

Abstract
Several gene therapy applications require the transfer and simultaneous expression of multiple genes in the same cell. In this study, we analyzed the potential for coordinated expression of an endogenous bidirectional promoter located on chromosome X, which controls the expression of the heterogeneous nuclear ribonucleoprotein H2 (HNRNPH2) and alpha-galactosidase (GLA) genes. The promoter was cloned in both transcriptional orientations in a foamy virus (FV) vector backbone, whereas the enhanced green fluorescent protein (EGFP) and low-affinity nerve growth factor receptor (ΔLNGFR) reporter genes were cloned in the 5′–3′ and 3′–5′ transcriptional orientations, respectively. In all the cell lines tested, both vectors showed high levels of transgene coexpression that reached 76% of total positive cells (range from 76 to 18%). Comparison of EGFP and ΔNGFR levels revealed that the side of the promoter that drives the expression of the HNRNPH2 gene in the genome was stronger and in accordance to its in situ activity. When tested with CD34+ cells, transgene coexpression reached 35.3% of all positive cells in progenitor assays and 16.8% of all positive cells after transplantation in NOD/severe combined immunodeficient mice. In summary, we show that the endogenous promoter used in this study holds bidirectional activity in the context of FV vectors and can be used in gene therapy applications requiring synchronized expression of two genes.
Keywords: FV, bidirectional promoter, gene transfer, hematopoietic stem cells
Introduction
The expression of multiple genes in gene transfer applications is a desirable property of viral vectors. Coexpression of a marker gene or an antibiotic resistance gene could be used to enrich gene-corrected cells before their in vivo administration. In applications in which regulated gene expression is required, co-delivery of a transcriptional activator or repressor to the same target cell could also be a useful feature of any vector design. In vivo selection of transduced cells can also be carried out by concomitant expression of the MGMT gene.1-5 Finally, the phenomenon of vector-mediated insertional mutagenesis could be tackled by simultaneous expression of conditionally cytotoxic genes.
A number of approaches have been used to generate bicistronic vectors capable of simultaneous transgene expression. A widely used method is the inclusion of viral internal ribosome entry site (IRES) elements that permit cap-independent mRNA translation. However, this technology has been characterized by reduced expression of the transgene downstream of the IRES element and cell type-dependent efficiency of balanced expression.6-8 Another method has been the construction of viral vectors with two transcription units cloned in tandem but this design has been characterized by transcriptional interference.9,10 This strategy has also been exploited in lentiviral vectors, but the published reports present conflicting data on the problem of promoter interference.11,12 Expression of multiple genes by a single vector can also be accomplished with artificial genes encoding multiple proteins linked by the 2A sequences of the foot-and-mouth disease virus in which cleavage of the expressed polyproteins is mediated by the 2A peptides.13-16 However, addition of the 2A peptide requires engineering of the proteins, although the expression of the cDNAs has been reported to be dependent on the nature of the expressed genes and the cloning order in the construct.3 Coordinated expression has also been achieved with the construction of a synthetic bidirectional promoter using elements from the viral CMV, the UBI-C and the PGK promoter sequences.17 Recently, a ubiquitously acting chromatin opening element (A2UCOE), located between the divergently transcribed heterogeneous nuclear ribonucleoprotein H2 (HNRPA2B1) and CBX3 genes, has shown more stable and position-independent expression compared with viral promoters.18 The A2UCOE element is a methylation-free CpG island that is present in divergently transcribed housekeeping genes, but it has not been tested for its bidirectional promoter activity in vitro.
Computational studies have identified a large number of divergently transcribed gene pairs, representing about 10% of all human genes, located on opposite strands with transcriptional start sites <1000 bp apart.19-21 The transcripts of many bidirectional gene pairs were simultaneously coexpressed, indicating that their shared cis-regulatory elements could initiate transcription at both directions.21 This bidirectional regulation system has been observed in the mammalian genome and it may reflect the requirement for tightly coordinated expression in certain cellular pathways, such as cell cycle,22 histone gene expression23 and heat shock responses.24 Such dual promoters are theoretically less likely to activate transgene silencing as they are often driving the expression of housekeeping genes.21 In this study, we have used such a putative bidirectional promoter which was identified in the course of a gene targeting experiment (DW Russell, unpublished data) and is located on chromosome Xq22.1. The promoter is driving the expression of the HNRNPH2 and the alpha-galactosidase (GLA) human genes, which are expressed in many tissues, including the hematopoietic system (HNRNPH2 (http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg18&position=chrX:100549847-100555773&hgsid=146016699&knownGene=full); GLA (http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg18&position=chrX:100539435-100549657&hgsid=146016699&knownGene=full)). We show that when cloned in a foamy virus (FV) vector backbone, the promoter holds bidirectional activity and can transcribe two reporter genes in a coordinated manner, recapitulating its endogenous activity.
Results
Both promoter orientations are active in diverse cellular environments
The putative bidirectional promoter, located at chromosome position Xq22.1, lies in the intragenic region between the GLA (NM_000169.2) and the HNRNPH2 genes (NM_001032393.1) (Figure 1a). The sequence was 402 bp long and included the region immediately upstream of the translation start site of the GLA gene and part of the HNRPH2 5′-untranslated region up to position +103 of the mRNA. The promoter was PCR isolated from human genomic DNA and cloned in an intermediate vector (see Materials and methods) from which it was further amplified with primers carrying ClaI or AgeI restriction enzyme sites, fitted to permit directional cloning into a deleted FV vector with an enhanced green fluorescent protein (EGFP) reporter gene (Figure 1b). The constructs were designated ΔΦ.GH.F and ΔΦ.HG.F to mark the sequence orientation towards the HNRNPH2 or the GLA gene respectively, followed by the EGFP gene (F). The mean titer for the vector (expressed as EGFP-transducing particles per ml of non-concentrated supernatant) with the GH orientation was 5 × 10E5 (n = 3), whereas the titer with the HG orientation was 3.3 × 10E5 (n = 3). The vectors showed high titers as those obtained with standard FV vectors with constitutive promoters, indicating that the inserted sequence did not exert any deleterious effect on viral vector packaging. To test whether the cloned sequence provided adequate promoter activity, we transduced HT1080 cells with the ΔΦ.HG.F, ΔΦ.GH.F and ΔΦ.PGK.EGFP vectors at similar multiplicity of infections (MOIs) (2.1, 3.0 and 2.5, respectively) and analyzed EGFP expression levels by flow cytometry. Transgene expression could be detected in cells transduced with either ΔΦ.GH.F or ΔΦ.HG.F vectors; in addition, it seems that the GH promoter has reasonable expression levels that can be compared with those obtained with the PGK promoter (Figure 1c). Specifically, the HG orientation produced a mean fluorescence intensity (MFI) = 22 that was 15.9% of the ΔΦ.PGK.EGFP vector (PGK MFI 124), whereas the GH orientation reached 33.3% of the PF expression levels (GH MFI 35). To test whether the promoter was active in diverse cellular environments, we transduced a series of established lines with both vectors and at similar MOIs and analyzed EGFP expression by flow cytometry. Preliminary data showed that EGFP expression was observed in various cell lines (human cervical epithelia (HeLa), human teratocarcinoma (HT1080), human embryonic kidney (293T), baby hamster kidney (BHK), human chronic myeloid leukemia (K562), including primary murine Lin- cells (data not shown). These results indicate that the promoter was functional in diverse cellular environments and its expression levels were comparable with those of the PGK promoter. To directly compare the potential of the promoter in a bicistronic vector design (see Figure 2a below), we compared the GFP levels from the bidirectional promoter to the GFP levels generated from a vector driving GFP expression off an IRES element. The results (Figure 1d) showed directly comparable levels for the HG orientation (HeLa: 8 ± 2.2; HT1080: 10.9 ± 2) and higher MFI levels for the GH orientation (HeLa: 13.1 ± 5.7; HT1080: 36.2 ± 10.6) relative to the IRES vector (HeLa: 9 ± 1.7; HT1080: 17.8 ± 5.8), indicating that the promoter was capable of potent GFP expression and could substitute the IRES element in bicistronic vector designs.
Figure 1.
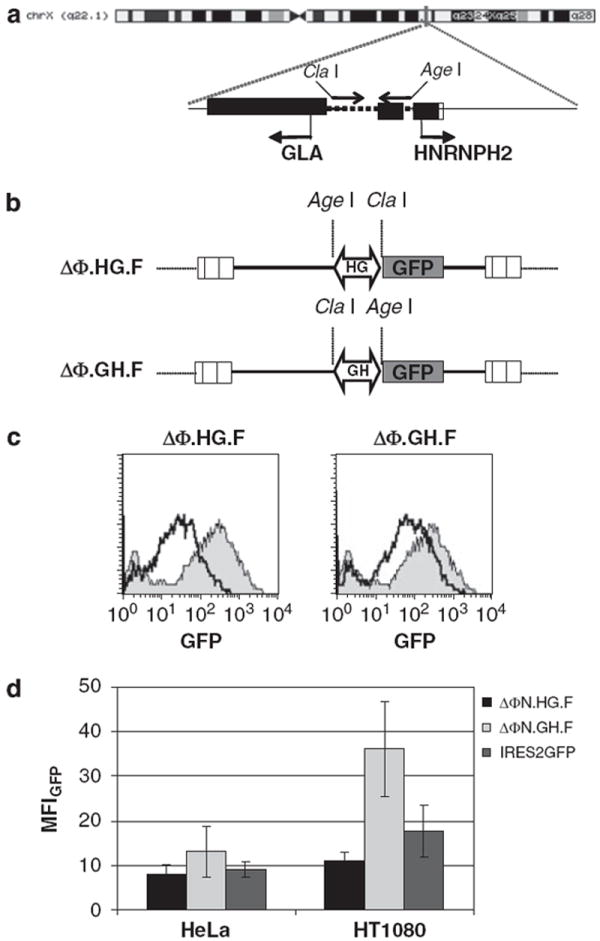
Construction and functional assays of foamy virus (FV) vectors with an endogenous bidirectional promoter (a) Genomic organization of the bidirectional promoter located on chromosome Xq22 in the intragenic region between the alpha-galactosidase (GLA) and the heterogeneous nuclear ribonucleoprotein H2 (HNRNPH2) genes. A 402-bp region (dotted line) was PCR amplified with primers carrying ClaI and AgeI restriction sites and subsequently cloned in the deleted foamy viral vector backbone ΔΦ. (b) Illustration of the FV viral constructs generated after insertion of the putative promoter in either orientation. HG denotes the transcription towards the GLA gene, whereas GH denotes transcription towards the HNRNPH2 gene. An enhanced green fluorescent protein (EGFP) reporter gene was placed downstream of the promoter in sense orientation relative to viral transcription. (c) Transduction of HT1080 cells with ΔΦN.HG.F and ΔΦN.GH.F vectors carrying the EGFP expression cassette. Flow cytometry analysis of EGFP expression in comparison with cells transduced with FV vectors carrying a standard PGK promoter (filled histogram). (d) GFP mean fluorescence intensity (MFI) levels from an IRES2GFP vector were compared with the MFI levels generated from the bicistronic ΔΦ.N.HG.F and ΔΦ.N.GH.F vectors. Results from at least three independent experiments with s.d. are shown.
Figure 2.
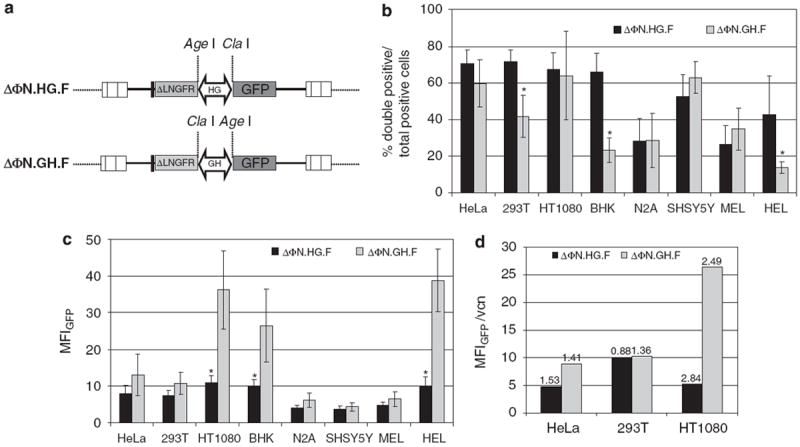
Gene expression efficiency of the bidirectional promoter (a) Diagram of the foamy virus (FV) vectors with the two reporter genes (low-affinity nerve growth factor receptor (ΔLNGFR) and enhanced green fluorescent protein (EGFP)) cloned on either end of the promoter. GH denotes the alpha-galactosidase (GLA) to heterogeneous nuclear ribonucleoprotein H2 (HNRNPH2) orientation and HG the HNRNPH2 to GLA orientation of the promoter. A rabbit β-globin polyA was cloned in the 5′ end of ΔLNGFR. (b) Cell lines of diverse cellular background were transduced with the ΔΦN.HG.F or ΔΦN.GH.F FV vectors and the expression of EGFP and ΔLNGFR was analyzed by flow cytometry. The percentage of double-positive cells (coexpressing EGFP and ΔLNGFR) over the total number of reporter-positive (EGFP single positive+ΔLNGFR single positive+EGFP and ΔLNGFR double positive) cells from at least three individual experiments were plotted. (c) Comparison of promoter strength for each vector as determined by the total mean fluorescence intensity (MFI) of the EGFP protein. Values for both single- and double-positive cells were plotted. Mean values ±s.d. of at least three different experiments are shown. (d) Correlation of vector copy number determined by quantitative Taqman PCR and MFI in HeLa, 293T and HT1080 cells transduced with ΔΦN.HG.F or ΔΦN.GH.F FV viral vectors. Values above bars refer to average provirus copies per cell. (*P≤0.05).
Dual reporter gene expression in vitro with FV vectors carrying the bidirectional promoter
To test the ability of the bidirectional promoter to drive the simultaneous expression of two transgenes, we cloned each orientation of the promoter in a dual reporter FV backbone that had the deleted low-affinity nerve growth factor receptor (ΔLNGFR) with a rabbit β-globin polyA upstream and the EGFP reporter downstream of the promoter sequence (Figure 2a). These vectors were designated ΔΦN.HG.F and ΔΦN.GH.F and were produced at high titers; concentrated vector stock titers were 3.24±1.33 × 106 TU ml−1 (n = 20) for ΔΦN. HG.F and 3.4 ± 2 × 106 TU ml−1 (n = 12) for ΔΦN.GH.F (quantitated as EGFP-transducing units on HeLa cells). These values were comparable with standard FV-EGFP vectors with constitutive promoters. We then explored the expression potential of the two vectors in diverse cellular environments by transducing a number of cells lines, namely, HeLa, 293T, BHK, HT1080, Neuro-2A, SHSY5Y, MEL and HEL. The cells were transduced at an MOI ranging from 1 to 4 and expression was analyzed by flow cytometry. We calculated the percentage of cells coexpressing EGFP and ΔLNGFR as a fraction of the total positive cells in each case to control for any transduction bias of the FV vectors in the different cell lines.
The bidirectional promoter was efficient in driving the simultaneous expression of both transgenes in the epithelial (HeLa, 293T, HT1080 and BHK), neuroblastoma (Neuro-2A and SHSY5Y) and hematopoietic cell lines (MEL and HEL). Overall, the dual promoter was active in both directions (ΔΦN.HG.F and ΔΦN.GH.F) as indicated by similar percentages of double-positive cells. In three out of eight cell lines (293T, BHK and HEL) the ΔΦN.HG.F vector gave statistically significant (P<0.05) higher numbers of positive cells coexpressing the two transgenes compared with the ΔΦN.GH.F vector (Figure 2b). A pattern of balanced expression was more evident in the epithelial cells compared with hematopoietic or neuroblastoma lines, with >60% of total positive cells coexpressing both EGFP and ΔLNGFR. These results indicate that the promoter showed bidirectional activity that was significantly pronounced in epithelial cell lines.
To assay for transgene expression levels, we calculated the total EGFP MFI for each vector in each cell line transduced at similar MOIs (range 1–4). As shown in Figure 2c, in three (HT1080, BHK and HEL) out of eight cell lines, a higher EGFP MFI was observed with the ΔΦN.GH.F vector (P<0.05) indicating that the HNRNPH2 side of the promoter, which drives EGFP expression in this vector, is stronger in these cell lines. No particular differences were observed for the ΔLNGFR MFI between ΔΦN.HG.F and ΔΦN.GH.F vectors (data not shown). To confirm that the higher EGFP MFI observed with the ΔΦN.GH.F vector was due to transcription imbalances between the two promoter sides rather than copy number variations, we determined the vector copy number by Taqman PCR in three cell lines (HeLa, 293T and HT1080). Expression of EGFP was then corrected for vector copy number and plotted in Figure 2d. The results show that in HT1080 cells, there is a clear bias of the GH promoter orientation, which is less prominent in HeLa and absent in 293T cells (Figure 2d). This indicated that the HNRNPH2 side of the promoter has stronger activity in this cell line and suggested that vector performance is, to some extent, cell line dependent.
In vitro transduction of primary hematopoietic stem/progenitor cells by FV vectors carrying the bidirectional promoter
We subsequently assessed the expression efficiency of the bidirectional promoter in primary human cord blood (CB) CD34+ cells by transducing them ex vivo with ΔΦN.HG.F and ΔΦN.GH.F vectors at MOI 20 in serum-free medium supplemented with cytokines, as described in the Materials and methods section. Each CD34+ sample was transduced in parallel with both vectors to facilitate direct comparison. Transgene expression was analyzed by flow cytometry 5–7 days after transduction in cell pools expanded in liquid cultures. We also analyzed the efficiency of the dual promoter on human short-term hematopoietic progenitors in methylcellulose cultures scored at 10 days after transduction. Overall, ΔΦN.HG.F vector gave a slightly higher—yet nonsignificantly different—percentage (35.3±19%) of total positive cells coexpressing EGFP and ΔLNGFR (Figure 3a). CB CD34+ cells transduced with ΔΦN.GH.F vector coexpressed the two transgenes at an average of 26.8±9.4% and presented with a skewed EGFP expression as seen by higher percentage of single EGFP-positive cells (Figure 3b). Analysis of promoter orientation strength was assayed by quantitation of the total MFI of each reporter gene in each vector. Cumulative analysis of the EGFP MFI illustrated higher levels with the ΔΦN.GH.F vector (Figure 3c), but owing to extensive variability, the difference from the values obtained with ΔΦN.HG.F vector was not significant (P = 0.14). In contrast to EGFP, no differences in the total ΔLNGFR MFI (Figure 3d) were detected between the two vectors, but this could be attributed to the nature of the ΔLNGFR protein or the different detection method. These findings were in accordance with what we had observed in the cell lines strongly indicating that the HNRNPH2 side of the promoter (ΔΦN.GH.F vector) was also more potent in human hematopoietic stem cells (HSCs).
Figure 3.
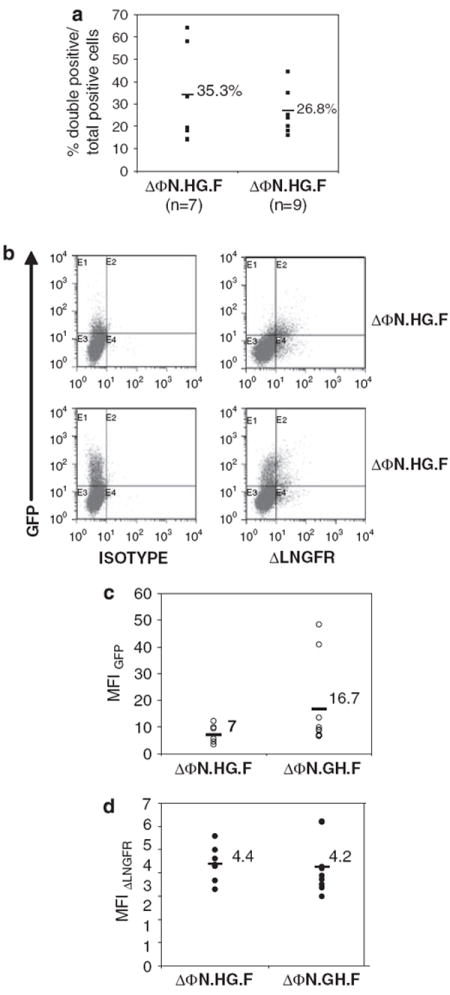
In vitro promoter performance in primary human hematopoietic stem cells (HSCs) (a) Human cord blood (CB) CD34+ were transduced with ΔΦN.HG.F or ΔΦN.GH.F foamy viral vectors and the number of cells coexpressing enhanced green fluorescent protein (EGFP) and low-affinity nerve growth factor receptor (ΔLNGFR) over the percentage of total positive cells are shown. (b) Representative fluorescent-activated cell sorting plots of CB CD34+ cells analyzed 7 days after transduction with ΔΦN.HG.F or ΔΦN.GH.F vectors. (c) The mean fluorescence intensities (MFIs) for EGFP and ΔLNGFR (d) of the transgene-positive population are shown for each vector. n indicates the number of individual experiments performed. The horizontal bar shown in each column represents the mean percentage value for each vector.
Dual expression by the bidirectional promoter in NOD/SCID repopulating cells
To test the long-term expression efficiency of the bidirectional promoter, we performed NOD/severe combined immunodeficient (SCID) bone marrow transplantation experiments with human CB CD34+ cells transduced with ΔΦN.HG.F or ΔΦN.GH.F FV vectors at MOI 20 as mentioned in the previous section. Busulfan-conditioned animals receiving transduced cells were analyzed 8–10 weeks after transplantation for the presence of human multilineage hematopoiesis in their bone marrow by flow cytometry. Human cell engraftment levels, as measured by the percentage of human CD45+ cells in the bone marrow, ranged from 0.1 to 94% of total cells depending on the conditioning regime and were unaffected by the vector used to transduce the transplanted cells (data not shown). We observed a mean of 16.8% (range 3.7–27.5%) and 13.6% (range 6.3–28%) of total human positive cells coexpressing EGFP and ΔLNGFR, with ΔΦN.HG.F and ΔΦN.GH.F vectors, respectively (P>0.3) (Figure 4a), originating from SCID repopulating cells containing comparable vector copies (average vector copy number: ΔΦN.HG.F: 2.6; ΔΦN.GH.F: 2). Both promoter orientations were active in the engrafted human cell population, whereas the total EGFP and ΔLNGFR MFI comparison reproduced the pattern observed for human HSCs in vitro. Specifically, the ΔΦN.GH.F vector produced higher EGFP MFI compared with the ΔΦN.HG.F vector (Figure 4b), although comparable levels of ΔLNGFR were seen with either vector (P>0.5) (Figure 4c). In addition, both vectors and hence promoter orientations exhibited nonstatistically significant different numbers of EGFP-positive cells in lymphoid (CD19+) and myeloid (CD33+) subpopulations (Figure 4d) strongly indicating the ability of the bidirectional promoter to function across different hematopoietic cell lineages. Finally, we tested our vector for the possibility of silencing; we analyzed colonies (both myeloid and erythroid) of human SCID repopulating cell collected at 8 weeks after BMT for the presence of the provirus and for EGFP mRNA. From a total of 50 individually picked colonies, 14 were tested positive for FV sequences, and in 8 of them (57%), we could detect GFP mRNA (data not shown). These data indicate that our vector was not subjected to strong silencing pressure.
Figure 4.
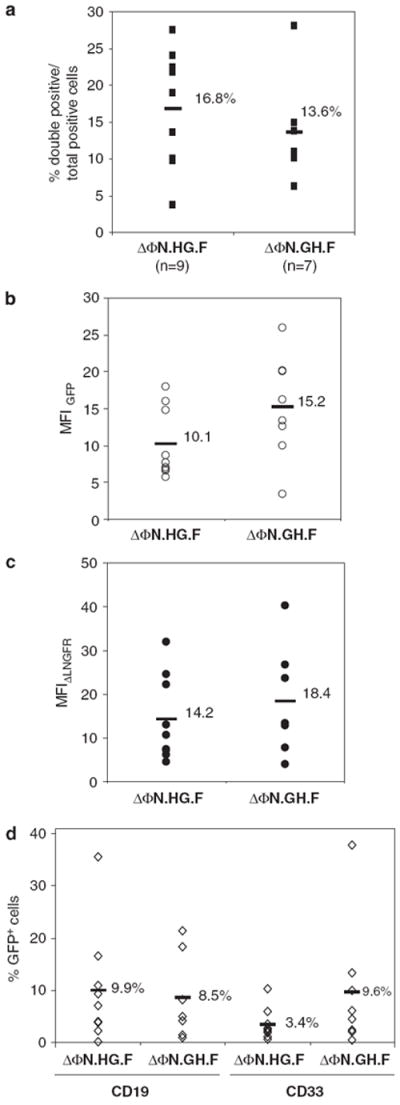
Transgene expression by the bidirectional promoter in human hematopoietic cells in vivo. Human cord blood (CB) CD34+ transduced with ΔΦN.HG.F or ΔΦN.GH.F foamy viral vectors were transplanted in busulfan-conditioned NOD/severe combined immunodeficient (SCID) animals and their bone marrow was analyzed 2 months after transplantation by flow cytometry. (a) The percentage of human CD45+ cells coexpressing enhanced green fluorescent protein (EGFP) and low-affinity nerve growth factor receptor (ΔLNGFR) in each mouse over the percentage of total positive cells is shown. n indicates the number of individual animals analyzed for each vector. (b) The mean fluorescence intensities (MFIs) for ΔLNGFR and EGFP (c) of the human CD45+ bone marrow cells is plotted for every individual animal. (d) Total percentage EGFP-positive cells in B (CD19+) and myeloid (CD33+) lineages for each mouse. Horizontal bars indicate the mean value in each column.
The bidirectional promoter retains its in situ transcriptional activity in the foamy vector
Overall, our gene transfer results with the bidirectional promoter from diverse cellular backgrounds indicate that the side of the promoter that drives transcription of HNRNPH2 gene in the genome is stronger. To investigate whether the dual promoter has retained its in situ activity when introduced randomly in the genome with an FV vector, we examined the mRNA expression levels of HNRNPH2 and GLA genes in the same cell lines used for gene transfer using a real-time PCR assay. As seen in Figure 5, a <1 relative expression ratio of GLA over HNRNPH2 was seen in the majority of the cells tested, indicating that these cells expressed higher mRNA levels of HNRNPH2 compared with GLA. Similar results were also obtained for human CB CD34+, implying that the transcriptional activity of the dual promoter in its genomic locus is also biased towards the HNRNPH2 gene. These results are in accordance with our EGFP transgene expression data, denoting that the transcriptional activity of the dual promoter in situ is reproduced when the latter was transferred into a cell in the context of an FV vector.
Figure 5.
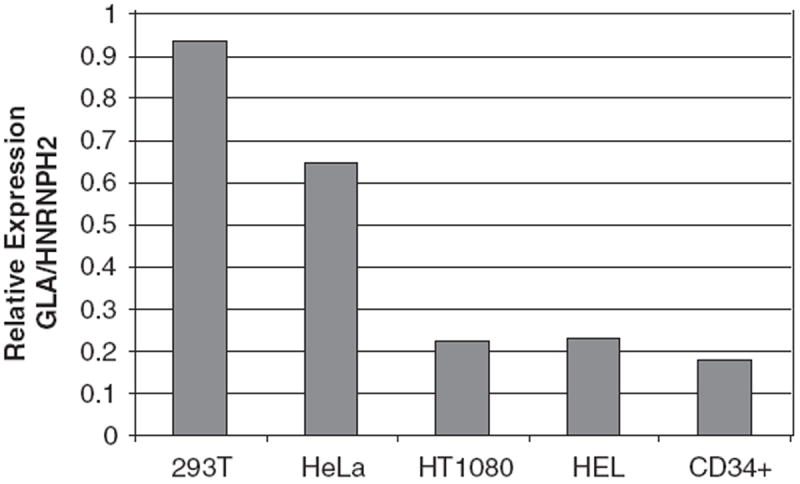
Endogenous expression of the alpha-galactosidase (GLA) and heterogeneous nuclear ribonucleoprotein H2 (HNRNPH2) genes. Relative expression of GLA and HNRNPH2 genes in human cord blood (CB) CD34+ and cell lines by quantitative Taqman PCR standardized for endogenous GAPDH. The 2-ΔΔCt values are plotted from one out of two representative experiments.
Discussion
There are ~ 1500 divergently transcribed gene pairs in the genome indicating the existence of numerous putative bidirectional promoters.25,26 Promoters capable of simultaneous transcription by RNA polymerases II and III have also been identified, which could be used for transgene and short hairpin RNA expression by a single vector.27 The widespread occurrence of such promoters probably reflects the requirement for tightly regulated gene expression. Such endogenous promoters should follow tissue-specific expression pattern and should offer a useful tool for selecting endogenous putative dual promoters with a particular tissue or cell specificity.
In this report, we describe a novel retroviral vector based on a deleted FV backbone that has the ability to express two cDNAs in a coordinated manner. This was accomplished with the use of an endogenous human promoter that is located on chromosome X, in the intragenic region between the GLA and the HNRNPH2 genes. We initially confirmed its bidirectional activity in a FV vector with a single reporter (EGFP) by cloning the promoter in either orientation. Both orientations of the promoter proved functional in various cell types transduced at low MOIs, with a propensity for superior efficiency in epithelial cells, denoting nonetheless its potential use in gene transfer applications. The promoter retained its activity regardless of its orientation in the FV vector. In a variety of cell lines, including mouse HSCs, the EGFP levels obtained with the bidirectional promoter as determined by MFI reached 20–70% of the MFI values seen with a standard PGK promoter.
We then assayed the ability of the promoter to drive the simultaneous expression of a pair of reporter genes cloned on either side, as similar endogenous promoters located between divergently transcribed genes often express the two genes in a discordant manner.21 We cloned the promoter in a FV vector carrying the EGFP and the ΔLNGFR reporters and we calculated the number of double positive over the total number of transgene-positive cells. Our results showed that the promoter retained its intrinsic bidirectional activity in most of the lines tested and the expression of the two transgenes was coordinately regulated. The dual promoter also proved functional, although slightly less efficient, in human HSCs with 35% of total positive cells coexpressing both transgenes when transduced with the ΔΦN.HG.F vector. Both sides of the promoter were active in SCID repopulating cells, with an average of 16% total positive human cells coexpressing EGFP and ΔLNGFR. Interestingly, the efficiency of the vector in the different hematopoietic lineages in our xenotransplantation model proved comparable with the results seen in mouse HSCs with a recently reported ubiquitously acting chromatin-opening element (UCOE) element.18 It is noteworthy that these levels were achieved with low vector copy numbers (~2), minimizing the risk for insertional mutagenesis. Importantly, stable expression of the two transgenes was observed after hematopoietic cell differentiation in vitro and in vivo, indicating not only that the promoter was not being silenced but also that divergent transcription remained unaltered through this process.
Comparison of promoter activity strength between the two vectors (ΔΦN.GH.F and ΔΦN.HG.F) was evaluated by scoring for total EGFP MFI; the promoter produced higher detectable levels of EGFP in the ΔΦN.GH.F vector, indicating that the direction that drives the expression of the HNRNPH2 gene in the genome was relatively stronger. Particularly, in HT1080, BHK and HEL cells, the effect was more pronounced and gave statistically significant deviations. This effect, however, was clearly cell line-dependent and not due to copy number variations. The enhanced activity of the HNRNPH2 side of the dual promoter was also observed in human HSCs and SCID repopulating cells. The fact that no such increase was observed when ΔLNGFR expression was evaluated (in the ΔΦN.HG.F vector) most likely reflects the nature of the latter protein and possible limitations of its detection method. However, when we tested for mRNA levels of the endogenous linked genes, we also observed higher levels of HNRNPH2 RNA, indicating that our vector-transferred promoter recapitulated its endogenous operational mode. This is an important feature of the vector as one can correctly predict the activity of the dual promoter by measuring the respective GLA and HNRNPH2 levels in the cells of choice.
Viral vector-mediated gene transfer into hematopoietic stem cells has proved a promising tool for the treatment of life-threatening hematological genetic disorders. However, following adverse reactions from gene therapy trials,28,29 the problem of insertional mutagenesis in gene transfer applications has become a priority. Dual expression vectors offer the possibility of combined gene expression from a single provirus, thus cutting the risk of provirus-mediated transformation. FV vectors are attractive vehicles for gene transfer as they can efficiently transduce quiescent cells, including SCID repopulating cells, with minor transgene silencing30-32 and read-through transcription.33
So far, few studies have used endogenous promoters for gene transfer applications probably due to their reduced strength compared with viral ones. Ongoing evidence, however, suggests that low transgene expression may be sufficient for particular applications of gene therapy.34,35 Synthetic bidirectional promoters are attractive and probably stronger alternatives to endogenous ones, but promoter interference may impair the expression of adjacent transcription units placed in the same vector.12 Endogenous promoters on the other hand have evolved with a built-in capacity for simultaneous expression of the linked genes and this was illustrated with the dual promoter used in this study. Apart from circumstances in which really high levels of protein expression are required, our dual promoter should offer an attractive tool for coexpression of two transgenes. In addition, its small size offers an advantage considering the size limitations posed by retroviral vector packaging.
Materials and methods
FV vector construction
The putative promoter sequence was amplified from genomic DNA using primers specific for the intragenic region between the GLA and HNRNPH2 genes and cloned into an intermediate vector (pA2AGloxPpA, DW Russell unpublished data). The plasmid was sequence-verified and used for amplifying the bidirectional promoter with the following primer sets: (1) forward 5′-CGACCGGTGGTAATTTTCCTCC-3′ and reverse 5′-C CATCGATTGTCACGGTGACCG-3′ fitted with AgeI and ClaI sites, respectively, to allow directional cloning in sense to the GLA gene transcription; and (2) forward 5′-C CATCGATGGTAATTTTCCTCC-3′ and reverse 5′-CG ACCGGTTGTCACGGTGACCG-3′ fitted with ClaI and AgeI sites, respectively, to allow directional cloning antisense to the GLA gene transcription. PCR conditions were 94 °C for 4 min, followed by four cycles at 94 °C for 1 min, 53 °C for 1 min, 72 °C for 1 min, and 30 cycles at 94 °C for 1 min, 60 °C for 1 min, 72 °C for 1 min. The product was purified, restriction enzyme-digested, gel purified, and cloned into promoter-less FV vector backbone36 carrying the EGFP reporter gene downstream. The resulting constructs pΔΦ-GH-F and pΔΦ-HG-F (with GH denoting the orientation towards the HNRPH2 gene and HG denoting the orientation towards the GLA gene) were sequenced to identify the intact clones. Clones with either promoter orientation were used for FV vector production. To make the dual reporter vector, we first inserted a rabbit β-globin polyA sequence37 upstream of the FV multiple cloning site and in reverse orientation to the FV transcription unit and then cloned the bidirectional promoter in either genomic orientation. We then cloned the deleted LNGFR gene from plasmid pBBSNF2′hmp (a kind gift from R Richards, University of Washington) and generated the final vectors pΔΦN.GH.F and pΔΦN.HG.F with N indicating ΔLNGFR and F the EGFP reporters. The IRES2GFP vector used for the comparison of the new promoter to the IRES2 element was a bicistronic ΔϕPGKgp91pHoxIRES2GFP vector. The results are from more than three independent transduction experiments at low MOIs (1–2) in HeLa and HT1080 cells.
FV vector production and cell line transduction
Foamy virus vector stocks were made by calcium phosphate transfection of vectors and helper plasmids in 293T cells plated on 10-cm dishes as previously described.38 The cells were treated overnight with 0.5 M sodium butyrate (Sigma Aldrich, Athens, Greece) and replenished with fresh medium the following day. The viral supernatant was concentrated by high-speed centrifugation (47 800 g for 4 h) and either frozen down in 5% dimethyl sulfoxide or used directly for transduction. Vector stock titers were assayed on HeLa cells and titer was expressed as EGFP-transducing units per ml of supernatant after fluorescent-activated cell sorting analysis for EGFP expression. All cell lines were grown in 5% CO2, 100% humidified atmosphere using Dulbecco’s modified Eagle’s medium-glutamine medium supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotics (penicilin–streptomycin) (all from Invitrogen, SanDiego, CA, USA). Cell line transduction was carried out in 6-well plates at MOIs 1–4.
Ex vivo human HSC transduction and mouse transplantation
Human CB CD34+ cells were isolated from total mononuclear cells after gradient centrifugation on Histopaque (Sigma) and hypotonic lysis of red blood cells. Positive selection was carried out using human CD34+-positive selection cocktail (Miltenyi Biotech, Cologne, Germany) according to the manufacturer’s recommendations. Transduction was carried out at MOI 20 for 18–20 h on retronectin-coated plates (TaKaRa Bio, Shiga, Japan) in StemSpan serum-free medium (Stem Cell Technologies, Vancouver, BC, Canada), supplemented with human stem cell factor FLT-3 ligand and human thrombopoietin at 100 ng ml−1 (all from Peprotech, London, UK). For liquid culture, CD34+ cells were expanded for 5–7 days in the presence of the above cytokines. For clonal analysis, transduced CD34+ cells were plated in human Methocult H4434 methylcellulose medium (Stem Cell Technologies) as previously described.39
The NOD/SCID animals were originally a kind gift from Dr Bonnet (Cancer Research UK, London). They were maintained at our institute’s animal facility in specific pathogen-free conditions. At 48 and 24 h before transplantation, 8–12-week-old animals were conditioned with two intraperitoneal doses of busulfan (Sigma) or Busilvex at 25 mg kg−1. Mice were intravenously transplanted with 0.5–1.7 × 105 transduced human CD34+ cells into the tail vein.
Transgene expression analysis by flow cytometry
The transduction efficiency of cell lines was determined by flow cytometry for the expression of EGFP and ΔLNGFR using an anti-NGFR antibody (BD Pharmingen, Athens, Greece). Cell viability was determined using 7AAD (Sigma). We calculated the percentage of cells coexpressing EGFP and ΔLNGFR using the following equation:
Bone marrow engraftment of animals was analyzed 8–10 weeks after transplantation by flow cytometry. The presence of human cells was detected using a mouse anti-human CD45 antibody, and transgene expression in mature hematopoietic lineages was assessed by staining with anti-human CD33–PE or anti-human CD19–PE (all antibodies from BD Pharmingen). Mouse isotype controls (BD Pharmingen) were also used. Dead cell exclusion was carried out using propidium iodide (Sigma) and data were acquired on BD FACScan (Beckton Dickinson) or on a Beckman Coulter Cytomics FC500 flow cytometer (Beckman Coulter, Fullerton, CA, USA). Fluorescent-activated cell sorting analysis was carried out using CXP analysis software.
Taqman PCR analysis
The vector copy number in the transduced cell lines, human CD34+ cells and bone marrow samples was analyzed by Quantitative TaqMan PCR performed on an ABI Prism 7000 detector (Applied Biosystems, Foster City, CA USA). Amplification of the foamy provirus was carried out using SYBR Green master mix (Applied Biosystems or Stratagene, Amsterdam Zuidoost, The Netherlands) and primers 5′-CTGGAATGTTACTCAAA GAGCTGTTT-3′ and 5′-TGGAACAGGATGCTGCATT CT-3′. The amount of human and mouse genomic DNA loaded per reaction was adjusted using a primer/probe (VIC dye) for RNase P and primer/probe for rodent GAPDH (VIC dye), respectively (all from Applied Biosystems). Samples were analyzed in duplicate 25-μl reactions. Standards on the basis of different amounts of pDWE37 plasmid (range 2.8–2.8E+05 vector copies) or genomic DNA (0.6–625 ng) of mouse or human origin were also analyzed and their threshold cycle (Ct) values were plotted in standard curves. These were then used to extrapolate the absolute vector copy number and number of cells per reaction, respectively. The FV vector copy number detection limit was ~28 copies per reaction, although the lower limit of RNase P or rodent GAPDH detection was in the region of 0.6 ng of human or mouse DNA per reaction.
Analysis of HNRNPH2 and GLA gene expression was performed on cDNA reverse transcribed from total RNA using MMLV RT (Promega, Manheim, Germany) according to the manufacturer’s standard protocol. Taqman PCR amplification was carried out using SYBR Green master mix as above and primers HNRNPH2-F: 5′-GGG CTTATCCAACCAGTC-3′, HNRNPH2-R: 5′-TTAGTGC TCCTTCTCTACC-3′, GLA-F: 5′-AGAGATTCAGAAGG CAGAC-3′ and GLA-R: 5′-GCATCAATGTCGTAGTAT CC-3′. The relative expression of GLA over HNRNPH2 expression was calculated using the 2-ΔΔCt method, after correction for the amount of template using GAPDH (forward: 5′-GGGAAGCTTGTCATCAATGG-3′ and reverse: 5′-CATCGCCCCACTTGATTTTG-3′).
Statistical analysis
Data were subjected to one-way analysis of variance using SPSS software to assess the significance of differences between means. Individual values from three or more independent experiments were analyzed per cell type.
Acknowledgments
This work was supported in part by the European IP CONSERT (FP6, 005242) and an ENTER Grant to ES (GSRT, Number 04ER022).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Sawai N, Zhou S, Vanin EF, Houghton P, Brent TP, Sorrentino BP. Protection and in vivo selection of hematopoietic stem cells using temozolomide, O6-benzylguanine, and an alkyltransferase-expressing retroviral vector. Mol Ther. 2001;3:78–87. doi: 10.1006/mthe.2000.0223. [DOI] [PubMed] [Google Scholar]
- 2.Gerull S, Beard BC, Peterson LJ, Neff T, Kiem HP. In vivo selection and chemoprotection after drug resistance gene therapy in a nonmyeloablative allogeneic transplantation setting in dogs. Hum Gene Ther. 2007;18:451–456. doi: 10.1089/hum.2006.039. [DOI] [PubMed] [Google Scholar]
- 3.Chinnasamy D, Milsom MD, Shaffer J, Neuenfeldt J, Shaaban AF, Margison GP, et al. Multicistronic lentiviral vectors containing the FMDV 2A cleavage factor demonstrate robust expression of encoded genes at limiting MOI. Virol J. 2006;3:14. doi: 10.1186/1743-422X-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai S, Ernstberger A, Wang H, Bailey BJ, Hartwell JR, Sinn AL, et al. In vivo selection of hematopoietic stem cells transduced at a low multiplicity-of-infection with a foamy viral MGMT(P140K) vector. Exp Hematol. 2008;36:283–292. doi: 10.1016/j.exphem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, et al. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 6.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 7.Hennecke M, Kwissa M, Metzger K, Oumard A, Kroger A, Schirmbeck R, et al. Composition and arrangement of genes define the strength of IRES-driven translation in bicistronic mRNAs. Nucleic Acids Res. 2001;29:3327–3334. doi: 10.1093/nar/29.16.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozak M. Alternative ways to think about mRNA sequences and proteins that appear to promote internal initiation of translation. Gene. 2003;318:1–23. doi: 10.1016/s0378-1119(03)00774-1. [DOI] [PubMed] [Google Scholar]
- 9.Emerman M, Temin HM. Genes with promoters in retrovirus vectors can be independently suppressed by an epigenetic mechanism. Cell. 1984;39:449–467. [PubMed] [Google Scholar]
- 10.Eszterhas SK, Bouhassira EE, Martin DI, Fiering S. Transcriptional interference by independently regulated genes occurs in any relative arrangement of the genes and is influenced by chromosomal integration position. Mol Cell Biol. 2002;22:469–479. doi: 10.1128/MCB.22.2.469-479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X, Zhan X, D’Costa J, Tanavde VM, Ye Z, Peng T, et al. Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol Ther. 2003;7:827–838. doi: 10.1016/s1525-0016(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 12.Curtin JA, Dane AP, Swanson A, Alexander IE, Ginn SL. Bidirectional promoter interference between two widely used internal heterologous promoters in a late-generation lentiviral construct. Gene Therapy. 2008;15:384–390. doi: 10.1038/sj.gt.3303105. [DOI] [PubMed] [Google Scholar]
- 13.Ryan MD, Drew J. Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 1994;13:928–933. doi: 10.1002/j.1460-2075.1994.tb06337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furler S, Paterna JC, Weibel M, Bueler H. Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Therapy. 2001;8:864–873. doi: 10.1038/sj.gt.3301469. [DOI] [PubMed] [Google Scholar]
- 15.de Felipe P. Skipping the co-expression problem: the new 2A ‘CHYSEL’ technology. Genet Vaccines Ther. 2004;2:13. doi: 10.1186/1479-0556-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD. E unum pluribus: multiple proteins from a self-processing polyprotein. Trends Biotechnol. 2006;24:68–75. doi: 10.1016/j.tibtech.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Amendola M, Venneri MA, Biffi A, Vigna E, Naldini L. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol. 2005;23:108–116. doi: 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Thornhill SI, Howe SJ, Ulaganathan M, Schambach A, Sinclair J, et al. Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood. 2007;110:1448–1457. doi: 10.1182/blood-2006-12-060814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adachi N, Lieber MR. Bidirectional gene organization: a common architectural feature of the human genome. Cell. 2002;109:807–809. doi: 10.1016/s0092-8674(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 20.Takai D, Jones PA. Origins of bidirectional promoters: computational analyses of intergenic distance in the human genome. Mol Biol Evol. 2004;21:463–467. doi: 10.1093/molbev/msh040. [DOI] [PubMed] [Google Scholar]
- 21.Trinklein ND, Aldred SF, Hartman SJ, Schroeder DI, Otillar RP, Myers RM. An abundance of bidirectional promoters in the human genome. Genome Res. 2004;14:62–66. doi: 10.1101/gr.1982804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guarguaglini G, Battistoni A, Pittoggi C, Di Matteo G, Di Fiore B, Lavia P. Expression of the murine RanBP1 and Htf9-c genes is regulated from a shared bidirectional promoter during cell cycle progression. Biochem J. 1997;325(Part 1):277–286. doi: 10.1042/bj3250277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn J, Gruen JR. The genomic organization of the histone clusters on human 6p213. Mamm Genome. 1999;10:768–770. doi: 10.1007/s003359901089. [DOI] [PubMed] [Google Scholar]
- 24.Hansen JJ, Bross P, Westergaard M, Nielsen MN, Eiberg H, Borglum AD, et al. Genomic structure of the human mitochondrial chaperonin genes: HSP60 and HSP10 are localised head to head on chromosome 2 separated by a bidirectional promoter. Hum Genet. 2003;112:71–77. doi: 10.1007/s00439-002-0837-9. [DOI] [PubMed] [Google Scholar]
- 25.Greene WK, Sontani Y, Sharp MA, Dunn DS, Kees UR, Bellgard MI. A promoter with bidirectional activity is located between TLX1/HOX11 and a divergently transcribed novel human gene. Gene. 2007;391:223–232. doi: 10.1016/j.gene.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 26.Yang MQ, Elnitski LL. Prediction-based approaches to characterize bidirectional promoters in the mammalian genome. BMC Genomics. 2008;9(Suppl 1):S2. doi: 10.1186/1471-2164-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hung CF, Cheng TL, Wu RH, Teng CF, Chang WT. A novel bidirectional expression system for simultaneous expression of both the protein-coding genes and short hairpin RNAs in mammalian cells. Biochem Biophys Res Commun. 2006;339:1035–1042. doi: 10.1016/j.bbrc.2005.11.113. [DOI] [PubMed] [Google Scholar]
- 28.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 29.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Josephson NC, Vassilopoulos G, Trobridge GD, Priestley GV, Wood BL, Papayannopoulou T, et al. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc Natl Acad Sci USA. 2002;99:8295–8300. doi: 10.1073/pnas.122131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vassilopoulos G, Trobridge G, Josephson NC, Russell DW. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood. 2001;98:604–609. doi: 10.1182/blood.v98.3.604. [DOI] [PubMed] [Google Scholar]
- 32.Trobridge G, Russell DW. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J Virol. 2004;78:2327–2335. doi: 10.1128/JVI.78.5.2327-2335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendrie PC, Huo Y, Stolitenko RB, Russell DW. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol Ther. 2008;16:534–540. doi: 10.1038/sj.mt.6300398. [DOI] [PubMed] [Google Scholar]
- 34.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 35.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 36.Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R, et al. Foamy virus vector integration sites in normal human cells. Proc Natl Acad Sci USA. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gil A, Proudfoot NJ. Position-dependent sequence elements downstream of AAUAAA are required for efficient rabbit betaglobin mRNA 3′ end formation. Cell. 1987;49:399–406. doi: 10.1016/0092-8674(87)90292-3. [DOI] [PubMed] [Google Scholar]
- 38.Vassilopoulos G, Josephson NC, Trobridge G. Development of foamy virus vectors. Methods Mol Med. 2003;76:545–564. doi: 10.1385/1-59259-304-6:545. [DOI] [PubMed] [Google Scholar]
- 39.Siapati EK, Bigger BW, Miskin J, Chipchase D, Parsley KL, Mitrophanous K, et al. Comparison of HIV- and EIAV-based vectors on their efficiency in transducing murine and human hematopoietic repopulating cells. Mol Ther. 2005;12:537–546. doi: 10.1016/j.ymthe.2005.01.022. [DOI] [PubMed] [Google Scholar]