

Abstract
HIV proteinase inhibitors reduce the levels of Leishmania parasites in vivo and in vitro, but their biochemical target is unknown. We have identified an ortholog of the yeast Ddi1 protein as the only member of the aspartic proteinase family in Leishmania parasites, and in this study we investigate this protein as a potential target for the drugs. To date, no enzyme assay has been developed for the Ddi1 proteins, but Saccharomyces cerevisiae lacking the DDI1 gene secrete high levels of protein into the medium. We developed an assay in which these knockout yeast were functionally complemented to low secretion by introduction of genes encoding Ddi1 orthologs from Leishmania major or humans. Plasmid alone controls gave no complementation. Treatment of the Ddi1 transformants with HIV proteinase inhibitors showed differential effects dependent on the origin of the Ddi1. Dose responses allowed calculation of IC50 values; e.g., for nelfinavir, of 3.4 μM (human Ddi1) and 0.44 μM (Leishmania Ddi1). IC50 values with Leishmania constructs mirror the potency of inhibitors against parasites. Our results show that Ddi1 proteins are targets of HIV proteinase inhibitors and indicates the Leishmania Ddi1 as the likely target for these drugs and a potential target for antiparasitic therapy.—White, R. E., Powell, D. J., Berry, C. HIV proteinase inhibitors target the Ddi1-Like protein of Leishmania parasites.
Keywords: proteasome, HAART
A growing literature has emerged recently that shows the effects of HIV proteinase inhibitors (HIVPrIs), at therapeutically relevant doses, on other potential pathogens. Effects have been reported against Plasmodium species, (1–10), Candida (11–14), Leishmania species (15–17), and a range of other opportunistic infections (18). In HIV therapy, the inhibitors act against the retroviral aspartic proteinase, a homodimeric viral enzyme of the A2 proteinase family (19), with low nanomolar or subnanomolar Ki values (20). Therapeutic doses might reach plasma concentrations in the tens of micromolar range (4) that are sufficient to inhibit known monomeric (A1 family) aspartic proteinases from Candida albicans (11–13) and Plasmodium species (6, 7, 21). In Plasmodium, only food vacuole aspartic proteinases have been tested for susceptibility, although 10 members of the A1 proteinases (plasmepsins) are present in Plasmodium falciparum (22). HIVPrIs appear to act on erythrocytic stage parasites and pre-erythrocytic stages (1), and the production of food vacuole plasmepsins by the latter has not been established. However, HIVPrI have different effects on parasites from the general aspartic proteinase inhibitor pepstatin and are equally effective against food vacuole plasmepsin knockouts (3), implying another target for these drugs, as suggested by other investigators (8). HIVPrI interact antagonistically with artemisinin and related compounds (23) but synergistically with chloroquine (8, 24, 25). The latter interactions might be mediated via glutathione metabolism (24). Other inhibitor effects have been reported, including changes in CD36-mediated cytoadherence that cause a reduction in macrophage phagocytosis of parasite-infected erythrocytes (5). From these observations, it is clear that the exact mechanism and actual parasite proteins that are inhibited by the HIVPrI therapy remain to be confirmed. Of particular interest in the context of parasite aspartic proteinases as possible targets for the HIVPrIs are the effects on Leishmania species. Aspartic proteinase activity has been reported in extracts from these parasites (26, 27) but inspection of the completed genomes of Leishmania species reveals that these parasites do not encode A1 family, monomeric aspartic proteinases. The only aspartic proteinase present in these parasites seems to be an A2 family, retroviral-like proteinase that is related to the yeast Ddi1 protein and is part of a family of proteins conserved throughout the eukaryotes (28). Clearly, as the only representative of the aspartic proteinases in Leishmania, particularly as a retroviral-like enzyme, this protein is a potential target for the HIVPrIs in these parasites.
The Ddi1 protein of the yeast Saccharomyces cerevisiae is involved in a wide range of functions, including protein targeting to the proteasome, control of cell cycle, and suppression of protein secretion from the cell (29). Ddi1 has several distinct regions: an N-terminal ubiquitin-like domain (UBL), the central retroviral-type aspartic proteinase (RVP domain), and a C-terminal region containing a t-SNARE binding region and a ubiquitin associated (UBA) domain. In the Leishmania major ortholog (accession number XP_001687454), while the RVP domain is well conserved, there is only a short (∼40 residue) extension at the N terminus and an ∼80 residue C-terminal region that shows no similarity to that of the yeast Ddi1. In this family of proteins, the RVP domain is best conserved across the Eukaryotes, with major differences in the flanking regions in different taxonomic groups. The human ortholog is predicted to have an N-terminal UBL domain (although with little identity to that of Ddi1) but, like the Leishmania ortholog, lacks the C-terminal UBA (30) and t-SNARE binding domains and has instead a short (∼30 residue) region following the RVP that is conserved in mammalian Ddi1 orthologs but with no known function.
In this study, we demonstrate that both the human and Leishmania Ddi1 orthologs can complement the protein secretion phenotype of a yeast DDI1 knockout, and we show the effects of a number of HIVPrI on both enzymes. This work suggests that inhibition of the Leishmania Ddi1 ortholog might be the mechanism by which HIVPrI therapy mediates its antiparasitic action.
MATERIALS AND METHODS
Strains and plasmids
S. cerevisiae strains used in this study were BY4742 (Matα; his3D1; leu2D0; lys2D0; ura3D0) and the DDI1-knockout derivative Y16141 (BY4742; Matα; his3D1; leu2D0; lys2D0; ura3D0; YER143w::kanMX4; hereafter referred to as DDI1 knockout) and were supplied by EUROSCARF (Frankfurt, Germany).
S. cerevisiae strains were cultured using YEPD medium (10 mg/ml yeast extract, 20 mg/ml glucose, 20 mg/ml bactopeptone, 0.1 mg/ml uracil, and 0.1 mg/ml adenine) or selective minimal medium (1.6 mg/ml yeast nitrogen base that does not contain ammonium chloride or amino acids, 20 mg/ml glucose, 5 mg/ml ammonium chloride, 20 μg/ml histidine, 20 μg/ml lysine, and 20 μg/ml uracil).
Ddi1 constructs
The construct encoding the full-length Ddi1 from S. cerevisiae has been described previously (31). The equivalent constructs encoding the human and Leishmania orthologs were produced as follows. The full-length human gene was amplified from a human cDNA library by PCR using EasyA DNA polymerase (Stratagene, La Jolla, CA, USA) and forward (CCATGGATGCTGATCACCGTGTACTGC) and reverse (GGATCCTTAATGTTCTTTTCGTCCTGAATC) primers, which contained NcoI or BamHI sites at their respective 5′ ends. The conditions used were 95°C for 5 min followed by 30 cycles of 95°C for 1 min, 60°C for 1 min, and 72°C for 1 min, prior to a final elongation step of 72°C for 5 min. The gene encoding the full-length L. major Ddi1 ortholog was amplified from L. major friedlin strain genomic DNA in the same way using forward (CCATGGATGGACGAGCGCCAGCTAG) and reverse (GGATCCTTACGTGTCGAAGAGGAGCG) primers, which also contained NcoI or BamHI sites at their respective 5′ ends. The resultant PCR products were ligated into the pGEM-T vector (Promega, Southampton, UK) and sequenced. The DNA inserts were excised with NcoI and BamHI and cloned into yeast expression vector pVTL260 (32). The pVTL260 constructs were then transformed into the S. cerevisiae DDI1-knockout strain Y16141 (a BY4742 derivative) by the lithium acetate transformation method (33). Point mutation of the L. major active site aspartate to alanine was performed by PCR using the QuickChange Site-Directed mutagenesis kit (Stratagene) using the forward and reverse primers AAAGCCTTCGTCGCGTCCGGCGCGCAG and CTGCGCCGGACGCGACGAAGGCTTTC.
Proteinase inhibitors
Proteinase inhibitors were obtained from a number of sources, and their structures are shown in Fig. 1. The HIVPrIs lopinavir, indinavir, and tipranavir were purchased from Toronto Research Chemicals (Ontario, ON, Canada). Ritonavir was obtained from MoleKula UK Ltd. (Dorset, UK). Nelfinavir was obtained from Pfizer Inc. (New York, NY, USA), and saquinavir was received from Roche Products Ltd. (Welwyn Garden City, UK). Amprenavir was provided by GlaxoSmithKline (Harlow, UK). The general aspartic proteinase inhibitor isovaleryl pepstatin was purchased from Sigma-Aldrich Chemicals (Poole, UK).
Figure 1.
Structures of inhibitors.
Proteinase inhibitor assay
Selective minimal medium (1.5 ml in a 24-well plate) was inoculated with a colony from a selective minimal plate, grown overnight at 30°C. The plates were incubated with shaking in a Microtherm plate shaker incubator (Camlab, Cambridge, UK), for 48 h at 30°C to an attenuance at 600 nm of ∼2 in the presence or absence of inhibitor. After this time, 1 ml of culture was removed into a preweighed tube, and the supernatant was separated from whole cells by centrifugation at 13,000 rpm for 5 min. The supernatant was removed, and the protein content was determined using the RC DC protein assay kit (Bio-Rad, Hemel Hempstead, UK) with BSA as the standard. The cell pellet was dried at 100°C and weighed so that the secreted protein concentrations determined could be normalized by expressing the protein concentration as milligrams of protein secreted per milligram of dry weight. Each experiment was repeated several times (as shown in the figure legends), and values for the means ± se were calculated. IC50 values were calculated from dose response titrations with IC50s generated with the GraFit 5.1 data analysis package (Erithacus Software Ltd., Surrey, UK). Inhibition of protein secretion by compound was determined using a 4-parameter logistic fit with Eq. 1:
| (1) |
where a is the fitted uninhibited value minus the background, x is the inhibitor concentration, s is the Hill slope factor, and B is the value of a noninhibited control sample.
Yeast cell morphology
Yeast cells were grown for 24 h at 30°C in selective minimal medium, either in the presence or absence of 25 μM inhibitors, and their morphology was examined using a Leitz Diaplan phase contrast microscope at ×100 magnification (Leica Microsystems, Wetzlar, Germany). Images were captured using a ProgRes C5 digital camera (Jenoptik, Jena, Germany).
RESULTS
We have confirmed previously an earlier report (34) that DDI1-knockout yeast show significantly higher levels of protein secretion than the wild-type parent and that lower levels of secretion are restored by complementation with the DDI1 gene on plasmid pVTL260 (31). In this study, we cloned the genes encoding the full-length Leishmania and human orthologs of Ddi1 into the same plasmid. Figure 2 shows that these constructs also can restore secretion to wild-type, low levels. Plasmid pVTL260 alone does not produce any effect. A mutant of the Leishmania protein, in which the putative active site aspartic acid residue (D58, functionally equivalent to D220 in S. cerevisiae Ddi1) was changed to an alanine, was incapable of reverting yeast to the low-secretion phenotype. This indicates that catalytic activity is likely to be important for this phenomenon (as we have previously demonstrated with S. cerevisiae Ddi1; ref. 31).
Figure 2.

Protein secretion by yeast strains. Levels of protein secretion per milligram dry weight of cells is shown for strain BY4742 (n=24), DDI1-knockout strain Y16141 (n=24), and strain Y16141 transformed with plasmid alone (pVTL260; n=15) or the constructs encoding Ddi1 orthologs from S. cerevisiae (Sc Ddi1; n=24), human (H Ddi1; n=6), L. major (Lm Ddi1; n=26), and the active site mutant of the L. major protein (Lm Ddi1 D58A; n=20). Bars represent means ± se.
We next investigated the ability of aspartic proteinase inhibitors (25 μM) to affect protein secretion, since these compounds might also inhibit the catalytic activity of the Ddi1-like proteins. The general aspartic proteinase inhibitor isovaleryl pepstatin was tested along with 7 HIVPrIs against wild-type BY4742 yeast and the Y16141-knockout yeast cells complemented with S. cerevisiae Ddi1, the human ortholog, or the Leishmania protein (Table 1 and Fig. 3). Several inhibitors produced high levels of secretion in the complemented yeast, clearly blocking the complementation by the Ddi1-like proteins. It should be noted that the level of the effect and the apparent relative potencies of individual inhibitors varies among the different constructs, thus indicating that the Ddi1-like proteins, rather than background factors in the yeast strain (including numerous yeast A1 family aspartic proteinases), are the targets of the inhibitors.
Table 1.
Percentage inhibition and IC50 values for wild-type S. cerevisiae and yeast complemented with Ddi1 family proteins
| Inhibitor | BY4742, inhibition at 25 μM (%) |
Y16141 Ddi1 |
Y16141 human |
Y16141 Leishmania |
|||
|---|---|---|---|---|---|---|---|
| Inhibition at 25 μM (%) | IC50 (μM) | Inhibition at 25 μM (%) | IC50 (μM) | Inhibition at 25 μM (%) | IC50 (μM) | ||
| Amprenavir | 91 | 95 | 2.8 ± 0.2 | 9 | − | 100 | 5.7 ± 0.7 |
| Indinavir | 36 | 24 | − | 38 | − | 100 | 3.0 ± 0.2 |
| Isovaleryl pepstatin | 7 | 8 | − | 14 | − | 100 | 14 ± 3 |
| Lopinavir | 6 | 6 | − | 0 | − | 38 | 56 ± 4 |
| Nelfinavir | 50 | 40 | − | 97 | 3.4 ± 0.4 | 100 | 0.44 ± 0.03 |
| Ritonavir | 1 | 0 | − | 3 | − | 22 | 380 ± 140 |
| Saquinavir | 31 | 24 | − | 18 | − | 100 | 2.8 ± 0.3 |
| Tipranavir | 58 | 86 | − | 90 | 1.5 ± 0.2 | 97 | 2.9 ± 0.2 |
Results for the wild-type strain BY4742 are shown along with the DDI1-knockout strain Y16141 complemented with the Ddi1 homologs from S. cerevisiae (Ddi1), human, or L. major. IC50 values are given as means ± se (n=6).
Figure 3.
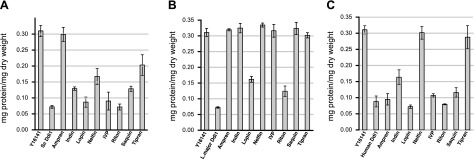
Effect of proteinase inhibitors on secretion. Proteinase inhibitors (25 μM) were assayed against Y16141 cells expressing Ddi1 orthologs from S. cerevisiae (A), L. major (B), and human (C). Knockout strain Y16141 and the strain complemented with the Ddi1 ortholog from each species are shown in the absence of inhibitors as the first two bars in each graph. Bars represent means ± se; n = 6 for all inhibitors. Ampren, amprenavir; Indin, indinavir; Lopin, lopinavir; Nelfin, nelfinavir; IVP, isovaleryl pepstatin; Riton, ritonavir; Saquin, saquinavir; Tipran, tipranavir.
For those inhibitors that gave knockout levels of secretion in the complemented strains, we assayed the effects over a range of concentrations in order to establish IC50 values for their activities. An example of such a titration for nelfinavir against the Leishmania complemented yeast is shown in Fig. 4, and the calculated IC50 values are shown in Table 1. Again, the differential activity of these inhibitors against the different target enzymes is evident.
Figure 4.

IC50 determination for nelfinavir against Y16141 expressing the L. major Ddi1 ortholog. Different concentrations of inhibitor were added, and protein secretion was measured as before. For each concentration, the assay was repeated 6 times. Bars represent means ± se. Using Eq. 1, the IC50 was calculated as 0.44 ± 0.03, Hill slope factor 0.72 ± 0.04.
In the course of these experiments, we also observed the yeast cultures microscopically. In cultures of DDI1-knockout yeast transformed with plasmid alone or plasmid encoding either the S. cerevisiae, Leishmania, or human Ddi1 orthologs, cell size showed little variation from that seen with the wild-type yeast (Fig. 5A–D, shown for Leishmania and human compared to wild-type and plasmid only). However, in cultures of constructs encoding the active site mutants of the Saccharomyces or Leishmania proteins, cells with greatly enlarged morphologies can be observed (∼80 and 80–85% of cells, respectively), and cells appear to have a tendency to clump together (Fig. 5E, F). Similar effects were seen when constructs expressing the equivalent nonmutant proteins were treated with concentrations of HIVPrIs expected to abolish their catalytic activity [e.g., 25 μM amprenavir (Saccharomyces; ∼75% of cells) and either amprenavir or nelfinavir (Leishmania; 85–90 and ∼95% of cells, respectively), as shown in Fig. 5G–I]. These inhibitors showed no effects on knockout yeast transformed with plasmid alone or on yeast transformed to express the human ortholog of Ddi1 (Fig. 5J–L).
Figure 5.

Morphology of yeast cells. Cells were grown in the presence or absence of amprenavir or nelfinavir (25 μM). A) Wild-type BY4742 cells. B–L) Y16141 cells transformed with pVTL260 plasmid (B), the L. major construct (C), the human construct (D), the Saccharomyces active site mutant D220A construct (E), the L. major active site mutant D58A construct (F), the Saccharomyces Ddi1 construct in the presence of 25 μM amprenavir (G), the Leishmania construct in the presence of 25 μM amprenavir (H), the Leishmania construct in the presence of 25 μM nelfinavir (I), pVTL260 plasmid in the presence of 25 μM amprenavir (J), pVTL260 plasmid in the presence of 25 μM nelfinavir (K), and the human construct in the presence of 25 μM nelfinavir (L). Scale bars = 20 μM.
DISCUSSION
Here we show that the human and L. major Ddi1 orthologs can complement the secretion phenotype of the DDI1-knockout yeast strain Y16141, to restore wild-type, low secretion levels. Previously, we have shown that for complementation by S. cerevisiae Ddi1 itself, both the putative active site aspartate residue (and thus the proposed catalytic activity) and the N-terminal 179 aa contribute to the low-secretion phenotype and that both are required for full complementation (31). The full complementation by the human ortholog is consistent with these previous results, since it possesses an N-terminal UBL domain and the RVP. However, the full complementation by the Leishmania protein, which lacks the features of the Ddi1 N terminus and has only an ∼40-aa sequence upstream of the RVP, is, perhaps, surprising. At this point, we can only speculate that other factors in the Leishmania protein permit this full complementation in the absence of the Ddi1-like N-terminal residues.
A mutant of the putative active site aspartic acid residue in the L. major Ddi1-like protein abolishes complementation and produces a high-secretion phenotype. The yeast cells expressing Saccharomyces or Leishmania active site mutants of the Ddi1 proteins also contain cells that differ in their morphology from wild-type yeast, DDI1-knockout yeast, and cells complemented with nonmutant Ddi1 orthologs. When the putative proteolytic activity is abolished by inhibition, rather than by mutation, a similar effect is observed with enlarged cells that appear to show a defect in septation and/or cytokinesis (Fig. 5). Thus, either the absence of Ddi1-like protein or the presence of orthologs likely to be active catalytically give a normal morphology, whereas the presence of catalytically inactivated Saccharomyces or Leishmania proteins can give rise to the abnormal morphology. At this point, we can only speculate on this phenomenon, but perhaps the enlarged phenotype results from an important interaction that is initiated in these cases, but which cannot proceed normally due to the lack of catalysis. Enlarged cells might be a result of a block in cell division, and yeast Ddi1 is known to have a role in cell-cycle control through UBL-mediated associations with the proteasome (35–38), but the lack of such UBL domains on the Leishmania protein makes this an unlikely factor here. However, in another study, mutations in the active site of yeast Ddi1 were implicated in checkpoint regulation (29). In addition, overexpression of the S. cerevisiae Ddi1 protein in yeast with a temperature-sensitive mutation in the Sec9 t-SNARE has been reported to result in the accumulation of low-density secretory vesicles (34). We cannot currently assess the relevance of this finding for our observations, but our enlarged cells continue to secrete proteins into the medium at a high rate (Figs. 2 and 3B) despite their swollen appearance. Morphological changes in Leishmania amazonensis parasites treated in culture with nelfinavir have been observed by transmission electron microscopy and exhibited cytoplasmic shrinking and an increase in the number of vesicles (17), which might show some parallels with the yeast morphology observed here. In addition, the effects of inhibitors on cellular proliferation (17), and indications that antiproliferative effects on promastigotes and axenic amastigoes might be due to a blockage in cell division (39) are consistent with their action on a Ddi1-like target.
However the complementation occurs, the Leishmania protein restores wild-type secretion levels to DDI1-knockout yeast, and this effect can be blocked by both the general aspartic proteinase inhibitor isovaleryl pepstatin and by HIVPrIs. To make a detailed assessment of the interactions of HIVPrIs with the Ddi1 family proteins, it would be desirable to assay the effects kinetically using purified target proteins. However, extensive attempts to establish such a kinetic assay using recombinant proteins have, thus far, proved unsuccessful. As a result, our indirect assay using the yeast secretion phenotype is currently the best-available test of inhibitor action against these proteins.
The inhibitory potencies, as shown in Table 1, show a general correlation with reported growth inhibition by HIVPrIs in L. major (IC50: saquinavir, 7 μM; indinavir, 8 μM; ref. 15) and, to a lesser extent, the effects on inhibition of cellular growth and multiplication with L. amazonensis (IC50; nelfinavir, 15 μM; lopinavir, 16 μM; amprenavir, 62 μM; ref. 17). Leishmania infantum has been reported to be relatively resistant to the action of HIVPrIs compared to L. major (17), but sensitivity of L. infantum and Leishmania donovani to nelfinavir in the tens of micromolar range has also been reported (16). Variations in the Ddi1-like proteins in the individual Leishmania species might produce corresponding differences in inhibitory sensitivity, but this condition is hard to assess. No sequence exists for the L. amazonensis protein, but the L. infantum Ddi1 ortholog differs from that of L. major by 11 residues, 3 of which lie in the RVP domain, while the Leishmania mexicana variant shows 19 changes compared to L. major (6 within the RVP domain), and the protein from Leishmania braziliensis is significantly longer at the N terminus and contains a number of short insertions relative to the other Leishmania proteins. Further work will be required to assess whether the differences in sequence are sufficient to explain any differences the inhibitor sensitivity of the parasites or whether other factors, for instance cellular uptake, might be involved.
Such considerations are also important in comparing previous experiments with parasites in culture (15, 17) with our experimental protocol using yeast transformants to assess inhibition of the individual target proteins that they express. The yeast cell wall will possess different compound permeability properties to the native Leishmania cell membrane, and Saccharomyces species also possess a range of ABC drug transporters in their cell membranes (40), which might result in intracellular exposure of the Ddi1 orthologs to concentrations of inhibitor that differ from those added to the yeast medium.
Given the caveats related to inhibitor uptake in our yeast system, the most potent inhibitors against the Leishmania protein appeared to be nelfinavir, followed by saquinavir, tipranavir, indinavir, and amprenavir (Table 1). Against the human protein, tipranavir appears the most potent, followed by nelfinavir, while S. cerevisiae Ddi1 is most susceptible to amprenavir. Thus, our results indicate a better therapeutic window for the Leishmania protein compared to the human ortholog for inhibitors such as saquinavir, indinavir, and amprenavir. Previous studies have indicated direct effects of HIVPrIs on the proteasome and its proteolytic activities (41–43). In our yeast assay system, the yeast proteasome, along with yeast pepsin family aspartic proteinases, are constant factors, so differential sensitivity indicates activity of the HIVPrIs against the Ddi1-like proteins. Our work would, therefore, indicate the possibility of a double action for HIVPrIs both on the proteasome directly (as previously reported) and on the Ddi1-like proteins that interact with the proteasome (29).
The HIVPrIs are well-established therapeutics that have undergone extensive biochemical, toxicity, and clinical testing that constitute major expenses in the development of new drugs. The extension of their use to new neglected disease targets, such as leishmaniasis, requires an understanding of their biochemical targets in the parasites. Our results show that the Leishmania Ddi1-like protein is susceptible to the action of HIVPrIs and leads us to propose this protein as a target of HIVPrIs in the parasites themselves. This proposition is based on the inhibition data presented here and on our bioinformatic analysis that confirms this protein as the only aspartic proteinase encoded by the L. major genome. By extension, since the RVP domain found in this protein is highly conserved throughout the eukaryotes (28), we would further suggest that the equivalent proteins from Plasmodium and Candida albicans are also possible targets for the HIVPrIs in these pathogens. Here we have highlighted the importance of these potential targets, established a simple inhibitor screening protocol and produced a human counterscreen using the human ortholog, that will be useful for assessment of further inhibitors that might be developed to target the parasite enzymes.
Acknowledgments
This work was supported by Biotechnology and Biological Sciences Research Council Collaborative Award in Science and Engineering BB/D526137/1 to R.E.W.
REFERENCES
- 1. Hobbs C. V., Voza T., Coppi A., Kirmse B., Marsh K., Borkowsky W., Sinnis P. (2009) HIV protease inhibitors inhibit the development of preerythrocytic-stage Plasmodium parasites. J. Infect. Dis. 199, 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peatey C. L., Andrews K. T., Eickel N., MacDonald T., Butterworth A. S., Trenholme K. R., Gardiner D. L., McCarthy J. S., Skinner-Adams T. S. (2010) Antimalarial asexual stage-specific and gametocytocidal activities of HIV protease inhibitors. Antimicrob. Agents Chemother. 54, 1334–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parikh S., Liu J., Sijwali P., Gut J., Goldberg D. E., Rosenthal P. J. (2006) Antimalarial effects of human immunodeficiency virus type 1 protease inhibitors differ from those of the aspartic protease inhibitor pepstatin. Antimicrob. Agents Chemother. 50, 2207–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skinner-Adams T. S., McCarthy J. S., Gardiner D. L., Hilton P. M., Andrews K. T. (2004) Antiretrovirals as antimalarial agents. J. Infect. Dis. 190, 1998–2000 [DOI] [PubMed] [Google Scholar]
- 5. Nathoo S., Serghides L., Kain K. C. (2003) Effect of HIV-1 antiretroviral drugs on cytoadherence and phagocytic clearance of Plasmodium falciparum-parasitised erythrocytes. Lancet 362, 1039–1041 [DOI] [PubMed] [Google Scholar]
- 6. Martins T. M., Domingos A., Berry C., Wyatt D. M. (2006) The activity and inhibition of the food vacuole plasmepsin from the rodent malaria parasite Plasmodium chabaudi. Acta Trop. 97, 212–218 [DOI] [PubMed] [Google Scholar]
- 7. Andrews K. T., Fairlie D. P., Madala P. K., Ray J., Wyatt D. M., Hilton P. M., Melville L. A., Beattie L., Gardiner D. L., Reid R. C., Stoermer M. J., Skinner-Adams T., Berry C., McCarthy J. S. (2006) Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria. Antimicrob. Agents Chemother. 50, 639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Skinner-Adams T. S., Andrews K. T., Melville L., McCarthy J., Gardiner D. L. (2007) Synergistic interactions of the antiretroviral protease inhibitors saquinavir and ritonavir with chloroquine and mefloquine against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 51, 759–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lek-Uthai U., Suwanarusk R., Ruengweerayut R., Skinner-Adams T. S., Nosten F., Gardiner D. L., Boonma P., Piera K. A., Andrews K. T., Machunter B., McCarthy J. S., Anstey N. M., Price R. N., Russell B. (2008) Stronger activity of human immunodeficiency virus type 1 protease inhibitors against clinical isolates of Plasmodium vivax than against those of P. falciparum. Antimicrob. Agents Chemother. 52, 2435–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Redmond A. M., Skinner-Adams T., Andrews K. T., Gardiner D. L., Ray J., Kelly M., McCarthy J. S. (2007) Antimalarial activity of sera from subjects taking HIV protease inhibitors. AIDS 21, 763–765 [DOI] [PubMed] [Google Scholar]
- 11. Cassone A., De Bernardis F., Torosantucci A., Tacconelli E., Tumbarello M., Cauda R. (1999) In vitro and in vivo anticandidal activity of human immunodeficiency virus protease inhibitors. J. Infect. Dis. 180, 448–453 [DOI] [PubMed] [Google Scholar]
- 12. Cassone A., Tacconelli E., De Bernardis F., Tumbarello M., Torosantucci A., Chiani P., Cauda R. (2002) Antiretroviral therapy with protease inhibitors has an early, immune reconstitution-independent beneficial effect on Candida virulence and oral candidiasis in human immunodeficiency virus-infected subjects. J. Infect. Dis. 185, 188–195 [DOI] [PubMed] [Google Scholar]
- 13. Cassone A., Cauda R. (2002) HIV proteinase inhibitors: do they really work against Candida in a clinical setting? Trends Microbiol. 10, 177–178 [DOI] [PubMed] [Google Scholar]
- 14. Skrbec D., Romeo D. (2002) Inhibition of Candida albicans secreted aspartic protease by a novel series of peptidomimetics, also active on the HIV-1 protease. Biochem. Biophys. Res. Commun. 297, 1350–1353 [DOI] [PubMed] [Google Scholar]
- 15. Savoia D., Allice T., Tovo P. A. (2005) Antileishmanial activity of HIV protease inhibitors. Int. J. Antimicrob. Agents 26, 92–94 [DOI] [PubMed] [Google Scholar]
- 16. Trudel N., Garg R., Messier N., Sundar S., Ouellette M., Tremblay M. J. (2008) Intracellular survival of Leishmania species that cause visceral leishmaniasis is significantly reduced by HIV-1 protease inhibitors. J. Infect. Dis. 198, 1292–1299 [DOI] [PubMed] [Google Scholar]
- 17. Santos L. O., Marinho F. A., Altoe E. F., Vitorio B. S., Alves C. R., Britto C., Motta M. C., Branquinha M. H., Santos A. L., d'Avila-Levy C. M. (2009) HIV aspartyl peptidase inhibitors interfere with cellular proliferation, ultrastructure and macrophage infection of Leishmania amazonensis. PLoS One 4, e4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pozio E., Morales M. A. (2005) The impact of HIV-protease inhibitors on opportunistic parasites. Trends Parasitol. 21, 58–63 [DOI] [PubMed] [Google Scholar]
- 19. Barrett A. J., Rawlings N. D., Woessner J. F. (2004) Handbook of Proteolytic Enzymes, Vol. 1, Elsevier Academic, London [Google Scholar]
- 20. Wilson S. I., Phylip L. H., Mills J. S., Gulnik S. V., Erickson J. W., Dunn B. M., Kay J. (1997) Escape mutants of HIV-1 proteinase: enzymic efficiency and susceptibility to inhibition. Biochim. Biophys. Acta 1339, 113–125 [DOI] [PubMed] [Google Scholar]
- 21. Parikh S., Gut J., Istvan E., Goldberg D. E., Havlir D. V., Rosenthal P. J. (2005) Antimalarial activity of human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 49, 2983–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coombs G. H., Goldberg D. E., Klemba M., Berry C., Kay J., Mottram J. C. (2001) Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 17, 532–537 [DOI] [PubMed] [Google Scholar]
- 23. He Z., Chen L., You J., Qin L., Chen X. (2010) In vitro interactions between antiretroviral protease inhibitors and artemisinin endoperoxides against Plasmodium falciparum. Int. J. Antimicrob. Agents 35, 191–193 [DOI] [PubMed] [Google Scholar]
- 24. He Z., Chen L., You J., Qin L., Chen X. (2009) Antiretroviral protease inhibitors potentiate chloroquine antimalarial activity in malaria parasites by regulating intracellular glutathione metabolism. Exp. Parasitol. 123, 122–127 [DOI] [PubMed] [Google Scholar]
- 25. He Z., Qin L., Chen L., Peng N., You J., Chen X. (2008) Synergy of human immunodeficiency virus protease inhibitors with chloroquine against Plasmodium falciparum in vitro and Plasmodium chabaudi in vivo. Antimicrob. Agents Chemother. 52, 2653–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alves C. R., Corte-Real S., Bourguignon S. C., Chaves C. S., Saraiva E. M. (2005) Leishmania amazonensis: early proteinase activities during promastigote-amastigote differentiation in vitro. Exp. Parasitol. 109, 38–48 [DOI] [PubMed] [Google Scholar]
- 27. Valdivieso E., Dagger F., Rascon A. (2007) Leishmania mexicana: identification and characterization of an aspartyl proteinase activity. Exp. Parasitol. 116, 77–82 [DOI] [PubMed] [Google Scholar]
- 28. Krylov D. M., Koonin E. V. (2001) A novel family of predicted retroviral-like aspartyl proteases with a possible key role in eukaryotic cell cycle control. Curr. Biol. 11, R584–587 [DOI] [PubMed] [Google Scholar]
- 29. Gabriely G., Kama R., Gelin-Licht R., Gerst J. E. (2008) Different domains of the UBL-UBA ubiquitin receptor, Ddi1/Vsm1, are involved in its multiple cellular roles. Mol. Biol. Cell 19, 3625–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fatimababy A. S., Lin Y. L., Usharani R., Radjacommare R., Wang H. T., Tsai H. L., Lee Y., Fu H. (2010) Cross-species divergence of the major recognition pathways of ubiquitylated substrates for ubiquitin/26S proteasome-mediated proteolysis. FEBS J. 277, 796–816 [DOI] [PubMed] [Google Scholar]
- 31. White R. E., Dickinson J. R., Semple C. A. M., Powell D. J., Berry C. (2011) The retroviral proteinase active site and the N-terminus of Ddi1 are required for repression of protein secretion. FEBS Lett. 585, 139–142 [DOI] [PubMed] [Google Scholar]
- 32. Melcher K. (2000) A modular set of prokaryotic and eukaryotic expression vectors. Anal. Biochem. 277, 109–120 [DOI] [PubMed] [Google Scholar]
- 33. Ito H., Fukuda Y., Murata K., Kimura A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lustgarten V., Gerst J. E. (1999) Yeast VSM1 encodes a v-SNARE binding protein that may act as a negative regulator of constitutive exocytosis. Mol. Cell. Biol. 19, 4480–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ivantsiv Y., Kaplun L., Tzirkin-Goldin R., Shabek N., Raveh D. (2006) Unique role for the UbL-UbA protein Ddi1 in turnover of SCFUfo1 complexes. Mol. Cell. Biol. 26, 1579–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Diaz-Martinez L. A., Kang Y., Walters K. J., Clarke D. J. (2006) Yeast UBL-UBA proteins have partially redundant functions in cell cycle control. Cell. Div. 1, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplun L., Tzirkin R., Bakhrat A., Shabek N., Ivantsiv Y., Raveh D. (2005) The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol. Cell. Biol. 25, 5355–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bertolaet B. L., Clarke D. J., Wolff M., Watson M. H., Henze M., Divita G., Reed S. I. (2001) UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Biol. 8, 417–422 [DOI] [PubMed] [Google Scholar]
- 39. Valdivieso E., Rangel A., Moreno J., Saugar J. M., Canavate C., Alvar J., Dagger F. (2010) Effects of HIV aspartyl-proteinase inhibitors on Leishmania sp. Exp. Parasitol. 126, 557–563 [DOI] [PubMed] [Google Scholar]
- 40. Rogers B., Decottignies A., Kolaczkowski M., Carvajal E., Balzi E., Goffeau A. (2001) The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 3, 207–214 [PubMed] [Google Scholar]
- 41. Andre P., Groettrup M., Klenerman P., de Giuli R., Booth B. L., Jr., Cerundolo V., Bonneville M., Jotereau F., Zinkernagel R. M., Lotteau V. (1998) An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl. Acad. Sci. U. S. A. 95, 13120–13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Piccinini M., Rinaudo M. T., Anselmino A., Buccinna B., Ramondetti C., Dematteis A., Ricotti E., Palmisano L., Mostert M., Tovo P. A. (2005) The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antivir. Ther. 10, 215–223 [PubMed] [Google Scholar]
- 43. Piccinini M., Rinaudo M. T., Chiapello N., Ricotti E., Baldovino S., Mostert M., Tovo P. A. (2002) The human 26S proteasome is a target of antiretroviral agents. AIDS 16, 693–700 [DOI] [PubMed] [Google Scholar]