

Abstract
We have previously demonstrated that a stable synthetic analog of 20-HETE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine (5,14-HEDGE), restores vascular reactivity, blood pressure, and heart rate in endotoxemic rats. The aim of this study was to determine whether decreased renal expression and activity of soluble epoxide hydrolase (sEH), MEK1, ERK1/2, IKKβ, IκB-α, and NF-κB as well as systemic and renal proinflammatory cytokine production associated with increased expression and activity of CYP2C23 contributes to the effect of 5,14-HEDGE to prevent hypotension, tachycardia, inflammation, and mortality in response to systemic administration of lipopolysaccharide (LPS). Blood pressure fell by 33 mmHg and heart rate rose by 57 beats/min in LPS (10 mg/kg, i.p.)-treated rats. Administration of LPS also increased mRNA and protein expression of sEH associated with a decrease in CYP2C23 mRNA and protein expression. Increased activity of sEH and p-MEK1, p-ERK1/2, p-IκB-α, NF-κB, and p-NF-κB protein levels as well as TNF-α and IL-8 production by LPS were also associated with a decreased activity of AA epoxygenases. These effects of LPS were prevented by 5,14-HEDGE (30 mg/kg, s.c.; 1 h after LPS). Treatment of endotoxemic mice with 5,14-HEDGE also raised the survival rate of animals from 84% to 98%. A competitive antagonist of vasoconstrictor effects of 20-HETE, 20-hydroxyeicosa-6(Z),15(Z)-dienoic acid, 20-HEDE (30 mg/kg, s.c.; 1 h after LPS) prevented the effects of 5,14-HEDGE on blood pressure, heart rate, expression and/or activity of sEH, CYP2C23, and ERK1/2 as well as TNF-α and IL-8 levels in rats treated with LPS. These results suggest that decreased expression and/or activity of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production associated with increased CYP2C23 expression and antiinflammatory mediator formation participate in the protective effect of 5,14-HEDGE against hypotension, tachycardia, inflammation, and mortality in the rodent model of septic shock.
Keywords: Endotoxin, Soluble epoxide hydrolase, CYP2C23, NF-κB, Hypotension, Inflammation, Mortality
1. Introduction
20-hydroxyeicosatetraenoic acid (20-HETE) is an ω-hydroxylation product of arachidonic acid (AA) that is produced by cytochrome P450 (CYP) enzymes, mainly by the CYP4A and CYP4F isoforms in the kidney, heart, liver, brain, lung, and vasculature [1–4]. In the vasculature, 20-HETE causes vasoconstriction in several vascular beds, including renal, cerebral, aortic, mesenteric, and coronary arteries [5–9]. Activation of protein kinases, such as mitogen-activated protein kinase (MAPK), MAPK kinase (MEK), and extracellular signal-regulated kinase (ERK) which contribute to the regulation of vascular tone, has been shown to mediate to the vasoconstrictor effect of 20-HETE [10–13]. As opposed to its vasoconstrictor effect, 20-HETE has also been reported to produce vasodilation in the vasculature including renal and coronary arteries [13–15]. These vasodilatory responses of 20-HETE have been attributed to nitric oxide (NO) release [16], conversion of 20-HETE to 20-OH-PGE2 and 20-OH-PGF2α by cyclooxygenase (COX) [7,13,17], and increased formation of PGE2 [17] and PGI2 [13–15,17]. In addition, 20-HETE has been shown to activate NF-κB signaling and induce expression of cellular adhesion molecules and cytokines, thereby promoting inflammation [18,19]. CYP4A- and CYP4F-derived 20-HETE is also involved in LPS-induced acute systemic inflammation as a proinflammatory mediator [20,21].
Cis-epoxyeicosatrienoic acids (EETs) are synthesized predominantly by CYP epoxygenases of the CYP2 family, including the 2C and 2J classes. In addition, some other mammalian CYP isoforms have been reported to generate EETs (e.g., CYP1A, CYP2B, and CYP2D) [22]. The primary epoxidation products are four regioisomers: 5,6-, 8,9-, 11,12-, and 14,15-EET. Although each enzyme is able to convert AA to all four EET regioisomers, 11,12- and 14,15-EET have been reported as the main products of these epoxygenases [23]. EETs can be further metabolized by several pathways including hydration by soluble epoxide hydrolase (sEH) [24,25], esterification to glycerophospholipids [26,27], and β-oxidation by CYPs and COXs [28–31]. EETs are rapidly hydrated in vivo by epoxide hydrolases, primarily sEH in the cytosol, to their more stable and less active corresponding diols, dihydroxyeicosatrienoic acids (DHETs) [24,25]. The three major CYP2C isoforms expressed in the rat kidney are the CYP2C24, CYP2C11, and CYP2C23, the latter being the most specific isoform to renal microvessels [32,33]. CYP2C23 has also been shown to be a major isoform of CYP epoxygenase responsible for 11,12-EET formation in the kidney [32,34]. EETs have been implicated in various biological processes, including vascular tone and blood pressure [35,36]. In addition, proinflammatory mediators like cytokines and lipopolysaccharide (LPS), also known as endotoxin, which is the most potent microbial mediator in the pathogenesis of septic shock [1], decrease endothelial epoxygenase enzyme expression and formation of EETs [37]. Therefore, in contrast to 20-HETE, EETs have potent antiinflammatory properties by attenuating cytokine-induced nuclear factor-κB (NF-κB) activation and leukocyte adhesion to the vascular wall [38]. However, to the best of our knowledge, there has been no previous attempt to examine contribution of EETs to changes in hemodynamic variables and mortality seen in septic shock.
Because of the divergent effects of the CYP epoxygenase and hydroxylase pathways in the regulation of vascular tone and inflammation, changes in the functional balance between these parallel pathways might contribute to the pathogenesis and progression of inflammatory disease, such as sepsis and septic shock. Our previous studies with the use of a stable synthetic analog of 20-HETE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine, 5,14-HEDGE, which mimics the effects of endogenously produced 20-HETE, and a competitive antagonist of vasoconstrictor effects of 20-HETE, 20-hydroxyeicosa-6(Z),15(Z)-dienoic acid, 20-HEDE, in cardiovascular and renal tissues of rats suggested that decreased formation of 20-HETE and EETs contributes to vascular hyporeactivity, hypotension, tachycardia, inflammation, and decreased survival in a rodent model of septic shock [39–42]. Therefore, the present study was conducted to determine whether decreased renal expression and activity of sEH, MEK1, ERK1/2, IκB kinase (IKK) β, inhibitor of κB (IκB)-α, and NF-κB as well as systemic and renal proinflammatory cytokine production associated with increased CYP2C23 expression and activity contribute to the protective effect of 5,14-HEDGE against hypotension, tachycardia, and inflammation in LPS-treated rats and mortality in endotoxemic mice. The results of this study have been presented in abstract form [41,42].
2. Materials and methods
2.1. Endotoxic shock model
Experiments were performed on Wistar rats (male; 200–330 g; n = 60) and Balb/c mice (male and female; 20–40 g; n = 210) (Research Center of Experimental Animals, Mersin University, Mersin, Turkey) fed a standard chow. They were synchronized by maintenance of controlled environmental conditions throughout the experiments. The circadian rhythmicity of the animals was entrained by a standardized 12 h light and 12 h dark cycle. All experiments were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Ethics Committee of Mersin University School of Medicine. Endotoxic shock was induced in rats and mice as previously described by Tunctan et al. [43,44]. Rats were randomly divided into saline (n = 10), LPS (n = 10), 5,14-HEDGE (n = 10), LPS + 5,14-HEDGE (n = 10), 20-HEDE (n = 10), and LPS + 5,14-HEDGE + 20-HEDE (n = 10) groups. In the mortality studies, mice were randomly divided into saline (n = 20), LPS (n = 50), 5,14-HEDGE (n = 20), LPS + 5,14-HEDGE (n = 50), 20-HEDE (n = 20), and LPS + 5,14-HEDGE + 20-HEDE (n = 50) groups. In the saline, 5,14-HEDGE, and 20-HEDE groups, animals received saline (4 ml/kg, i.p.) at time 0. Animals in the LPS, LPS + 5,14-HEDGE, and LPS + 5,14-HEDGE + 20-HEDE groups were treated with LPS (Escherichia coli LPS, O111:B4; Sigma Chemical Co., St. Louis, MO, USA) (10 mg/kg, i.p.; sublethal dose) at time 0. In the 5,14-HEDGE, LPS + 5,14-HEDGE, and LPS + 5,14-HEDGE + 20-HEDE groups, animals were treated with a stable synthetic analog of 20-HETE, 5,14-HEDGE (30 mg/kg, s.c.) [39,40] and/or a competitive antagonist of vasoconstrictor effects of 20-HETE, 20-HEDE (30 mg/kg, s.c.) [39,40] 1 h after injection of saline or LPS, respectively. 5,14-HEDGE and 20-HEDE were synthesized in the Department of Biochemistry University of Texas Southwestern Medical Center, Dallas, Texas, US. Mean arterial pressure (MAP) and heart rate (HR) of the rats were measured using a tail-cuff device (MAY 9610 Indirect Blood Pressure Recorder System, Commat Ltd., Ankara, Turkey) during a control period at time 0 and 1, 2, 3, and 4 h. All rats survived in the experiments. In the mortality studies, survival rate was recorded every 6 h for 3 days after the administration of saline or LPS to mice. Rats were euthanized 4 h after the administration of saline or LPS, and blood samples and kidneys were collected from all animals. Detailed method about preparation of serum and tissue samples is reported in the supplementary material.
2.2. Messenger ribonucleic acid (mRNA) isolation and reverse transcription-polymerase chain reaction (RT-PCR)
Complementary deoxyribonucleic acids (cDNAs) for sEH, CYP2C23, and β-actin were synthesized followed by mRNAs isolation from the frozen tissue powders as given in detail in the supplementary material.
2.3. Immunoblotting
Immunoblotting for sEH, CYP2C23, MEK1, ERK1/2, IκB-α, phosphorylated IκB-α, NF-κB, phosphorylated NF-κB, and β-actin proteins were performed according to the method reported in the supplementary material.
2.4. Measurement of MEK1 and ERK1/2 activities
Phosphorylated protein levels of MEK1 and ERK1/2 (as an index for MEK1 and ERK1/2 activity, respectively) in the tissue homogenates were measured by ELISA according to the manufacturer’s instructions in the RayBio®Phospho-MEK1 (Ser217/221) ELISA Kit (RayBiotech Inc., Norcross, GA, USA) and ERK1/2 (pTpY185/187) ELISA Kit (Invitrogen Co., Camarillo, CA, USA), respectively.
2.5. Measurement of activities of AA epoxygenases and sEH
Activities of AA epoxygenasesas (an index for CYP2C23 activity) and sEH were measured in the renal microsomal and cytosolic fractions, respectively, according to the method reported in the supplementary material.
2.6. Measurement of tumor necrosis factor (TNF)-α and interleukin (IL)-8 levels
TNF-α and IL-8 levels in the sera and tissue samples were measured by ELISA according to the manufacturer’s instructions in the Rat TNF-α Platinum ELISA Kit (Bender Medsystems, Vienna, Austria) and Rat Interleukin 8 (IL-8) ELISA Kit (Cusabio Biotech Co., Ltd., Hubei Province, P.R. China), respectively.
2.7. Statistical analysis
Data except mortality are expressed as means ± S.E.M. Data were analyzed by one-way ANOVA followed by Student–Newman–Keuls test for multiple comparisons, Kruskal–Wallis test followed by Dunns test for multiple comparisons and Student’s t or Mann-Whitney U tests when appropriate. The survival data were analyzed by Fisher’s exact test. A P value < 0.05 was considered to be statistically significant.
3. Results
3.1. Effect of 5,14-HEDGE on the cardiovascular response to LPS
LPS caused a gradual fall in MAP and an increase in HR over the 4 h course of the experiment (p < 0.05) (Table 1). The change in MAP and HR reached a maximum 4 h after the administration of LPS. MAP fell by 33 mmHg and HR rose by 57 bpm in rats treated with LPS. A synthetic analog of 20-HETE, 5,14-HEDGE, which mimics the effects of endogenously produced 20-HETE, prevented the fall in MAP and the increase in HR in rats given LPS (p < 0.05). A competitive antagonist of the vasoconstrictor effects of 20-HETE, 20-HEDE, reversed the ability of 5,14-HEDGE to oppose the effects of endotoxin on MAP and HR (p < 0.05). 5,14-HEDGE or 20-HEDE had no effect on MAP or HR in rats treated with vehicle (p > 0.05).
Table 1.
Time course of the effects of 5,14-HEDGE and 20-HEDE on MAP and HR following administration of saline (vehicle) or LPS to conscious rats.
| Time after LPS injection (h) |
|||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| Vehicle | |||||
| MAP | 124.90 ± 0.48 | 125.70 ± 0.83 | 126.70 ± 0.99 | 124.20 ± 0.68 | 126.90 ± 0.48 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| HR | 312.90 ± 6.19 | 309.60 ± 6.93 | 307.10 ± 4.35 | 312.70 ± 4.63 | 309.90 ± 7.52 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| LPS | |||||
| MAP | 125.30 ± 0.80 | 95.10 ± 0.61*,‡ | 93.80 ± 0.55*,‡ | 94.90 ± 0.38*,‡ | 94.30 ± 0.82*,‡ |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| HR | 315.50 ± 6.40 | 350.90 ± 8.74*,‡ | 359.10 ± 7.42*,‡ | 376.50 ± 8.34*,‡ | 367.20 ± 3.82*,‡ |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| 5,14-HEDGE | |||||
| MAP | 124.10 ± 0.82 | 125.70 ± 0.62 | 124.40 ± 0.83 | 124.40 ± 0.79 | 123.90 ± 0.59 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| HR | 312.90 ± 6.54 | 314.20 ± 4.79 | 314.90 ± 4.56 | 312.40 ± 4.23 | 312.70 ± 7.25 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| LPS + 5,14-HEDGE | |||||
| MAP | 125.20 ± 0.96 | 95.00 ± 0.65‡ | 125.00 ± 0.99#,+ | 123.80 ± 0.81#,+ | 124.10 ± 0.48#,+ |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| HR | 309.00 ± 5.29 | 361.20 ± 8.19‡ | 309.30 ± 8.94#,+ | 310.10 ± 8.56#+ | 315.00 ± 4.54#,+ |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| 20-HEDE | |||||
| MAP | 125.00 ± 0.83 | 126.00 ± 0.80 | 125.20 ± 0.66 | 126.80 ± 0.55 | 126.40 ± 0.82 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| HR | 314.70 ± 3.73 | 314.00 ± 6.43 | 314.20 ± 6.48 | 312.80 ± 4.96 | 308.30 ± 8.38 |
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| LPS + 5,14-HEDGE + 20-HEDE | |||||
| MAP | 125.10 ± 0.78 | 95.80 ± 0.68‡ | 126.20 ± 0.57#,+ | 95.78 ± 0.76§,‡,◆ | 95.00 ± 0.57§,‡,◆ |
| (n = 10) | (n = 10) | (n = 10) | (n = 9) | (n = 8) | |
| HR | 310.20 ± 6.75 | 347.20 ± 4.21‡ | 349.80 ± 5.00§,‡ | 363.90 ± 8.29§,‡ | 371.90 ± 9.79§,‡ |
| (n = 10) | (n = 10) | (n = 10) | (n = 9) | (n = 8) | |
5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.). Data are expressed as means ± S.E.M. of 10 animals.
Significant difference from the corresponding value seen in rats treated with saline (vehicle) (p < 0.05).
Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05).
Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
Significant difference from the time 0 h value within a group (p < 0.05).
Significant difference from the time 1 h value within a group (p < 0.05).
Significant difference from the time 2 h value within a group (p < 0.05).
3.2. Effect of 5,14-HEDGE on LPS-induced increase in sEH expression and activity
LPS increased sEH mRNA (Fig. 1A) and protein (Fig. 1B) expression as well as its activity (Fig. 1C) in the renal tissue (p < 0.05). The increase in sEH expression and activity was prevented in the tissues of rats treated with LPS and 5,14-HEDGE (p < 0.05) (Fig. 1). 20-HEDE reversed the effect of 5,14-HEDGE on sEH expression, but not its activity, in LPS-treated rats (p < 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal sEH expression and activity in vehicle-treated rats (p > 0.05).
Fig. 1.

Effects of 5,14-HEDGE and 20-HEDE on changes in renal sEH expression of (A) mRNA and (B) protein and (C) activity measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. sEH mRNA and protein expression in tissue homogenates and activity in renal cytosolic fractions were measured by RT-PCR, immunoblotting, and HPLC, respectively. Data are expressed as means ± S.E.M of 4 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
3.3. Effect of 5,14-HEDGE on LPS-induced decrease in CYP2C23 expression and activity
Expression of CYP2C23 mRNA (Fig. 2A) and protein (Fig. 2B) as well as activity of AA epoxygenases (Fig. 2C) was decreased in the renal tissue of endotoxemic rats (p < 0.05). The decrease in CYP2C23 expression and activity of AA epoxygenases in tissues of rats caused by LPS was prevented by concurrent treatment with 5,14-HEDGE (p < 0.05) (Fig. 2). 20-HEDE reversed the effect of 5,14-HEDGE on activity of AA epoxygenases, but not CYP2C23 expression, in LPS-treated rats (p < 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal CYP2C23 expression and activity of AA epoxygenases in vehicle-treated rats (p > 0.05).
Fig. 2.

Effects of 5,14-HEDGE and 20-HEDE on changes in renal CYP2C23 expression of (A) mRNA and (B) protein and (C) activity of AA epoxygenases measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. CYP2C23 mRNA and protein expression in tissue homogenates and activity in renal microsomal fractions were measured by RT-PCR, immunoblotting, and HPLC, respectively. Data are expressed as means ± S.E.M of 4 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
3.4. Effect of 5,14-HEDGE on MEK1 expression and activity in endotoxemic rats
To determine the effect of 5,14-HEDGE on LPS-induced changes in MEK1 expression and activity, unphosphorylated MEK1 protein expression and phosphorylated MEK1 protein levels were measured in the renal tissue of endotoxemic rats. Although LPS did not alter the unphosphorylated protein expression of MEK1 (p > 0.05), phosphorylated MEK1 protein levels were increased (p < 0.05) (Fig. 3). The increase in MEK1 phosphorylation in the tissues of rats caused by LPS was prevented by 5,14-HEDGE treatment (p < 0.05). 20-HEDE did not reverse the effect of 5,14-HEDGE on MEK1 activity in LPS-treated rats (p > 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal unphosphorylated and phosphorylated MEK1 protein levels in vehicle-treated rats (p > 0.05).
Fig. 3.
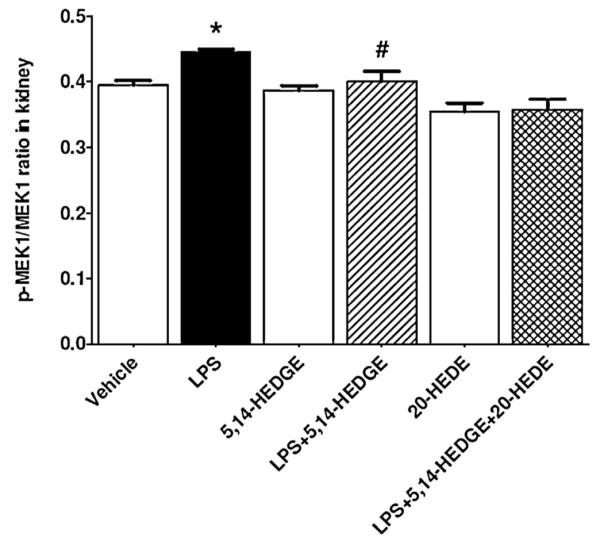
Effects of 5,14-HEDGE and 20-HEDE on changes in renal MEK1 activity measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. MEK1 protein expression and p-MEK1 (Ser217/221) protein levels in tissue homogenates were measured by immunoblotting and ELISA Kit, respectively. MEK1 activity was calculated as the ratio of p-MEK1 (Ser217/221)/MEK1 protein levels. Data are expressed as means ± S.E.M of 4–7 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
3.5. Effect of 5,14-HEDGE on LPS-induced increase in ERK1/2 expression and activity in endotoxemic rats
To examine the effect of 5,14-HEDGE on LPS-induced changes in ERK1/2 expression and activity, unphosphorylated ERK1/2 protein expression and phosphorylated ERK1/2 protein levels were measured in the renal tissue of endotoxemic rats. Although LPS did not alter the unphosphorylated protein expression of ERK1/2 (p > 0.05), phosphorylated ERK1/2 protein levels were increased (p < 0.05) (Fig. 4). The increase in ERK1/2 phosphorylation was prevented in the tissues of rats treated with 5,14-HEDGE (p < 0.05). 20-HEDE reversed the effect of 5,14-HEDGE on ERK1/2 activity in LPS-treated rats (p < 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal unphosphorylated and phosphorylated ERK1/2 protein levels in vehicle-treated rats (p > 0.05).
Fig. 4.
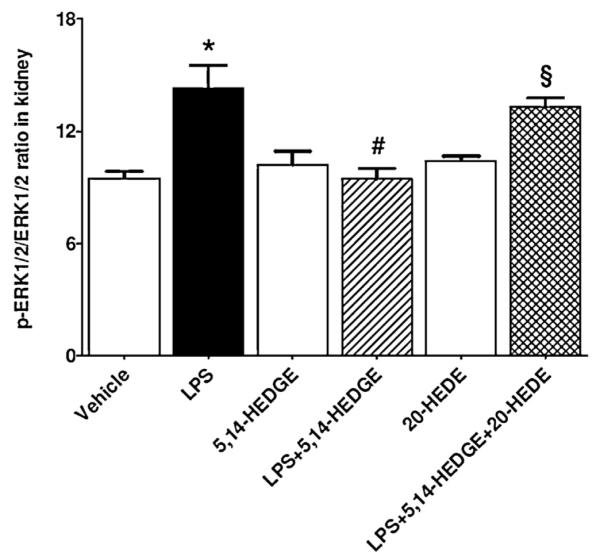
Effects of 5,14-HEDGE and 20-HEDE on changes in renal ERK1/2 activity measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. ERK1/2 protein expression and p-ERK1/2 (pTpY185/187) protein levels in tissue homogenates were measured by immunoblotting and ELISA Kit, respectively. ERK1/2 activity was calculated as the ratio of p-ERK1/2 (pTpY185/187)/ERK1/2 protein levels. Data are expressed as means ± S.E.M of 4–10 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
3.6. Effect of 5,14-HEDGE on LPS-induced increase in IκB-α expression and phosphorylation in endotoxemic rats
To demonstrate the effect of 5,14-HEDGE on LPS-induced changes in IκB-α expression and phosphorylation, unphosphorylated and phosphorylated IκB-α protein expressions were measured in the renal tissue of endotoxemic rats. LPS treatment increased phosphorylated (p < 0.05), but not unphosphorylated protein expression of IκB-α (p > 0.05) (Fig. 5). The increase in IκB-α phosphorylation caused by LPS was prevented in the tissues of rats treated with 5,14-HEDGE (p < 0.05). 20-HEDE did not reverse the effect of 5,14-HEDGE on IκB-α phosphorylation in LPS-treated rats (p > 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal unphosphorylated and phosphorylated IκB-α protein expression in vehicle-treated rats (p > 0.05).
Fig. 5.
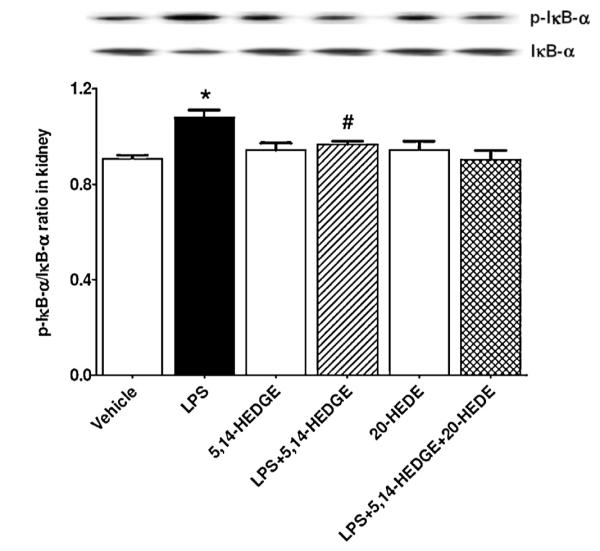
Effects of 5,14-HEDGE and 20-HEDE on changes in renal IκB-α protein expression and phosphorylation measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. IκB-α and p-IκB-α (Ser32) protein levels in tissue homogenates were measured by immunoblotting. Data are expressed as means ± S.E.M of 4 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05).
3.7. Effect of 5,14-HEDGE on LPS-induced increase in NF-κB expression and phosphorylation
To demonstrate the effect of 5,14-HEDGE on LPS-induced changes in NF-κB expression and phosphorylation, unphosphorylated and phosphorylated NF-κB protein expressions were measured in the renal tissue of endotoxemic rats. LPS increased unphosphorylated and phosphorylated NF-κB protein expression (p < 0.05) (Fig. 6). The increase in both unphosphorylated and phosphorylated NF-κB expressions produced by LPS was prevented in the tissues of rats treated with 5,14-HEDGE (p < 0.05). 20-HEDE did not reverse the effect of 5,14-HEDGE on NF-κB expression and phosphorylation in LPS-treated rats (p > 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal unphosphorylated and phosphorylated NF-κB protein expression in vehicle-treated rats (p > 0.05).
Fig. 6.
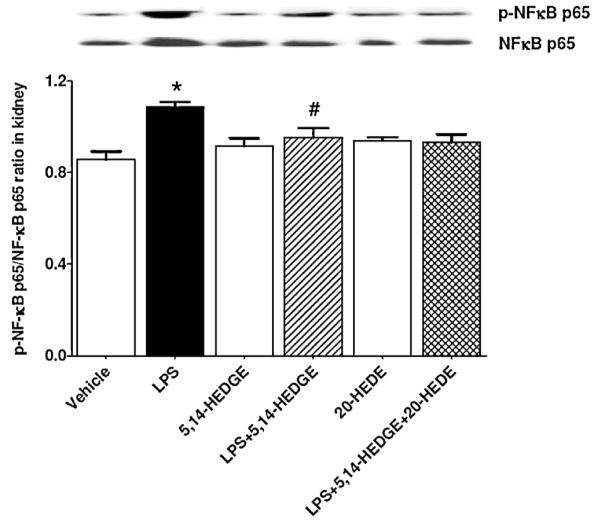
Effects of 5,14-HEDGE and 20-HEDE on changes in renal NF-κB protein expression and phosphorylation measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. NF-κB and p-NF-κB (Ser536) protein levels in tissue homogenates were measured by immunoblotting. Data are expressed as means ± S.E.M of 4 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05).
3.8. Effect of 5,14-HEDGE on LPS-induced increase in TNF-α and IL-8 levels
To investigate the effect of 5,14-HEDGE on LPS-induced changes in proinflammatory cytokine production, TNF-α and IL-8 levels were measured in the sera and renal tissues of endotoxemic rats. LPS caused an increase in systemic and renal levels of TNF-α (Fig. 7) and IL-8 (Fig. 8) (p < 0.05). The increased production of TNF-α and IL-8 was prevented in the tissues of rats treated with LPS and 5,14-HEDGE (p < 0.05) (Figs. 7 and 8). 20-HEDE reversed the effect of 5,14-HEDGE on the cytokine production in LPS-treated rats (p > 0.05). 5,14-HEDGE or 20-HEDE had no effect on the basal cytokine levels in vehicle-treated rats (p > 0.05).
Fig. 7.

Effects of 5,14-HEDGE and 20-HEDE on changes in TNF-α levels in (A) serum and (B) kidney measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. TNF-α levels in sera and tissue homogenates were measured by ELISA Kit. Data are expressed as means ± S.E.M of 5 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
Fig. 8.

Effects of 5,14-HEDGE and 20-HEDE on changes in IL-8 levels in (A) serum and (B) kidney measured 4 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious rats. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. IL-8 levels in sera and tissue homogenates were measured by ELISA Kit. Data are expressed as means ± S.E.M of 5 animals. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05). §Significant difference from the corresponding value seen in the rats treated with LPS and 5,14-HEDGE (p < 0.05).
3.9. Effect of 5,14-HEDGE on LPS-induced mortality
To determine whether 5,14-HEDGE improves LPS-induced mortality in the rodent model of septic shock, mice were administered the same dose of LPS that was given to rats intraperitoneally and survival was monitored for up to 72 h. The mortality rates at 24, 30, 36, and 72 h after LPS challenge were 2%, 4%, 10%, and 16%, respectively. The subcutaneous administration of 5,14-HEDGE at 30 mg/kg prevented the mortality when it was started at 24 h and repeated at 30, 36, 48, 54, and 72 h after administration of LPS (p < 0.05). However, only one of the mice treated with 5,14-HEDGE died at 60 h after injection of LPS. The overall survival was significantly improved in endotoxemic mice treated with 5,14-HEDGE (p < 0.05) (Fig. 9). In endotoxemic mice treated with 5,14-HEDGE and 20-HEDE (30 mg/kg, s.c.), the mortality rates were 1%, 8%, and 10% at 36, 60, and 72 h after LPS challenge, respectively. None of the mice died during the 72 h observation period after saline and/or 5,14-HEDGE or 20-HEDE administration alone.
Fig. 9.
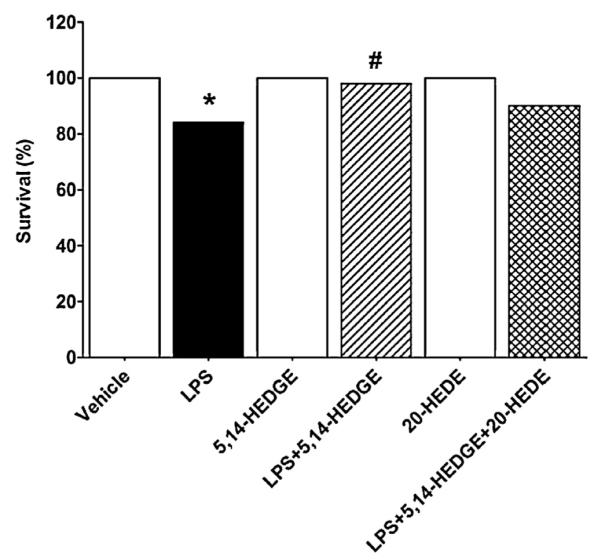
Effects of 5,14-HEDGE and 20-HEDE on survival 72 h after saline (vehicle) (4 ml/kg, i.p.) or LPS (10 mg/kg, i.p.) injection to conscious mice. 5,14-HEDGE (30 mg/kg, s.c.) and/or 20-HEDE (30 mg/kg, s.c.) were given 1 h after administration of saline or LPS. Data are obtained from 20–50 observations. *Significant difference from the corresponding value seen in rats treated with saline (p < 0.05). #Significant difference from the corresponding value seen in the rats treated with LPS (p < 0.05).
4. Discussion
The results of the present study indicate that increased expression and/or activity of sEH and MEK1, ERK1/2, IKKβ, IκB-α, and NF-κB, and TNF-α and IL-8 production associated with a decrease in expression and activity of CYP2C23 participate in the fall in blood pressure and tachycardia in rats treated with LPS and mortality in endotoxemic mice. These data also demonstrate that 5,14-HEDGE, a 20-HETE mimetic, restores blood pressure, prevents tachycardia and inflammation, and improves survival which may be due to decreased expression and/or activity of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production associated with increased CYP2C23 expression and antiinflammatory mediator formation in the rodent model of septic shock.
In the present study, we measured changes in the expression and/or activity of sEH, CYP2C23, and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production at 4 h after LPS administration. LPS produced a fall in MAP and increase in HR within 1 h that was sustained for 4 h. Administration of 5,14-HEDGE prevented the LPS-induced fall in blood pressure and increase in HR within 1 h. Compensatory mechanisms, including activation of the renin-angiotensin-aldosteron system, increased sensitivity of baroreceptor reflex mechanisms, and increased production of endothelin-1 and catecholamines, which are known to activate phospholipase A2 and release AA from tissue lipids, and results in prostaglandin synthesis, and also stimulate production of reactive nitrogen and oxygen species, have also been reported to be responsible for the changes in the formation of vasoregulatory molecules that could contribute to the fall in blood pressure during LPS-induced endotoxemia [1]. Our previous studies suggested that systemic administration of LPS to rats causes a decrease in CYP4A1 mRNA and protein expression in the kidney, heart, thoracic aorta, and superior mesenteric artery associated with reduced systemic and tissue levels of 20-HETE [40–42,45,46]. In contrast to our findings, Anwar-Mohamed et al. [20] demonstrated that CYP4A1 mRNA expression was increased in the heart of inflamed animals at 6, 12, and 24 h by 400%, 900%, and 6000%, respectively, after injection of LPS (E. coli LPS, O127:B8, 1 mg/kg, i.p.) to Sprague-Dawley rats. Similarly, but with a lower magnitude, renal mRNA expression of CYP4A1 was increased only at 24 h by 100% after injection of LPS (E. coli LPS, O127:B8, 1 mg/kg, i.p.) to rats. The LPS-induced changes in the expression of CYP4A1 as well as enhanced expression of TNF-α and IL-6 genes in cardiac and renal tissues were also associated with increased production of 20-HETE in the heart microsomes of inflamed animals by 40% ex vivo. The authors suggested that acute inflammation causes alteration in cardiac CYP-mediated AA metabolism in favor of 20-HETE formation. However, Theken et al. [21] reported that 20-HETE formation in the kidney, but not in the heart, decreased 6 or 24 h after injection of LPS (E. coli LPS, O111:B4, 1 mg/kg, i.p.) to C57BL/6 mice leading to the conclusion that acute activation of the innate immune response alters CYP expression and eicosanoid metabolism in an isoform-, tissue-, and time-dependent manner. Thus, the contradiction between previously published studies and our results could be attributed to differences in type and strain of animals, dose regimen, strain of LPS, and time points for measurement of enzyme expression and activity which might reflect the differences in the response to LPS treatment.
NF-κB has been implicated in the regulation of multiple biological phenomena and disease states, including inflammation and septic shock [1,47]. Activation of MEK1/ERK1/2 pathway in response to endotoxin or cytokines has been reported to act as an important regulator of NF-κB activation and NF-κB-dependent proinflammatory gene expression. It has been demonstrated that endotoxin activates ERK1/2, as well as inhibitory subunit of NF-κB (IκB) degradation and ensuing NF-κB activation in rat vascular smooth muscle cells [48,49]. Subsequent activation of NF-κB by ERK1/2 has shown to increase inflammatory mediator production [50–52]. These observations suggest that MEK1/ERK1/2 pathway involves in the vascular hyporeactivity to vasoconstrictor agents and decreased blood pressure in endotoxemic rats. Indeed, as shown by our group previously, a selective inhibitor of phosphorylation of ERK1/2 by MEK1, 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadien (U0126) prevented the endotoxin-induced fall in MAP and increase in HR in vivo as well as vascular hyporeactivity to norepinephrine associated with increased phosphorylation of ERK1/2 in vitro [53] and ex vivo [54,55]. These findings suggest that MEK1/ERK1/2 pathway-induced inhibition of the formation of vasoconstrictor mediators, such as 20-HETE, and removal of its influence on vascular tone may contribute to the hypotension, tachycardia, and vascular hyporeactivity in endotoxic shock. Supporting this view were our findings that administration of a synthetic analog of 20-HETE, 5,14-HEDGE prevented the increase in 20-HETE levels in the systemic circulation as well as cardiovascular and renal tissues contribute to the effect of 5,14-HEDGE to prevent the vascular hyporeactivity, hypotension, and tachycardia during rat endotoxemia [39–42]. In the present study, the LPS-induced fall in MAP and rise in HR were associated with increased sEH mRNA and protein expression. On the other hand, CYP2C23 mRNA and protein expression was decreased in the renal tissues of endotoxemic rats. Increased activities of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway associated with the production of proinflammatory cytokines, TNF-α and IL-8, were also associated with a decreased activity of AA epoxygenases. These effects of LPS on sEH and CYP2C23 expression and activity were prevented by 5,14-HEDGE given 1 h after injection of LPS. 5,14-HEDGE also prevented the LPS-induced increase in the activity of MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway associated with increased systemic and renal levels of proinflammatory cytokines in endotoxemic rats. Moreover, the overall survival was significantly improved in endotoxemic mice treated with 5,14-HEDGE. A competitive antagonist of vasoconstrictor effects of 20-HETE, 20-HEDE, reversed the effects of 5,14-HEDGE on sEH mRNA and protein expression, but not its activity, in rats treated with LPS. On the other hand, 20-HEDE reversed the effect of 5,14-HEDGE on AA epoxygenase activities, but not CYP2C23 expression. In addition, LPS-induced increase in ERK1/2 activity, but not phosphorylated MEK1, IκB-α, and NF-κB levels, was reversed by 20-HEDE in the renal tissue. These observations together with previous studies [37,39–46,53–57] suggest that increased production of EETs as well as decreased expression and/or activities of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway participates in the effect of 5,14-HEDGE to prevent the LPS-induced inflammatory response associated with a decrease in MAP and rise in HR in endotoxemic rats and mortality in mice. In addition, it seems that transcriptional (e.g., gene expression), translational (e.g., mRNA expression and stability), and posttranslational mechanisms (e.g., protein expression and stability) as well as changes in enzyme activity are involved in the regulation of sEH and CYP2C23 expression by 5,14-HEDGE in the rodent model of septic shock. Although our results demonstrated that 5,14-HEDGE directly increases CYP2C23 expression at translational, transcriptional, and posttranscriptional levels and activity of AA epoxygenases leading to EET production in endotoxemic rats, it is also possible that 5,14-HEDGE might increase CYP2C23 expression and activity indirectly by inhibiting expression and activity of sEH and/or decreasing the effects of 20-HETE, cytokines, and/or transcription factors (such as NF-κB) on the expression and activity of CYP2C23. However, additional experiments need to be conducted to demonstrate the validity of these hypotheses. Further characterization of the molecular mechanisms of interactions between sEH, CYP2C23, MEK1, ERK1/2, and IKKβ enzymes at molecular level will provide the framework for extension of this work into understanding the role of AA products in the inflammatory response associated with hypotension, tachycardia, and mortality during endotoxemia.
In conclusion, the present study provides evidence that endotoxin induces expression and/or activity of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production associated with decreased CYP2C23 expression and formation of antiinflammatory eicosanoids, EETs, during endotoxemia leading to hypotension, tachycardia, inflammation, and mortality in the rodent model of septic shock (Fig. 10). Moreover, it is likely that increased sEH activity suppresses conversion of EETs to DHETs by CYP2C23 in endotoxemic rats. Our findings also suggest that 5,14-HEDGE, a 20-HETE mimetic, prevents hypotension, tachycardia, inflammation, and mortality during endotoxemia presumably due to decreased expression and/or activity of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production associated with increased CYP2C23 expression and antiinflammatory mediator formation. Therefore, we speculate that 5,14-HEDGE could have antihypotensive effect due to its 20-HETE mimetic activity and inhibition of vasodilator mediator formation as well as anti-inflammatory effect which could be due to increased availability of EETs and decreased proinflammatory cytokine production. The current management of septic shock relies on immediate treatment with antibiotics and strong supportive care to control hypotension, tachycardia, cardiac output, and tissue oxygenation to maintain organ function. However, the failure of conventional therapy is that the pathophysiology of septic shock is the result of a highly complex set of processes in which the host response becomes dysregulated and causes cellular damage, tissue damage, and, ultimately, organ failure. In the light of the important role of 20-HETE and EETs in the regulation of cardiovascular hemostasis and inflammatory process, further studies with 20-HETE mimetics in experimental models of endotoxemia could provide a novel approach to treat hypotension, tachycardia, inflammation, and mortality which lead to multiple organ failure and death in septic shock.
Fig. 10.
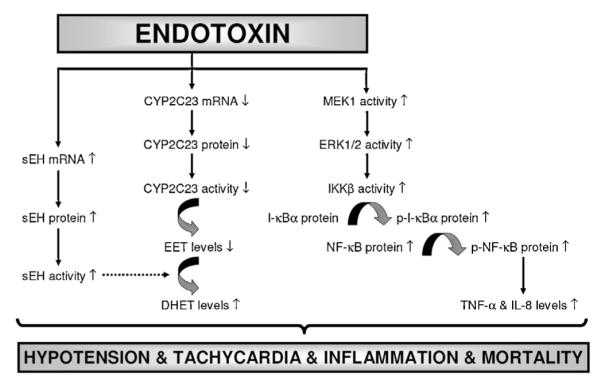
Schematic diagram showing the involvement of sEH, CYP2C23, MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway, and proinflammatory cytokine production in endotoxin-induced hypotension, tachycardia, inflammation, and survival based on the current study. Endotoxin, the lipid A part of LPS which is the most potent microbial mediator of the pathogenesis of sepsis and septic shock, induces sEH expression and activity associated with decreased expression and activity of CYP2C23. In addition, endotoxin increases activity of MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway associated with proinflammatory cytokine production leading to hypotension, tachycardia, inflammation, and mortality in the rodent model of septic shock. 5,14-HEDGE prevents the effects of endotoxin on the increase in expression and/or activity of sEH, MEK1, ERK1/2, IKKβ, IκB-α, and NF-κB associated with proinflammatory mediator production, and thus, restores blood pressure, prevents tachycardia and proinflammatory mediator formation, and improves survival during rodent endotoxemia. It should be noted that a competitive antagonist of vasoconstrictor effects of 20-HETE, 20-HEDE, prevents the effects of 5,14-HEDGE on blood pressure and HR associated with expression and/or activity of sEH, CYP2C23, and ERK1/2 as well as TNF-α and IL-8 levels in rats treated with LPS. It can be concluded that decreased expression and/or activity of sEH and MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway as well as proinflammatory cytokine production associated with increased CYP2C23 expression and antiinflammatory mediator formation (e.g., EETs) participate in the protective effect of 5,14-HEDGE against hypotension, tachycardia, inflammation, and mortality in the rodent model of septic shock. ( ) stimulation; (
) stimulation; ( ) inhibition.
) inhibition.
Supplementary Material
Acknowledgements
Financial support was provided by grants from Mersin University (BAP-ECZ F FB [BT] 2010-5 B), TUBITAK (SBAG-109S121), NIH (19134-37 and GM31278), the Robert A. Welch Foundation (GL625910), and The German Research Foundation.
Abbreviations
- 20-HEDE
20-hydroxyeicosa-6(Z),15(Z)-dienoic acid
- AA
arachidonic acid
- 20-HETE
20-hydroxyeicosatetraenoic acid
- 5,14-HEDGE
N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine
- cDNA
complementary deoxyribonucleic acid
- COX
cyclooxygenase
- CYP
cyctochrome P450
- EET
epoxyeicosatrienoic acid
- DHET
dihydroxyeicosatrienoic acid
- ERK
extracellular signal-regulated kinase
- HR
heart rate
- IKK
IκB kinase
- IκB
inhibitor of κB
- IL
interleukin
- i.p.
intraperitoneally
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- MAPK
mitogen-activated protein kinase kinase
- MEK
mitogen-activated protein kinase
- mRNA
messenger ribonucleic acid
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- PG
prostaglandin
- RT-PCR
reverse transcription-polymerase chain reaction
- s.c.
subcutaneously
- sEH
soluble epoxide hydrolase
- TNF
tumor necrosis factor
- U0126
1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadien
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.prostaglandins.2013.01.005.
References
- [1].Tunctan B, Korkmaz B, Sari AN, Kacan M, Unsal D, Serin MS, et al. A novel treatment strategy for sepsis and septic shock based on the interactions between prostanoids, nitric oxide, and 20-hydroxyeicosatetraenoic acid. Antiinflamm Antiallergy Agents Med Chem. 2012;11:121–50. doi: 10.2174/187152312803305759. [DOI] [PubMed] [Google Scholar]
- [2].Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omegahydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol. 2005;45:413–38. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- [3].Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res. 2005;41:175–93. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- [4].Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- [5].Escalante B, Omata K, Sessa W, Lee SG, Falck JR, Schwartzman ML. 20-hydroxyeicosatetraenoic acid is an endothelium-dependent vasoconstrictor in rabbit arteries. Eur J Pharmacol. 1993;235:1–7. doi: 10.1016/0014-2999(93)90812-v. [DOI] [PubMed] [Google Scholar]
- [6].Escalante B, Sessa WC, Falck JR, Yadagiri P, Schwartzman ML. Vasoactivity of 20-hydroxyeicosatetraenoic acid is dependent on metabolism by cyclooxygenase. J Pharmacol Exp Ther. 1989;248:229–32. [PubMed] [Google Scholar]
- [7].Randriamboavonjy V, Busse R, Fleming I. 20-HETE-induced contraction of small coronary arteries depends on the activation of Rho-kinase. Hypertension. 2003;41:801–6. doi: 10.1161/01.HYP.0000047240.33861.6B. [DOI] [PubMed] [Google Scholar]
- [8].Schwartzman ML, Falck JR, Yadagiri P, Escalante B. Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygeanse: formation and identification of novel endothelium-dependent vasoconstrictor metabolites. J Biol Chem. 1989;264:11658–62. [PubMed] [Google Scholar]
- [9].Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, et al. 20-HETE is an endogenous inhibitor of the large-conductance Ca(2+)-activated K+ channel in renal arterioles. Am J Physiol. 1996;270:R228–37. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- [10].Akbulut T, Regner KR, Roman RJ, Avner ED, Falck JR, Park F. 20-HETE activates the Raf/MEK/ERK pathway in renal epithelial cells through an EGFR- and c-Src-dependent mechanism. Am J Physiol. 2009;297:F662–70. doi: 10.1152/ajprenal.00146.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ponnoth DS, Nayeem MA, Kunduri SS, Tilley SL, Zeldin DC, Ledent C, et al. Role of ω-hydroxylase in adenosine-mediated aortic response through MAP kinase using A2A-receptor knockout mice. Am J Physiol. 2012;302:R400–8. doi: 10.1152/ajpregu.00481.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun CW, Falck JR, Harder DR, Roman RJ. Role of tyrosine kinase and PKC in the vasoconstrictor response to 20-HETE in renal arterioles. Hypertension. 1999;33:414–8. doi: 10.1161/01.hyp.33.1.414. [DOI] [PubMed] [Google Scholar]
- [13].Carroll MA, Capparelli MF, Doumand AB, Cheng MK, Jiang H, McGiff JC. Renal vasoactive eicosanoids: interactions between cytochrome P450 and cyclooxygenase metabolites during salt depletion. Am J Hypertens. 2001;14:159A. [Google Scholar]
- [14].Carroll MA, Garcia MP, Falck JR, McGiff JC. Cyclooxygenase dependency of the renovascular actions of cytochrome P450-derived arachidonate metabolites. J Pharmacol Exp Ther. 1992;260:104–9. [PubMed] [Google Scholar]
- [15].Pratt PF, Falck JR, Reddy KM, Kurian JB, Campbell WB. 20-HETE relaxes bovine coronary arteries through the release of prostacyclin. Hypertension. 1998;31:237–41. doi: 10.1161/01.hyp.31.1.237. [DOI] [PubMed] [Google Scholar]
- [16].Yu M, McAndrew RP, Al-Saghir R, Maier KG, Medhora M, Roman RJ, et al. Nitric oxide contributes to 20-HETE-induced relaxation of pulmonary arteries. J Appl Physiol. 2002;93:1391–9. doi: 10.1152/japplphysiol.00247.2002. [DOI] [PubMed] [Google Scholar]
- [17].Fang X, Faraci FM, Kaduce TL, Harmon S, Modrick ML, Hu S, et al. 20-Hydroxyeicosatetraenoic acid is a potent dilator of mouse basilar artery: role of cyclooxygenase. Am J Physiol Heart Circ Physiol. 2006;291:H2301–7. doi: 10.1152/ajpheart.00349.2006. [DOI] [PubMed] [Google Scholar]
- [18].Cheng J, Wu CC, Gotlinger KH, Zhang F, Falck JR, Narsimhaswamy D, et al. 20-Hydroxy-5,8,11,14-eicosatetraenoic acid mediates endothelial dysfunction via IκB kinase-dependent endothelial nitric-oxide synthase uncoupling. J Pharmacol Exp Ther. 2010;332:57–65. doi: 10.1124/jpet.109.159863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-κB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–10. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- [20].Anwar-mohamed A, Zordoky BN, Aboutabl ME, El-Kadi AO. Alteration of cardiac cytochrome P450-mediated arachidonic acid metabolism in response to lipopolysaccharide-induced acute systemic inflammation. Pharmacol Res. 2010;61:410–8. doi: 10.1016/j.phrs.2009.12.015. [DOI] [PubMed] [Google Scholar]
- [21].Theken KN, Deng Y, Kannon MA, Miller TM, Poloyac SM, Lee CR. Activation of the acute inflammatory response alters cytochrome P450 expression and eicosanoid metabolism. Drug Metab Dispos. 2011;39:22–9. doi: 10.1124/dmd.110.035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grasso E, Longo V, Coceani F, Giovanni Gervasi P. Cytochrome P450 expression and catalytic activity in coronary arteries and liver of cattle. Biochim Biophys Acta. 2005;1722:116–23. doi: 10.1016/j.bbagen.2004.11.018. [DOI] [PubMed] [Google Scholar]
- [23].Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–81. [PubMed] [Google Scholar]
- [24].Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li PL. 14,15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of K(Ca) channels. Am J Physiol. 2002;282:H1656–64. doi: 10.1152/ajpheart.00597.2001. [DOI] [PubMed] [Google Scholar]
- [25].Zeldin DC, Wei S, Falck JR, Hammock BD, Snapper JR, Capdevila JH. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: substrate structural determinants of asymmetric catalysis. Arch Biochem Biophys. 1995;316:443–51. doi: 10.1006/abbi.1995.1059. [DOI] [PubMed] [Google Scholar]
- [26].Chen J, Capdevila JH, Zeldin DC, Rosenberg RL. Inhibition of cardiac L-type calcium channels by epoxyeicosatrienoic acids. Mol Pharmacol. 1999;55:288–95. doi: 10.1124/mol.55.2.288. [DOI] [PubMed] [Google Scholar]
- [27].Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoylphospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem. 1991;266:7561–9. [PubMed] [Google Scholar]
- [28].Capdevila JH, Mosset P, Yadagiri P, Lumin S, Falck JR. NADPH-dependent microsomal metabolism of 14,15-epoxyeicosatrienoic acid to diepoxides and epoxyalcohols. Arch Biochem Biophys. 1988;261:122–33. doi: 10.1016/0003-9861(88)90111-7. [DOI] [PubMed] [Google Scholar]
- [29].Carroll MA, McGiff JC. A new class of lipid mediators: cytochrome P450 arachidonate metabolites. Thorax. 2000;55:S13–6. doi: 10.1136/thorax.55.suppl_2.S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, et al. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276:14867–74. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- [31].Fang X, Kaduce TL, Weintraub NL, VanRollins M, Spector AA. Functional implications of a newly characterized pathway of 11,12-epoxyeicosatrienoic acid metabolism in arterial smooth muscle. Circ Res. 1996;79:784–93. doi: 10.1161/01.res.79.4.784. [DOI] [PubMed] [Google Scholar]
- [32].Holla VR, Makita K, Zaphiropoulos PG, Capdevila JH. The kidney cytochrome P 450 2C23 arachidonic acid epoxygenase is upregulated during dietary salt loading. J Clin Invest. 1999;104:751–60. doi: 10.1172/JCI7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karara A, Makita K, Jacobson HR, Falck JR, Guengerich FP, DuBois RN, et al. Molecular cloning, expression, and enzymatic characterization of the rat kidney cytochrome P 450 arachidonic acid epoxygenase. J Biol Chem. 1993;268:13565–70. [PubMed] [Google Scholar]
- [34].Imaoka S, Wedlund PJ, Ogawa H, Kimura S, Gonzalez FJ, Kim HY. Identification of CYP2C23 expressed in rat kidney as an arachidonic acid epoxygenase. J Pharmacol Exp Ther. 1993;267:1012–6. [PubMed] [Google Scholar]
- [35].Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pfister SL, Gauthier KM, Campbell WB. Vascular pharmacology of epoxyeicosatrienoic acids. Adv Pharmacol. 2010;60:27–59. doi: 10.1016/B978-0-12-385061-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kessler P, Popp R, Busse R, Schini-Kerth VB. Proinflammatory mediators chronically downregulate the formation of the endothelium-derived hyperpolarizing factor in arteries via a nitric oxide/cyclic GMP-dependent mechanism. Circulation. 1999;99:1878–84. doi: 10.1161/01.cir.99.14.1878. [DOI] [PubMed] [Google Scholar]
- [38].Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tunctan B, Korkmaz B, Buharalioglu CK, Sahan Firat S, Anjaiah S, Falck J, et al. A 20-HETE agonist N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine, opposes the fall in blood pressure and vascular reactivity in endotoxin-treated rats. Shock. 2008;30:329–35. doi: 10.1097/SHK.0b013e31816471c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cuez T, Korkmaz B, Buharalioglu CK, Sahan-Firat S, Falck J, Malik KU, et al. A synthetic analogue of 20-HETE, 5,14-HEDGE, reverses endotoxin-induced hypotension via increased 20-HETE levels associated with decreased iNOS protein expression and vasodilator prostanoid production in rats. Basic Clin Pharmacol. 2010;106:378–88. doi: 10.1111/j.1742-7843.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tunctan B, Korkmaz B, Cuez T, Sari AN, Kacan M, Gilik D, et al. Contribution of iNOS COX-2 and CYP4A1 to the protective effect of a synthetic analog of 20-HETE, 5,14-HEDGE, against endotoxin-induced hypotension and mortality in experimental model of septic shock in rats and mice. Inflamm Res. 2011;60:S55. [Google Scholar]
- [42].Tunctan B, Korkmaz B, Sari AN, Kacan M, Gilik U, Serin MS, et al. A synthetic analog of 20-HETE, 5,14-HEDGE, reverses endotoxin-induced hypotension and mortality via increased expression and activities of CYP4A1 and CYP2C23 in a rodent model of septic shock: contribution of MEK1/ERK1/2/IKKβ/IκB-α/NF-κB pathway and soluble epoxide hydrolase. Sepsis. 2012;4:158–9. [Google Scholar]
- [43].Tunctan B, Korkmaz B, Yildirim H, Tamer L, Atik U, Buharalioglu CK. Increased production of nitric oxide contributes to renal oxidative stress in endotoxemic rat. Am J Infect Dis. 2005;1:111–5. [Google Scholar]
- [44].Tunctan B, Altug S, Uludag O, Demirkay B, Abacioglu N. Effects of cyclooxygenase inhibitors on nitric oxide production and survival in a mice model of sepsis. Pharmacol Res. 2003;48:37–48. [PubMed] [Google Scholar]
- [45].Buharalioglu CK, Korkmaz B, Cuez T, Sahan-Firat S, Sari AN, Malik KU, et al. Piroxicam reverses endotoxin-induced hypotension in rats: contribution of vasoactive eicosanoids and nitric oxide. Basic Clin Pharmacol Toxicol. 2011;109:186–94. doi: 10.1111/j.1742-7843.2011.00708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tunctan B, Sari AN, Kacan M, Unsal D, Buharalioglu CK, Sahan-Firat S, et al. NS-398 reverses hypotension in endotoxemic rats: contribution of eicosanoids NO, and peroxynitrite. Prostaglandins Other Lipid Mediat. 2012 Sep; doi: 10.1016/j.prostaglandins.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol. 2006;290:L622–45. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- [48].Son YH, Jeong YT, Lee KA, Choi KH, Kim SM, Rhim BY, et al. Roles of MAPK and NF-κB in interleukin-6 induction by lipopolysaccharide in vascular smooth muscle cells. J Cardiovasc Pharmacol. 2008;51:71–7. doi: 10.1097/FJC.0b013e31815bd23d. [DOI] [PubMed] [Google Scholar]
- [49].Yamakawa T, Eguchi S, Matsumoto T, Yamakawa Y, Numaguchi K, Miyata I, et al. Intracellular signaling in rat cultured vascular smooth muscle cells: roles of nuclear factor-κB and p38 mitogen-activated protein kinase on tumor necrosis factor-alpha production. Endocrinology. 1999;140:3562–72. doi: 10.1210/endo.140.8.6914. [DOI] [PubMed] [Google Scholar]
- [50].Jiang B, Brecher P, Cohen RA. Persistent activation of nuclear factor-κB by interleukin-1β and subsequent inducible NO synthase expression requires extracellular signal-regulated kinase. Arterioscler Thromb Vasc Biol. 2001;21:1915–20. doi: 10.1161/hq1201.099424. [DOI] [PubMed] [Google Scholar]
- [51].Kim SH, Kim J, Sharma RP. Inhibition of p38 and ERK MAP kinases blocks endotoxin-induced nitric oxide production and differentially modulates cytokine expression. Pharmacol Res. 2004;49:433–9. doi: 10.1016/j.phrs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [52].Suh SJ, Chung TW, Son MJ, Kim SH, Moon TC, Son KH, et al. The naturally occurring biflavonoid, ochnaflavone, inhibits LPS-induced iNOS expression, which is mediated by ERK1/2 via NF-κB regulation, in RAW264.7 cells. Arch Biochem Biophys. 2006;447:136–46. doi: 10.1016/j.abb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- [53].Korkmaz B, Ozveren E, Buharalioglu CK, Tunctan B. Extracellular signal-regulated kinase (ERK1/2) contributes to endotoxin-induced hyporeactivity via nitric oxide and prostacyclin production in rat aorta. Pharmacology. 2006;78:123–8. doi: 10.1159/000095962. [DOI] [PubMed] [Google Scholar]
- [54].Korkmaz B, Buharalioglu K, Sahan-Firat S, Cuez T, Demiryurek TA, Tunctan B. Activation of MEK1/ERK1/2/iNOS/sGC/PKG pathway associated with peroxinitrite formation contributes to hypotension and vascular hyporeactivity in endotoxemic rats. Nitric Oxide. 2011;24:160–72. doi: 10.1016/j.niox.2011.02.004. [DOI] [PubMed] [Google Scholar]
- [55].Tunctan B, Korkmaz B, Dogruer ZN, Tamer L, Atik U, Buharalioglu CK. Inhibition of extracellular signal-regulated kinase (ERK1/2) activity reverses endotoxin-induced hypotension via decreased nitric oxide production in rats. Pharmacol Res. 2007;56:56–64. doi: 10.1016/j.phrs.2007.03.006. [DOI] [PubMed] [Google Scholar]
- [56].Fife KL, Liu Y, Schmelzer KR, Tsai HJ, Kim IH, Morisseau C, et al. Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. J Pharmacol Exp Ther. 2008;327:707–15. doi: 10.1124/jpet.108.142398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Oyekan A. Nitric oxide inhibits renal cytochrome P450-dependent epoxygenases in the rat. Clin Exp Pharmacol Physiol. 2002;29:990–5. doi: 10.1046/j.1440-1681.2002.03762.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.