratio, an index of cellular phosphorylation potential. The relevance of ATP:oxygen ratio for determining energy efficiency remains controversial, as oxygen supply does not limit ATP production in normal heart, except at the highest workloads [48]. In fact, FAs produce substantially more ATP per molecule oxidized than do carbohydrates. Our study supports this contention by showing elevations in [PCr]/[ATP] concurrent with increases in FA oxidation relative to other substrates. These changes in high energy phosphates occur without elevation in MVO2, and therefore challenge the concept of LCFA oxidation promoting mitochondrial uncoupling in vivo. Although our measurements of [NADH]/[NAD] represent a composite of mitochondrial and cytosolic metabolites, they are consistent with prior studies which showed substrate induced elevations in cytosolic [PCr]/[ATP] and mitochondrial specific [NADH]/[NAD] [47, 49]. Thus, the immature heart retains the capacity of switching to more energy efficient FA oxidation during mechanical unloading or high dose substrate loading.
The energy modifications caused by increased free FA uptake and oxidation relative to carbohydrate have direct clinical importance. As carbohydrates promote protein loss during ECMO [25], lipid delivery represents another potential route for protein sparing. Lipid emulsions such as Intralipid are frequently used for nutritional support during ECMO [15]. Prior studies in vivo evaluated oxidation of individual free FA such as oleate [45]. Thus, previous data was incomplete with regard to the robustness of the free FA response and whether MCFAs have an impact on LCFA metabolism. We report for the first time in vivo use of emulsions containing multiple 13C labeled LCFAs as well as the MCFA, octanoate. This labeling strategy provides a physiological mixture of free FA which also emulates Intralipid, providing results more directly relevant to clinical practice. Our results show that the unloaded heart compared to the normal loaded heart has an increased preference for LCFAs, and that their increased utilization does not cause a reduction in [PCr]/[ATP] ratio.
ECMO causes cardiac stunning even when no prior direct myocardial injury occurs [4, 5]. Accordingly, we employed a previously uninjured and non-hypertrophied heart in order to define ECMO influence on substrate utilization and myocardial energy state without the confounding effects of hypoxia, ischemia or reperfusion. These factors, frequently encountered by the heart during surgical repair of congenital heart defects in infants, can modify hemodynamics during ECMO. In particular, ECMO markedly reduces both LV end diastolic volume and systolic LV wall stress in normal hearts, while increasing LV wall stress in hearts subjected to prior ischemic injury [50]. These factors may also cause changes in substrate metabolism. For instance, we previously showed that ischemia during cardiopulmonary bypass and subsequent reperfusion reduced substrate flux through PDH in the same immature pig model [17, 18]. Because of variability and additive effects caused by prior injury, we considered it imperative to first define substrate oxidation changes induced by ECMO alone. Although we have defined the early changes in substrate metabolism caused by ECMO, it remains unclear whether promotion of LCFA oxidation represents an advantage or a disadvantage. Some evidence exists suggesting that maintenance or promotion of free FA oxidation supports compensated hypertrophy [51-53]. However, this area is controversial and directional changes in substrate metabolism and their risk/benefit balance may vary according to the clinical condition. As noted, our model emulates unloading of immature hearts, which normally undergo physiological hypertrophy associated with rapid growth. We studied piglets just beyond the neonatal age, consistent with the developmental state for most human infants and children undergoing ECMO for cardiac failure. Further studies are ongoing to determine if similar alterations in substrate metabolism by ECMO occur after myocardial injury, stunning, and hypertrophy.
Conclusions
Our data show that ECMO induces a shift in substrate preference towards FA in the heart and maintains metabolic flexibility and myocardial energy state when supplied high dose free FA. Thus, this study suggests that nutritional strategies during ECMO, which include free FA supplementation are appropriate. Future studies are required to determine if substrate manipulation will improve cardiac function after weaning from the ECMO circuit.
Supplementary Material
Highlights.
Extracorporeal membrane oxygenation (ECMO) supports infants and children with severe cardiopulmonary compromise.
ECMO promotes a shift in substrate preference towards utilization of fatty acids.
High dose fatty acid loading during ECMO elevates the myocardial energy state.
The heart under ECMO demonstrates metabolic flexibility.
Acknowledgements
This work was supported by the National Institutes of Health R01HL60666 to M. A. Portman. A portion of the research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Footnotes
Disclosures None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Duncan BW. Pediatric mechanical circulatory support in the United States: past, present, and future. Asaio J. 2006;52:525–9. doi: 10.1097/01.mat.0000237588.44529.d1. [DOI] [PubMed] [Google Scholar]
- [2].Haines NM, Rycus PT, Zwischenberger JB, Bartlett RH, Undar A. Extracorporeal Life Support Registry Report 2008: neonatal and pediatric cardiac cases. ASAIO J. 2009;55:111–6. doi: 10.1097/MAT.0b013e318190b6f7. [DOI] [PubMed] [Google Scholar]
- [3].Rosenberg EM, Cook LN. Electromechanical dissociation in newborns treated with extracorporeal membrane oxygenation: an extreme form of cardiac stun syndrome. Crit Care Med. 1991;19:780–4. doi: 10.1097/00003246-199106000-00009. [DOI] [PubMed] [Google Scholar]
- [4].Martin GR, Short BL, Abbott C, O’Brien AM. Cardiac stun in infants undergoing extracorporeal membrane oxygenation. J Thorac Cardiovasc Surg. 1991;101:607–11. [PubMed] [Google Scholar]
- [5].Hirschl RB, Heiss KF, Bartlett RH. Severe myocardial dysfunction during extracorporeal membrane oxygenation. J Pediatr Surg. 1992;27:48–53. doi: 10.1016/0022-3468(92)90103-e. [DOI] [PubMed] [Google Scholar]
- [6].Pyles LA, Gustafson RA, Fortney J, Einzig S. Extracorporeal membrane oxygenation induced cardiac dysfunction in newborn lambs. J Cardiovasc Transl Res. 2010;3:625–34. doi: 10.1007/s12265-010-9215-5. [DOI] [PubMed] [Google Scholar]
- [7].Shen I, Levy FH, Benak AM, Rothnie CL, O’Rourke PP, Duncan BW, et al. Left ventricular dysfunction during extracorporeal membrane oxygenation in a hypoxemic swine model. Ann Thorac Surg. 2001;71:868–71. doi: 10.1016/s0003-4975(00)02281-5. [DOI] [PubMed] [Google Scholar]
- [8].Fortenberry JD, Bhardwaj V, Niemer P, Cornish JD, Wright JA, Bland L. Neutrophil and cytokine activation with neonatal extracorporeal membrane oxygenation. J Pediatr. 1996;128:670–8. doi: 10.1016/s0022-3476(96)80133-8. [DOI] [PubMed] [Google Scholar]
- [9].Adrian K, Mellgren K, Skogby M, Friberg LG, Mellgren G, Wadenvik H. Cytokine release during long-term extracorporeal circulation in an experimental model. Artif Organs. 1998;22:859–63. doi: 10.1046/j.1525-1594.1998.06121.x. [DOI] [PubMed] [Google Scholar]
- [10].Mc IRB, Timpa JG, Kurundkar AR, Holt DW, Kelly DR, Hartman YE, et al. Plasma concentrations of inflammatory cytokines rise rapidly during ECMO-related SIRS due to the release of preformed stores in the intestine. Lab Invest. 2010;90:128–39. doi: 10.1038/labinvest.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Priddy CM, Kajimoto M, Ledee DR, Bouchard B, Isern N, Olson AK, et al. Myocardial oxidative metabolism and protein synthesis during mechanical circulatory support by extracorporeal membrane oxygenation. Am J Physiol Heart Circ Physiol. 2013;304:H406–14. doi: 10.1152/ajpheart.00672.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shew SB, Keshen TH, Jahoor F, Jaksic T. The determinants of protein catabolism in neonates on extracorporeal membrane oxygenation. J Pediatr Surg. 1999;34:1086–90. doi: 10.1016/s0022-3468(99)90572-9. [DOI] [PubMed] [Google Scholar]
- [13].Itoi T, Lopaschuk GD. The contribution of glycolysis, glucose oxidation, lactate oxidation, and fatty acid oxidation to ATP production in isolated biventricular working hearts from 2-week-old rabbits. Pediatr Res. 1993;34:735–41. doi: 10.1203/00006450-199312000-00008. [DOI] [PubMed] [Google Scholar]
- [14].Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta. 1994;1213:263–76. doi: 10.1016/0005-2760(94)00082-4. [DOI] [PubMed] [Google Scholar]
- [15].Pettignano R, Heard M, Davis R, Labuz M, Hart M. Total enteral nutrition versus total parenteral nutrition during pediatric extracorporeal membrane oxygenation. Crit Care Med. 1998;26:358–63. doi: 10.1097/00003246-199802000-00041. [DOI] [PubMed] [Google Scholar]
- [16].Jaksic T, Hull MA, Modi BP, Ching YA, George D, Compher C. A.S.P.E.N. Clinical guidelines: nutrition support of neonates supported with extracorporeal membrane oxygenation. JPEN J Parenter Enteral Nutr. 2010;34:247–53. doi: 10.1177/0148607110369225. [DOI] [PubMed] [Google Scholar]
- [17].Olson AK, Hyyti OM, Cohen GA, Ning XH, Sadilek M, Isern N, et al. Superior cardiac function via anaplerotic pyruvate in the immature swine heart after cardiopulmonary bypass and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2315–20. doi: 10.1152/ajpheart.00739.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Olson AK, Bouchard B, Ning XH, Isern N, Rosiers CD, Portman MA. Triiodothyronine increases myocardial function and pyruvate entry into the citric acid cycle after reperfusion in a model of infant cardiopulmonary bypass. Am J Physiol Heart Circ Physiol. 2012;302:H1086–93. doi: 10.1152/ajpheart.00959.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Des Rosiers C, Lloyd S, Comte B, Chatham JC. A critical perspective of the use of (13)C-isotopomer analysis by GCMS and NMR as applied to cardiac metabolism. Metab Eng. 2004;6:44–58. doi: 10.1016/j.ymben.2003.10.004. [DOI] [PubMed] [Google Scholar]
- [20].Weljie AM, Newton J, Mercier P, Carlson E, Slupsky CM. Targeted profiling: quantitative analysis of 1H NMR metabolomics data. Anal Chem. 2006;78:4430–42. doi: 10.1021/ac060209g. [DOI] [PubMed] [Google Scholar]
- [21].Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys. 2000;381:1–7. doi: 10.1006/abbi.2000.1946. [DOI] [PubMed] [Google Scholar]
- [22].Sellick CA, Hansen R, Stephens GM, Goodacre R, Dickson AJ. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat Protoc. 2011;6:1241–9. doi: 10.1038/nprot.2011.366. [DOI] [PubMed] [Google Scholar]
- [23].Portman MA, Panos AL, Xiao Y, Anderson DL, Alfieris GM, Ning XH, et al. Influence of the pH of cardioplegic solutions on cellular energy metabolism and hydrogen ion flux during neonatal hypothermic circulatory arrest and reperfusion: a dynamic 31P nuclear magnetic resonance study in a pig model. J Thorac Cardiovasc Surg. 1997;114:601–8. doi: 10.1016/s0022-5223(97)70050-3. [DOI] [PubMed] [Google Scholar]
- [24].Portman MA, Panos AL, Xiao Y, Anderson DL, Ning X. HOE-642 (cariporide) alters pH(i) and diastolic function after ischemia during reperfusion in pig hearts in situ. Am J Physiol Heart Circ Physiol. 2001;280:H830–4. doi: 10.1152/ajpheart.2001.280.2.H830. [DOI] [PubMed] [Google Scholar]
- [25].Keshen TH, Miller RG, Jahoor F, Jaksic T. Stable isotopic quantitation of protein metabolism and energy expenditure in neonates on- and post-extracorporeal life support. J Pediatr Surg. 1997;32:958–62. doi: 10.1016/s0022-3468(97)90377-8. discussion 62-3. [DOI] [PubMed] [Google Scholar]
- [26].Agus MS, Javid PJ, Ryan DP, Jaksic T. Intravenous insulin decreases protein breakdown in infants on extracorporeal membrane oxygenation. J Pediatr Surg. 2004;39:839–44. doi: 10.1016/j.jpedsurg.2004.02.013. discussion - 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Agus MS, Javid PJ, Piper HG, Wypij D, Duggan CP, Ryan DP, et al. The effect of insulin infusion upon protein metabolism in neonates on extracorporeal life support. Ann Surg. 2006;244:536–44. doi: 10.1097/01.sla.0000237758.93186.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Labarthe F, Gelinas R, Des Rosiers C. Medium-chain fatty acids as metabolic therapy in cardiac disease. Cardiovasc Drugs Ther. 2008;22:97–106. doi: 10.1007/s10557-008-6084-0. [DOI] [PubMed] [Google Scholar]
- [29].Depre C, Shipley GL, Chen W, Han Q, Doenst T, Moore ML, et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med. 1998;4:1269–75. doi: 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- [30].Doenst T, Goodwin GW, Cedars AM, Wang M, Stepkowski S, Taegtmeyer H. Load-induced changes in vivo alter substrate fluxes and insulin responsiveness of rat heart in vitro. Metabolism. 2001;50:1083–90. doi: 10.1053/meta.2001.25605. [DOI] [PubMed] [Google Scholar]
- [31].Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104:805–12. doi: 10.1161/CIRCRESAHA.108.189951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC. Cardiac anaplerosis in health and disease: food for thought. Cardiovasc Res. 2011;90:210–9. doi: 10.1093/cvr/cvr055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lewandowski ED, Johnston DL, Roberts R. Effects of inosine on glycolysis and contracture during myocardial ischemia. Circ Res. 1991;68:578–87. doi: 10.1161/01.res.68.2.578. [DOI] [PubMed] [Google Scholar]
- [34].Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- [35].Hildebrandt AL, Pilegaard H, Neufer PD. Differential transcriptional activation of select metabolic genes in response to variations in exercise intensity and duration. Am J Physiol Endocrinol Metab. 2003;285:E1021–7. doi: 10.1152/ajpendo.00234.2003. [DOI] [PubMed] [Google Scholar]
- [36].Nordsborg N, Bangsbo J, Pilegaard H. Effect of high-intensity training on exercise-induced gene expression specific to ion homeostasis and metabolism. J Appl Physiol. 2003;95:1201–6. doi: 10.1152/japplphysiol.00257.2003. [DOI] [PubMed] [Google Scholar]
- [37].Pilegaard H, Ordway GA, Saltin B, Neufer PD. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab. 2000;279:E806–14. doi: 10.1152/ajpendo.2000.279.4.E806. [DOI] [PubMed] [Google Scholar]
- [38].Holness MJ, Kraus A, Harris RA, Sugden MC. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes. 2000;49:775–81. doi: 10.2337/diabetes.49.5.775. [DOI] [PubMed] [Google Scholar]
- [39].Puthanveetil P, Wang Y, Wang F, Kim MS, Abrahani A, Rodrigues B. The increase in cardiac pyruvate dehydrogenase kinase-4 after short-term dexamethasone is controlled by an Akt-p38-forkhead box other factor-1 signaling axis. Endocrinology. 2010;151:2306–18. doi: 10.1210/en.2009-1072. [DOI] [PubMed] [Google Scholar]
- [40].King KL, Okere IC, Sharma N, Dyck JR, Reszko AE, McElfresh TA, et al. Regulation of cardiac malonyl-CoA content and fatty acid oxidation during increased cardiac power. Am J Physiol Heart Circ Physiol. 2005;289:H1033–7. doi: 10.1152/ajpheart.00210.2005. [DOI] [PubMed] [Google Scholar]
- [41].Zhou L, Huang H, Yuan CL, Keung W, Lopaschuk GD, Stanley WC. Metabolic response to an acute jump in cardiac workload: effects on malonyl-CoA, mechanical efficiency, and fatty acid oxidation. Am J Physiol Heart Circ Physiol. 2008;294:H954–60. doi: 10.1152/ajpheart.00557.2007. [DOI] [PubMed] [Google Scholar]
- [42].Zhou L, Huang H, McElfresh TA, Prosdocimo DA, Stanley WC. Impact of anaerobic glycolysis and oxidative substrate selection on contractile function and mechanical efficiency during moderate severity ischemia. Am J Physiol Heart Circ Physiol. 2008;295:H939–H45. doi: 10.1152/ajpheart.00561.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–74. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- [44].Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–83. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. 2000;278:H1345–51. doi: 10.1152/ajpheart.2000.278.4.H1345. [DOI] [PubMed] [Google Scholar]
- [46].Hassinen I, Ito K, Nioka S, Chance B. Mechanism of fatty acid effect on myocardial oxygen consumption. A phosphorus NMR study. Biochim Biophys Acta. 1990;1019:73–80. doi: 10.1016/0005-2728(90)90126-o. [DOI] [PubMed] [Google Scholar]
- [47].Scholz TD, Laughlin MR, Balaban RS, Kupriyanov VV, Heineman FW. Effect of substrate on mitochondrial NADH, cytosolic redox state, and phosphorylated compounds in isolated hearts. Am J Physiol. 1995;268:H82–91. doi: 10.1152/ajpheart.1995.268.1.H82. [DOI] [PubMed] [Google Scholar]
- [48].Zhang J, Murakami Y, Zhang Y, Cho YK, Ye Y, Gong G, et al. Oxygen delivery does not limit cardiac performance during high work states. Am J Physiol Heart Circ Physiol. 1999;277:H50–7. doi: 10.1152/ajpheart.1999.277.1.H50. [DOI] [PubMed] [Google Scholar]
- [49].Kim DK, Heineman FW, Balaban RS. Effects of beta-hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260:H1767–73. doi: 10.1152/ajpheart.1991.260.6.H1767. [DOI] [PubMed] [Google Scholar]
- [50].Jugdutt BI. Ischemia/Infarction. Heart Fail Clin. 2012;8:43–51. doi: 10.1016/j.hfc.2011.08.006. [DOI] [PubMed] [Google Scholar]
- [51].Duda MK, O’Shea KM, Lei B, Barrows BR, Azimzadeh AM, McElfresh TE, et al. Low-carbohydrate/high-fat diet attenuates pressure overload-induced ventricular remodeling and dysfunction. J Card Fail. 2008;14:327–35. doi: 10.1016/j.cardfail.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ito M, Jaswal JS, Lam VH, Oka T, Zhang L, Beker DL, et al. High levels of fatty acids increase contractile function of neonatal rabbit hearts during reperfusion following ischemia. Am J Physiol Heart Circ Physiol. 2010;298:H1426–37. doi: 10.1152/ajpheart.00284.2009. [DOI] [PubMed] [Google Scholar]
- [53].Olson AK, Ledee D, Iwamoto K, Kajimoto M, O’Kelly Priddy C, Isern N, et al. C-Myc induced compensated cardiac hypertrophy increases free fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol. 2013;55:156–64. doi: 10.1016/j.yjmcc.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.