

Abstract
Prion diseases characterize a category of fatal neurodegenerative diseases. Although reports have increasingly shown that oxidative stress plays an important role in the progression of prion diseases, little is known about whether oxidative stress is a cause or a consequence of a prion disease. The mechanism of prion disease development also remains unclear. The purpose of this study was to investigate three things: the possible mechanisms of neuron cell damage, the conformation of anti-protease K (PK) PrPSc, and the role of oxidative stress in the progression of prion diseases. The study results demonstrated that normal PrPC transformed into a PK-resistant protein under oxidative stress in the presence of PrP106–126. Further, the protein misfolding cyclic amplification procedure may have accelerated this process. Mitochondrial damage and dysfunction in prion disease progression were also observed in this study. Our results suggested that neuron cell damage, and particularly mitochondrial damage, was induced by oxidative stress. This damage may be the initial cause of a given prion disease.
Keywords: Prion, ROS, Mitochondria, Oxidative stress, PMCA, Iron
Introduction
Prion diseases characterize a series of fatal neurodegenerative diseases, which include human Creutzfeldt–Jakob disease, bovine spongiform encephalopathy, and Scrapie in sheep. In transmissible spongiform encephalopathies (TSEs), the infectious agent or “prion” is thought to be PrPres (Prusiner 1982). PrPres is a protease-resistant conformer of the cell protein PrP, and it plays a key role in a group of fatal neurodegenerative disorders that have a unique property of being infectious, sporadic, or genetic in origin. These diseases are believed to be the consequence of conformational conversion of the cellular prion protein into an abnormal isoform. The exact pathogenic mechanism of these diseases remains uncertain, but it is believed that oxidative stress plays a central role in disease progression (Milhavet and Lehmann 2002).
Oxidative stress may be one of the many triggers of degeneration in the brain. Oxidative damage of macromolecules leads to a progressive decline in proper cell and tissue function. Mitochondria also constitute a major cellular source of reactive oxygen species (ROS) generation. At the same time, mitochondria are the main targets of ROS-induced oxidative damage (Kozlowski et al. 2009; Saretzki 2009). So, we postulated that oxidative stress might also be responsible for the neuronal cell death that is associated with prion infectious disease. However, whether oxidative stress is a cause or a consequence of prion disease is not well known.
It is well known that some metal ions, such as Cu2+, Zn2+, and Fe2+/Fe3+, are involved in cell oxidative stress. It has been reported that the concentrations of the total iron (Fe), Fe3+, and Fe2+/Fe3+ ratios were increased in the brains of scrapie-infected mice, demonstrating that iron-induced oxidative stress partly contributed to neurodegeneration in TSEs (Kim et al. 2000).
Residues 106–126 of the human PrP protein (PrP106–126) is a highly conserved sequence located in the important domain concerning membrane that is related to conformational conversion of PrP (Zheng et al. 2009). It has been shown that the PrP106–126 peptide exhibited the potentials of β-sheet formation and fibrillogenesis, possessed many other properties of PrPSc (PrPres), and was frequently used to study PrPSc pathogenesis (Seo et al. 2010).
The purpose of this study was to investigate the following: the possible mechanism of neuron cell damage, the conformation of anti-protease K (PK) PrPSc, and the role of oxidative stress in prion disease progression.
Materials and Methods
Cell Culture
The neuroblastoma cell lines (N2a) were obtained from the American Type Culture Collection. Cells were cultured in Dulbecco's modified Eagle medium (DMEM) (Hyclone Laboratories, Thermo, USA) that contained 10 % fetal bovine serum (Invitrogen-Gibco, USA) and gentamycin (0.1 mg/ml). Cells were cultured in a cell incubator and maintained at 5 % CO2 and 37 °C.
PrP106–126 Treatment
Synthetic PrP106–126 (sequence, L-T-N-M-K-H-M-A-G-A-A-A-A-G-A-V-V-G-G-L-G) was synthesized from Sangon Biotech Co., Ltd. (Shanghai, China). The peptides were dissolved in sterile DMSO at a concentration of 500 mM and stored at −80 °C.
Protease K Assay
Protease K assay was performed as described by Luana (Fioriti et al. 2005). The final concentration of PK was 50 μg/ml.
Preparation of PrPC and Cell Treatment
All PrPC assays used the normal brain homogenate (NBH) of the wild Golden Hamster; NBH preparation was performed as described by Chen (Chen et al. 2010). The ferric chlorides were dissolved in phosphate buffer saline (PBS). In order to treat the PrPC, Fe3+ (final concentrations 300/500 μM) and PrP106–126 (final concentration 50 μM) were diluted in NBH. All NBH were performed for protein misfolding cyclic amplification (PMCA) procedure.
N2a cells were cultured on cover slips in a six-well plate for 12 h, and then treated with Fe3+ (final concentrations 300/500 μM), and PrP106–126 (final concentration 50 μM) for 12 h. Cells were harvested via centrifugation, and resuspended in lysis buffer as previously described (Chen et al. 2010; Bocharova et al. 2005). The lysate was centrifuged at 20,000 × g for 20 min. The resulting pellet was dissolved in PBS for the Western blot assay.
PMCA Procedure
PMCA was performed as described previously (Castilla et al. 2006; Saá et al. 2005). Briefly, samples were loaded onto 0.2-ml PCR tubes. Tubes were positioned on an adaptor that was placed on the plate holder of a microsonicator (Misonix model 4000); then, samples were subjected to cycles of 30 min with incubation at 37 °C, followed by a 20 s pulse of sonication with a set amplitude of 75. Samples were incubated without shaking and immersed in the water of the sonicator bath. Standard PMCA rounds consisted of 72 cycles. For one sample, 10 μl Fe3+ (final concentrations 300/500 μM) and PrP106–126 (final concentration 50 μM) were diluted into normal brain homogenate when the PMCA cycles were performed.
Western Blot
Protein immunoblotting was performed as described previously. Equal amounts of lysate protein were resolved on a 10 % SDS-polyacrylamide gel and electrophoretically transformed to a nitrocellulose membrane. Immunoreactivity was detected through sequential incubation with horseradish peroxidase-conjugated secondary antibodies. The antibodies used for immunoblotting were 3F4 (Sigma, USA) and β-actin (Sigma, USA).
DCFH-DA Assay and Visual Detection
N2a cells were cultured on cover slips in a 12-well plate for 12 h, then treated with Fe3+ (final concentrations 300/500 μM), and PrP106–126 (final concentration 50 μM) for 12 h as described above. After incubation in DMEM containing 10 μM 2,7-dichlorodihydrofluorescein diacetate (H2-DCFDA, Sigma, USA) at 37 °C for 20 min, cells were washed with PBS. Finally, cells were mounted with DakoCytomation fluorescent medium and visualized via fluorescence microscopy.
NADH Dehydrogenase Assay Protocol
The concentration of NADH dehydrogenase was measured using the NADH dehydrogenase quantification kit (BioVison, USA). The N2a cells were then washed with cold PBS. Pellet 2 × 105 cells were used for each assay in a microcentrifuge tube (2,000 rpm for 5 min) and manipulated following manufacturer instructions.
Mitochondrial Damage
N2a cells were cultured and treated as described in section “Preparation of PrPC and Cell Treatment”, harvested by centrifugation, and fixed with glutaraldehyde. For transmission electron microscopy (TEM) using JEM-1230, all cells were treated in accordance with the required steps, and then underwent negative staining with 2 % uranyl acetate.
Results
The cellular Prion Protein (PrPC) Transformed to an Anti-PK Protein
A key event in the progression of prion diseases is the conversion of the cellular isoform of the prion protein (PrPC) into the pathogenic isoform PrPSc (Choi et al. 2006). PrPC is membrane bound, helix-rich, soluble, and sensitive to degradation by proteases. The refolded isoform PrPSc, however, is β-sheet-rich, insoluble, and partially resistant to degradation by proteases. When PrPSc is digested with PK, an N-terminal truncated form is generated. The new form is designated as PrP27–30; it consists of residues 90–231 and has an approximate molecular mass of 27–30 kDa (Panza et al. 2008). In TSEs-prion-seeded amyloid formation, resistance to PK is one notable property of PrPSc (Ma and Lindquist 1999).
Some researchers have reported that oxidative stress may be one of the many triggers of degeneration in the brain. Further, PrP106–126 shares several characteristics with PrPSc. The results of the present study demonstrated that after treatment by ferric ions and PrP106–126, PrPC transformed into an abnormal form with a protease K-resistance property. Further, the PMCA procedure may have accelerated this process. The PK-resistant protein concentrations were closely related to the Fe3+ concentrations (Fig. 1).
Fig. 1.
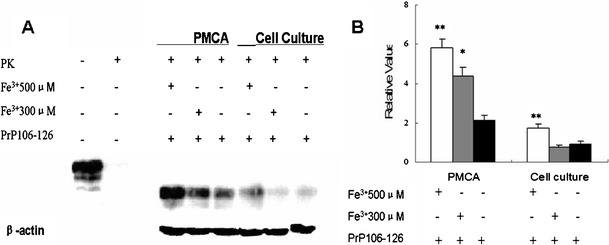
The PK-resistant PrPC after treatment with ferric ions and PrP106–126. a Ferric ions (final concentrations 300/500 μM) and PrP106–126 (final concentration 50 μM) were diluted into normal brain homogenates for 72 PMCA cycles. The PrPC were tested with 3F4 after digestion with protease K. The control is NBH without treatment by PK. Remnants of PrPC bands showed the PK-resistant property. N2a cells were cultured on cover slips in a six-well plate for 12 h, and then treated with Fe3+ (final concentrations 300/500 μM) and PrP106–126 (final concentration 50 μM) for 12 h. Cells were harvested, lysed, and dissolved in PBS for the Western blot assay. b The mean and standard error for densitometric analyses of blots from different treatment groups. Values are compared to those of the β-actins as fold. * p < 0.05 vs. PrP106–126 treatment only; ** p < 0.01 vs. PrP106–126 treatment
N2a Cells Presented Anti-PK PrPC After Treatment with Fe3+ and PrP106–126
PrPC is a glycosylphosphatidylinositol-anchored membrane protein, which is expressed in many cells. To analyze and compare how the ferric ions and PrP106–126 may affect neurons, we exposed neuroblastoma cells (N2a cells) to conditioned DMEM media containing Fe3+ and PrP106–126 as described above. When cells were harvested and lysated, PrPC were isolated and analyzed by Western blot.
The results showed that the PrPC, which was expressed by N2a cells, exhibited PK-resistance after treatment with ferric ions and PrP106–126. After digestion by the protease K, these proteins retained bands and were tested by antibody 3F4 (Fig. 1a). The concentrations of the PK-resistant-PrPC, which N2a cells were treated with the high concentrations ferric ion (500 μM Fe3+), were higher than the other one significantly (Fig. 1b). However, if there were no PrP106–126 additions, the PrPC did not show the PK-resistant property after the N2a cells were exposed to the ferric ions (data not shown).
N2a Cells Showed Increased ROS and Closely Correlated with Fe3+ and PrP106–126 Concentrations
A main pathological feature of prion diseases is neuronal loss, several proposed mechanisms for which include increased ROS and consequent oxidative stress (Haigh et al. 2011). The abnormal conformers of prion proteins can generally be detected in the brain in animal models. Detection occurs through identification of increased protease resistance, and the abnormal conformers can be identified well before overt disease is evident. At the earliest time when these abnormal protease-resistant conformers can be weakly detected, a significant lipid peroxidation level, a marker of increased ROS, can also be detected (Brazier et al. 2006). The increase of intracellular ROS in cells is concurrent with increased PrPres and activation of caspases (Haigh et al. 2011). The structures simultaneously show the highest intensity of oxidative damage. But the processes underlying ROS production in a prion disease and the precise relationship to the given prion disease remain obscure.
We attempted to identify whether the increase of intracellular ROS production was concomitant with the interaction of ferric ions and PrP106–126. To address this, DCFH-DA was included in cell growth media, which were treated with ferric ions and PrP106–126 as described above. The results showed that the ROS increased significantly following exposure to Fe3+ and PrP106–126 (Fig. 2) when compared to the control group.
Fig. 2.
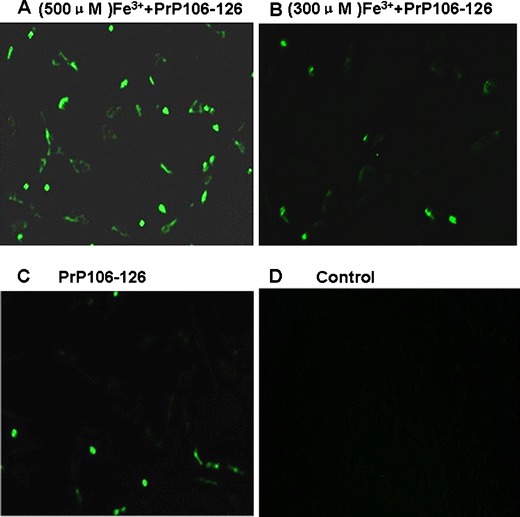
The treated cells were used for measuring ROS release with the DCFH-DA assay, as described in Section “DCFH-DA Assay and Visual Detection.” N2a cells were cultured on cover slips in a 12-well plate for 12 h, then treated with Fe3+ (final concentrations 500 μM) and PrP106–126 (final concentration 50 μM) for 12. a The fluorescence intensity had increased significantly (500 μM Fe3+ + PrP106–126). b Some fluorescence had emerged (300 μM Fe3+ + PrP106–126). c Some fluorescence had emerged (PrP106–126 only). d There is no fluorescence (control)
The Mitochondria of N2a Cells were Damaged Significantly under Oxidative Stress
Because the brain contains low levels of antioxidant enzymes and high levels of easily oxidized substrates, the brain is highly susceptible to oxidative damage (Sun et al. 2007). Oxidative stress, ubiquitination defects, and mitochondrial dysfunction are commonly associated with neurodegeneration. Also, NADH dehydrogenase is an essential component of the mitochondrial respiratory chain; it catalyzes the first step of intramitochondrial or intracellular NADH oxidation (or NAD+ reduction). NADH dehydrogenase participates not only in cell respiration, but also in cellular/organismal reactive oxygen species homeostasis, apoptosis initiation, or modulation (Dlasková et al. 2008).
To evaluate the damage of N2a cells after treatment with Fe3+ and PrP106–126, we focused on changes in the mitochondrial respiratory chain. The NADH dehydrogenase concentration was measured using a NADH dehydrogenase quantification kit. Further investigation involved extensive electron microscopic morphometric analyses of mitochondria. We thus studied the mitochondrial structure damage using a TEM.
The results proved that when N2a cells were treated with ferric ions and the PrP106–126 peptide, mitochondrial NADH dehydrogenase concentrations decreased significantly (Fig. 3). Also, the relative NADH dehydrogenase concentration decreased to nearly 60 % of its previous concentration in the mitochondria of cells that were treated with 500 μM Fe3+ and 50 μM PrP106–126 peptide. In other words, nearly 40 % of the mitochondrial respiratory chain demonstrated a loss of function. This may be one of causes of cell damage, particularly mitochondrial damage in cells damage, neuron cells apoptosis, or cell death. Morphometric analyses of the mitochondria confirmed these results. All mitochondria in the treated cells had been damaged to various degrees. Many mitochondria demonstrated a loss of internal structure, as well as poorly defined and sparse cristae (Fig. 4). The mitochondria from treated cells were also significantly larger than the control group counterparts. The mitochondria in B, C, and D groups (Fig. 4) were approximately 28.6–42.9 % larger than those in the control group (data not shown).
Fig. 3.
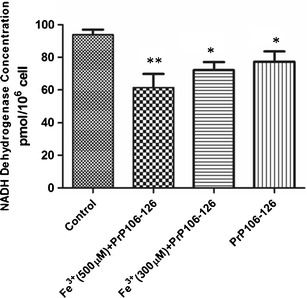
The NADH dehydrogenase concentrations in mitochondria. The NADH dehydrogenase concentrations were measured using the NADH dehydrogenase quantification kit. The NADH dehydrogenase concentrations of N2a cell mitochondria had decreased in different groups, respectively. * p < 0.05 vs. control; ** p < 0.01 vs. control
Fig. 4.

Morphometric analyses of mitochondria. N2a cells were fixed with glutaraldehyde, followed by negative staining with 2 % uranyl acetate. All micrographs were caught by transmission electron microscope (TEM) JEM-1230. The arrow shows the mitochondria. The scale bar represents 500 nm
Discussion
This study has shown that after treatment with ferric ions and PrP106–126, normal PrPC could be transformed into an abnormal form with protease K-resistance (Fig. 1). It is well known that resistance to PK digestion is an important property of PrPSc as an infectious factor in prion diseases. So, this PK-resistant protein may make the accumulation of more abnormal protein in cells possible. This protein may be also a cause of problematic cell damage. In this study, normal PrPC was transformed into a PK-resistant form due to the addition of Fe3+ and PrP106–126. Notably, the concentrations of the PK-resistant proteins were closely related to the concentrations of Fe3+ (Fig. 1a). The PMCA procedure may be an accelerator in this process. Similar results also appeared in N2a cells that were treated with Fe3+ and PrP106–126 (Fig. 1a). The DCFH-DA assay proved that intracellular ROS production had increased (Fig. 2a). Further, the amount of ROS, which manifested in fluorescence intensity in this study, was also correlated with the high Fe3+ concentration (Fig. 2). A prior study proposed that iron-induced oxidative stress may have partly contributed to neurodegeneration in TSEs (Kim et al. 2000). Our results proved that it was possible that iron-induced oxidative stress was one of the many triggers that transformed PrPC into PrPSc in prion diseases. This is so despite the fact that in this study, the PrP106–126 peptide was needed as an artificial PrPSc agent.
To further investigate the possible relationship between oxidative stress and neurodegeneration in prion diseases, we studied the molecular mechanism of N2a cell damage under the oxidative stress induced by Fe3+. In prior research, mitochondrial dysfunction has been proposed as a major factor in the pathogenesis of sporadic and familial PD (Valente et al. 2004). Further, NADH dehydrogenase is an essential component in the mitochondrial respiratory chain (Dlasková et al. 2008). So we focused here on the change in the mitochondrial respiratory chain and the mitochondrial structure damage to evaluate the damage of N2a cells after treatment with Fe3+ and PrP106–126. The results proved that the NADH dehydrogenase concentrations in the mitochondria decreased significantly (Fig. 3). The relative NADH dehydrogenase concentration decreased to nearly 60 % of the previous concentration in cell mitochondria. In other words, nearly 40 % of the mitochondrial respiratory chain lost its function. This loss may be one of the causes of cell damage, particularly mitochondrial damage, neuron cells apoptosis, or cell death. The morphometric analyses of mitochondria confirmed these results. Many mitochondria demonstrated damage to varying degrees, which included a loss of internal structure, as well as poorly defined and sparse cristae (Fig. 4).
In conclusion, oxidative stress may cause normal PrPC to transform into a PK-resistant protein, inducing further damage to cell mitochondria. When the mitochondrial damage appeared, the function of the mitochondrial respiratory chain also decreased. As a result, the ROS accumulated further. This cycle may be the cause of cell death or apoptosis. In other words, oxidative stress is one of the factors that induces prion disease. Further, the mitochondrial damage caused by oxidative stress marks the initial progression of the disease.
Acknowledgments
This work was supported by the Natural Science Foundation of China (project no. 31001048 and no. 31172293), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP, project no. 20100008120002), and the Foundation of Chinese Ministry of Science and Technology (project no. 2011BAI15B01), and the Program for Cheung Kong Scholars and Innovative Research Team in University of China (no. IRT0866).
References
- Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP Sc. J Mol Biol. 2005;346(2):645–659. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- Brazier MW, Lewis V, Ciccotosto GD, Klug GM, Lawson VA, Cappai R, Ironside JW, Masters CL, Hill AF, White AR. Correlative studies support lipid peroxidation is linked to PrPres propagation as an early primary pathogenic event in prion disease. Brain Res Bull. 2006;68(5):346–354. doi: 10.1016/j.brainresbull.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Castilla J, Saá P, Morales R, Abid K, Maundrell K, Soto C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Method Enzymol. 2006;412:3–21. doi: 10.1016/S0076-6879(06)12001-7. [DOI] [PubMed] [Google Scholar]
- Chen B, Morales R, Barria MA, Soto C. Estimating prion concentration in fluids and tissues by quantitative PMCA. Nat methods. 2010;7(7):519–520. doi: 10.1038/nmeth.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG. Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. NeuroToxicology. 2006;27(5):777–787. doi: 10.1016/j.neuro.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Dlasková A, Hlavatá L, Ježek P. Oxidative stress caused by blocking of mitochondrial Complex I H+ pumping as a link in aging/disease vicious cycle. Int J Biochem Cell Biol. 2008;40(9):1792–1805. doi: 10.1016/j.biocel.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Fioriti L, Quaglio E, Massignan T, Colombo L, Stewart RS, Salmona M, Harris DA, Forloni G, Chiesa R. The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci. 2005;28(1):165–176. doi: 10.1016/j.mcn.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Haigh CL, McGlade AR, Lewis V, Masters CL, Lawson VA, Collins SJ. Acute exposure to prion infection induces transient oxidative stress progressing to be cumulatively deleterious with chronic propagation in vitro. Free Radical Bio Med. 2011;51:594–608. doi: 10.1016/j.freeradbiomed.2011.03.035. [DOI] [PubMed] [Google Scholar]
- Kim NH, Park SJ, Jin JK, Kwon MS, Choi EK, Carp RI, Kim YS. Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Brain Res. 2000;884(1–2):98–103. doi: 10.1016/S0006-8993(00)02907-3. [DOI] [PubMed] [Google Scholar]
- Kozlowski H, Janicka-Klos A, Brasun J, Gaggelli E, Valensin D, Valensin G. Copper, iron, and zinc ions homeostasis and their role in neurodegenerative disorders (metal uptake, transport, distribution and regulation) Coordin Chem Rev. 2009;253(21):2665–2685. doi: 10.1016/j.ccr.2009.05.011. [DOI] [Google Scholar]
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol. 1999;1(6):358–361. doi: 10.1038/14053. [DOI] [PubMed] [Google Scholar]
- Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Rev. 2002;38(3):328–339. doi: 10.1016/S0165-0173(01)00150-3. [DOI] [PubMed] [Google Scholar]
- Panza G, Stöhr J, Dumpitak C, Papathanassiou D, Weiß J, Riesner D, Willbold D, Birkmann E. Spontaneous and BSE-prion-seeded amyloid formation of full length recombinant bovine prion protein. Biochem Bioph Res Co. 2008;373(4):493–497. doi: 10.1016/j.bbrc.2008.06.059. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Saá P, Castilla J, Soto C. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol. 2005;299:53–65. doi: 10.1385/1-59259-874-9:053. [DOI] [PubMed] [Google Scholar]
- Saretzki G. Telomerase, mitochondria and oxidative stress. Exp Gerontol. 2009;44(8):485–492. doi: 10.1016/j.exger.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Seo JS, Seol JW, Moon MH, Jeong JK, Lee YJ, Park SY. Hypoxia protects neuronal cells from human prion protein fragment–induced apoptosis. J Neurochem. 2010;112(3):715–722. doi: 10.1111/j.1471-4159.2009.06496.x. [DOI] [PubMed] [Google Scholar]
- Sun K, Johnson BS, Gunn TM. Mitochondrial dysfunction precedes neurodegeneration in mahogunin (Mgrn1) mutant mice. Neurobiol Aging. 2007;28(12):1840–1852. doi: 10.1016/j.neurobiolaging.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Zheng W, Wang L, Hong Y, Sha Y. PrP106-126 peptide disrupts lipid membranes: influence of C-terminal amidation. Biochem Bioph Res Co. 2009;379(2):298–303. doi: 10.1016/j.bbrc.2008.12.049. [DOI] [PubMed] [Google Scholar]