

Abstract
Hybridization plays an important evolutionary role in several groups of organisms. A phylogenetic approach to detect hybridization entails sequencing multiple loci across the genomes of a group of species of interest, reconstructing their gene trees, and taking their differences as indicators of hybridization. However, methods that follow this approach mostly ignore population effects, such as incomplete lineage sorting (ILS). Given that hybridization occurs between closely related organisms, ILS may very well be at play and, hence, must be accounted for in the analysis framework. To address this issue, we present a parsimony criterion for reconciling gene trees within the branches of a phylogenetic network, and a local search heuristic for inferring phylogenetic networks from collections of gene-tree topologies under this criterion. This framework enables phylogenetic analyses while accounting for both hybridization and ILS. Further, we propose two techniques for incorporating information about uncertainty in gene-tree estimates. Our simulation studies demonstrate the good performance of our framework in terms of identifying the location of hybridization events, as well as estimating the proportions of genes that underwent hybridization. Also, our framework shows good performance in terms of efficiency on handling large data sets in our experiments. Further, in analysing a yeast data set, we demonstrate issues that arise when analysing real data sets. Although a probabilistic approach was recently introduced for this problem, and although parsimonious reconciliations have accuracy issues under certain settings, our parsimony framework provides a much more computationally efficient technique for this type of analysis. Our framework now allows for genome-wide scans for hybridization, while also accounting for ILS. [Phylogenetic networks; hybridization; incomplete lineage sorting; coalescent; multi-labeled trees.]
Hybridization is believed to be an important evolutionary mechanism for several groups of eukaryotic organisms (Arnold 1997; Rieseberg 1997; Barton 2001; Mallet 2005, 2007). Evolutionary histories of species and genomes that involve hybridization are best modeled by phylogenetic networks, which account for both vertical and non-vertical evolutionary events (Nakhleh 2010). Additionally, trees that trace the evolution of different segments of the genome, also known as gene trees, grow within the branches of a phylogenetic network (Maddison 1997). This intertwined relationship between phylogenetic networks and the trees they contain naturally gave rise to a phylogeny-based approach to inferring phylogenetic networks from gene trees. In this approach, gene trees are compared, typically using a metric such as the subtree prune and redraft (SPR) distance, and the differences are taken as proxies for the amount and location of hybridization events (Nakhleh 2010).
However, in addition to hybridization, the incongruence among gene trees may be partly caused by incomplete lineage sorting (ILS), or deep coalescence events (Maddison 1997). Ignoring the presence of ILS could result in an over- or underestimation of the amount of hybridization events and/or wrong inference of the location of these events. Recent studies have documented large extents of ILS in groups of organisms across the Tree of Life (Syring et al. 2005; Pollard et al. 2006; Kuo et al. 2008; Than et al. 2008b; Cranston et al. 2009; White et al. 2009; Hobolth et al. 2011; Takuno et al. 2012). A wide array of methods have been developed for species-tree inference from gene-tree topologies when all incongruence is assumed to be due to ILS (see [Rannala and Yang 2008; Degnan and Rosenberg 2009; Liu et al. 2009] for recent surveys of such methods).
Relevant to this study are methods for inference under hybridization alone and under ILS alone that follow a parsimony approach: inferring the phylogenetic network with minimum number of reticulations in the former case, and inferring the phylogenetic tree that minimizes the amount of ILS in the latter case. This approach in both cases was proposed by Maddison (1997) and much progress has been made on developing methods for parsimonious reconciliations ever since, both in the case of hybridization (Bordewich and Semple 2004; MacLeod et al. 2005; Nakhleh et al. 2005; Beiko and Hamilton 2006) and ILS (Maddison and Knowles 2006; Than and Nakhleh 2009, 2010; Yu et al. 2011b, c). Nonetheless, the first class of methods does not account for ILS, and the latter does not account for hybridization. Accounting for both kinds of events is a very challenging task (Mallet 2005). Several attempts have been made in the last 5 years to handle both reticulation and ILS. Than et al. (2007) introduced a stochastic framework for computing the probability of a gene tree given a species tree under the coalescent, and in the presence of a single horizontal gene transfer (HGT) event. Meng and Kubatko (2009) introduced methods for estimating the contribution of hybridization using a model that allows for both hybridization and ILS. Kubatko (2009) further proposed using model selection with standard information criteria to identify hybridization in the presence of ILS. Joly et al. (2009) introduced a statistical approach for the same task based on genetic distances between sequences. Yu et al. (2011a) proposed extending the MDC (Minimize Deep Coalescences) criterion of (Maddison 1997) to detect hybridization despite ILS. However, these methods all focused on very limited cases: fewer than five taxa, one or two hybridization events, and a single allele samples per species.
It is important to note that another ubiquitous cause of gene tree incongruence is gene duplication and loss. Recent efforts have emerged for combining gene duplication/loss with ILS (Rasmussen and Kellis 2012) and for combining gene duplication/loss with HGT (Bansal et al. 2012), but incorporating duplication/loss with ILS and hybridization is beyond the scope of this work.
Most recently, Yu et al. (2012) proposed a method for computing the probability of gene-tree topologies given a phylogenetic network that is applicable to arbitrary numbers of taxa, arbitrary configurations of hybridization events, and any number of alleles sampled per species. Although this general framework allows for inference of hybridization in the presence of ILS, it currently suffers from two issues. First, to turn the work of Yu et al. into an inference method, there is a need to develop methods for searching the phylogenetic network space and optimizing branch lengths and inheritance probabilities. Second, the method is computationally very expensive; developing new algorithmic techniques to achieve scalability is imperative.
In this article, we present a parsimony framework for inferring hybridization in the presence of ILS that extends Maddison's proposal (Maddison 1997) to phylogenetic networks, and extend in novel ways the work of Yu et al. (2011a) to general networks. The computational contribution of this work is 2-fold: A parsimony criterion for reconciling a gene tree within the branches of a phylogenetic network so as to account for both hybridization and ILS, and a phylogenetic network search heuristic to enable inference of evolutionary histories from sets of gene-tree topologies. The framework only assumes knowledge of gene-tree topologies, which can be inferred by the method of choice of the practitioner, and infers a phylogenetic network with inheritance probabilities that correspond to proportions of genes involved in each of the hybridization events inferred. Our framework is general enough that it allows for multiple hybridizations (in any configuration), multiple alleles sampled per species, arbitrary divergence patterns following hybridizations, and methodologically no bounds on the numbers of leaves in the gene trees.
We demonstrate the performance of our framework in terms of estimating the hybridization events and inheritance probabilities on simulated data under different evolutionary settings. For most cases, the framework exhibits very good performance from a small number of loci. Further, we reanalyse a yeast data set and show the performance of the framework on biological data. We highlight two important issues: how to deal with uncertainty in the input gene trees, and the model selection problem that naturally arises when inferring phylogenetic networks. The speed of this parsimony framework makes it a good candidate for unrestricted analyses of multi-locus data sets, where hybridization is suspected, at least in order to obtain a first approximation to the true evolutionary history. Although parsimonious reconciliation of species/gene trees and inference under such a criterion is known to have consistency issues under certain settings (Than and Rosenberg 2011), parsimony remains a powerful approach in this domain, given its speed and good accuracy in many cases. We believe that our framework here can help in identifying good evolutionary hypotheses, which can be further analysed with more detailed approaches such as the one of Yu et al. (2012).
We have implemented our method and made it available in open-source form in the software package PhyloNet (Than et al. 2008a). The software package, as well as supporting documentation and a tutorial on its use, can be accessed at http://bioinfo.cs.rice.edu/PhyloNet.
Methods
Here we describe our parsimony criterion for reconciling gene trees within the branches of phylogenetic networks, and our heuristic search for inferring phylogenetic networks and inheritance probabilities under this criterion.
Phylogenetic Networks and Gene Trees
The coalescent model (Kingman 1982) views the evolution of multiple alleles of a locus backward in time. The multispecies coalescent generalizes the model to a phylogenetic tree that captures the evolution of multiple populations (Degnan and Rosenberg 2009). Under this model, a gene tree may disagree with a species tree due to ILS (Fig. 1A). Here, each gene tree models the evolution of a set of alleles at a single locus in multiple species, and all incongruence is assumed to be due to ILS.
Figure 1.
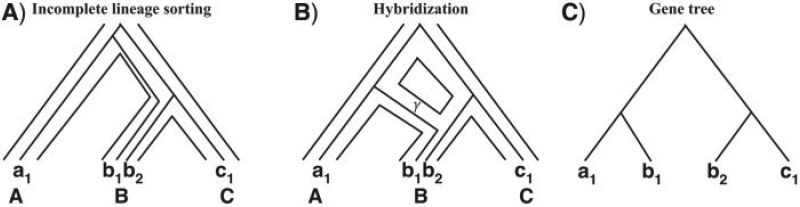
Gene trees within species trees and species networks. A) Under the multispecies coalescent model, a gene tree may be incongruent with the species tree due to ILS. B) When hybridization occurs between two species (or, populations), the species phylogeny takes the shape of a network, and a gene tree “grows” within the branches of the network. The variable γ corresponds to the probability of a lineage in the hybrid population being inherited from the “left” parent (1 − γ is the probability of inheritance from the “right” parent). C) ILS and hybridization can give rise to the same gene tree shape (or, topology).
When hybridization between two populations occurs, the evolutionary history of the species takes the form of a phylogenetic network, rather than a tree, so as to capture the contributions of genetic material from two parents (Fig. 1B). A phylogenetic network is a rooted, directed, acyclic graph whose leaves are labeled uniquely by a set of species. A phylogenetic network contains a unique node of in-degree 0 and out-degree 2 (the root), a set of nodes of in-degree 1 and out-degree 0 (the leaves), a set of nodes of in-degree 1 and out-degree 2 (the tree nodes), and a set of nodes of in-degree 2 and out-degree 1 (the reticulation nodes). Associated with every pair of reticulation edges (e1,e2) that are incident into a reticulation node are two real numbers γe1 and γe2 , respectively, such that γe1 + γe2 = 1. These parameters are interpreted as the inheritance probabilities: Given a lineage x at a reticulation node, it is inherited from one parent with probability γe1 and from other parent with probability γe2 (see Section 1 in the Supplementary Material for more details; doi:10.5061/dryad.sr534). If γe1 = 0 for a reticulation edge e1 incident into node x, this indicates that e1 is redundant and that no hybridization involves x.
The evolution of a gene within the branches of a phylogenetic network can be viewed backward in time, such that whenever a reticulation node is encountered, the gene traces one of the two parents with a certain probability (the inheritance probability). Figure 1 shows examples of a phylogenetic network on three species A, B, and C, and a gene tree with one allele sampled from A, two alleles sampled from B, and one allele sampled from C. An inheritance probability that is estimated at a value different from 0 and 1 indicates hybridization at the reticulation node, whereas a value of 0 or 1 imply that the reticulation node is redundant and can be replaced by a tree node attached to one of the two parents only.
Coalescent Histories and the MDC Criterion
We denote by V(g) and E(g) the sets of nodes and edges, respectively, of graph g, and by Ct(v) the subtree of tree t that is rooted at node v. For a phylogenetic network N, we denote by CN (v) the subgraph of N that is induced by all the nodes reachable (or, “under”) from v.
Given gene tree gt and species phylogeny ST (tree or network), a coalescent history is a function f : V(gt) → V(ST) such that the following conditions hold: 1) if w is a leaf in gt that is labeled by an allele from species x, then f (w) is the leaf in ST labeled with x; and 2) if w is a node in Cgt(v), then f (w) is a node in CST (f (v)). Figure 1A shows a coalescent history of the gene tree in Figure 1C within the branches of a species tree, whereas Figure 1B shows a coalescent history of the same gene tree yet within the branches of a species network.
Given a gene tree gt and a species phylogeny ST, and given a function f defining a coalescent history of gt within ST, the number of lineages in each branch in ST can be computed by inspection. For example, in Figure 1A, the number of lineages in the branch leading directly to taxon B is 2, whereas the number of lineages in the branch leading directly to C is 1. Given a coalescent history of a gene tree within the branches of a species tree, the number of extra lineages on a branch of the species tree is the number of lineages “exiting” the branch minus one. For example, the number of extra lineages on the branch incident with species B in Figure 1A is 1, since two lineages exit the branch. In fact, given the gene tree in Figure 1C and species tree in Figure 1A, the reconciliation given in panel A is the one with the smallest number of extra lineages (for that fixed species tree). Given a set of coalescent histories for a set of gene trees, the total number of extra lineages is obtained by summing the number of extra lineages over all gene trees. More formally, let us denote by XL(ST,gt) the number of extra lineages within an optimal coalescent history of gt within ST. For a set  of gene trees, we have
of gene trees, we have
| (1) |
Under parsimony, a reconciliation of the set of gene trees within the branches of a species tree that minimizes the total number of extra lineages over all gene trees provides an optimal evolutionary history of the gene genealogies for the given species tree; this is the minimizing deep coalescences (MDC) criterion proposed in Maddison (1997). Given a collection  of gene trees, the MDC (Minimizing Deep Coalescences) criterion (Maddison 1997) seeks the species tree ST* where
of gene trees, the MDC (Minimizing Deep Coalescences) criterion (Maddison 1997) seeks the species tree ST* where
For the inference problem, a species tree is sought so as to MDC over all possible (species) tree candidates. Efficient algorithms for solving this inference problem were recently introduced (Than and Nakhleh 2009, 2010; Yu et al. 2011b, 2011c).
ILS and Hybridization: MDC on Phylogenetic Networks
Although Maddison defined this criterion for species trees, we extend it naturally to phylogenetic networks, given that we defined the concept of coalescent histories on phylogenetic networks above. Than and Nakhleh (2009) defined a mapping between a species tree and a gene tree that yields the optimal coalescent history that results in the minimum number of extra lineages. The key principle is to let the gene lineages coalesce as “low” as possible as long as they are consistent with the topologies of the gene and species trees. However, a similar idea for obtaining the optimal coalescent history of a gene tree within the branches of a phylogenetic network does not work; we illustrate this issue in Section 2 in the Supplementary Material. Further, Than and Nakhleh (2009) devised exact algorithms for inferring species trees from collections of rooted, binary gene trees under the MDC criterion, which were later extended to handle cases of unrooted or non-binary gene trees and with arbitrary numbers of alleles sampled per species (Than and Nakhleh 2009; Yu et al. 2011b, 2011c). However, none of these algorithms apply directly to the case where the species phylogeny is a network with at least one reticulation node.
We recently introduced an approach for reconciling a gene tree within the branches of a species network based on the concept of a multi-labeled tree, or MUL-tree (Yu et al. 2012). A phylogenetic network can be converted to a MUL-tree by proceeding in a bottom-up fashion (leaves to root), replicating the subtree at a reticulation node every time such a node is encountered. Upon termination of this process (when the root is reached), the resulting structure is a rooted tree whose leaves are not necessarily uniquely labeled. Each of the four panels in Figure 2 shows the single MUL-tree that corresponds to the phylogenetic network in Figure 1B. Once the MUL-tree is obtained, the evolution of a gene tree is modeled by mapping the alleles it contains to the respective species from which they were sampled. Figure 2 shows the four possible allele mappings for the gene tree in Figure 1C. Given an allele mapping, the multispecies coalescent then proceeds in the standard manner within the branches of the MUL-tree. Every coalescent history of a set of alleles on a phylogenetic network corresponds to a coalescent history of an allele mapping on the corresponding MUL-tree. Consequently, the optimal number of extra lineages arising from reconciling a gene tree within the branches of a species network can be computed using a slightly modified application of the MDC criterion on the MUL-tree and the set of allele mappings (Yu et al. 2012). Our method is illustrated in Figure 3 and its full details are given in Section 3 in the Supplementary Material.
Figure 2.
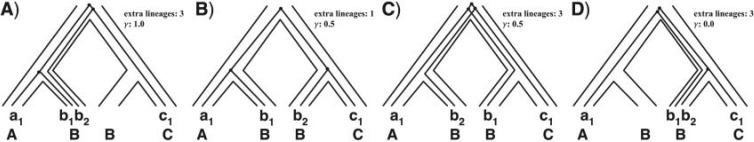
Phylogenetic networks and MUL-trees. The MUL-tree that corresponds to the phylogenetic network in Figure 1B. The four panels show the four possible allele mappings of the gene-tree in Figure 1C. The allele mapping in (B) corresponds to the coalescent history in Figure 1B. Values of 1.0 and 0.0 for γ in panels (A) and (D), respectively, indicate no support for hybridization in these two cases. The MUL-tree in panel (B) has the lowest number of extra lineages, and hence corresponds to the optimal reconciliation of the gene tree within the branches of the phylogenetic network.
Figure 3.

A schematic illustration of our method for computing an optimal coalescent history of a gene-tree within the branches of a phylogenetic network. The phylogenetic network is converted to a MUL-tree, and the alleles sampled are mapped in every possible way to the leaves of the MUL-tree. For each allele mapping, the number of extra lineages is computed on the MUL-tree, and the mapping that yields the minimum overall number corresponds to an optimal coalescent history.
Given a collection 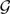 of gene trees, once the optimal coalescent histories for all of them are computed within the branches of a phylogenetic network N (using the MUL-tree approach), the inheritance probabilities associated with the reticulation nodes are estimated as follows. Let x be a reticulation node in N. Given the optimal coalescent histories computed, let lx be the number of lineages that trace the left parent in all the coalescent histories, and let rx be the number of lineages that trace the right parent in all the coalescent histories. Then, the probability associated with the left reticulation edge incident with x is lx/(lx + rx) and the probability associated with the right reticulation edge incident with x is rx/(lx + rx) (see Section 4 in the Supplementary Material for details of special cases).
of gene trees, once the optimal coalescent histories for all of them are computed within the branches of a phylogenetic network N (using the MUL-tree approach), the inheritance probabilities associated with the reticulation nodes are estimated as follows. Let x be a reticulation node in N. Given the optimal coalescent histories computed, let lx be the number of lineages that trace the left parent in all the coalescent histories, and let rx be the number of lineages that trace the right parent in all the coalescent histories. Then, the probability associated with the left reticulation edge incident with x is lx/(lx + rx) and the probability associated with the right reticulation edge incident with x is rx/(lx + rx) (see Section 4 in the Supplementary Material for details of special cases).
Searching the Network Space
The space of phylogenetic networks is very large, and it is infeasible to enumerate all networks in order to identify the optimal one under the MDC score. Instead, we employ a local search heuristic that searches the space of phylogenetic networks, while scoring them based on Equation (1). We denote by Ω(n, k) the space of phylogenetic networks that contain n taxa and k reticulation nodes. Suppose an optimal phylogenetic network with at most m reticulation nodes is sought. Our search strategy first searches the space Ω(n, 0) until some (potentially local) optimum is reached. The search then proceeds to Ω(n, 1), searches in that space until an optimum is reached, and then jumps to Ω(n, 2). This strategy continues until either an optimal network is reached in Ω(n, m), or the locally optimal score in Ω(n, k + 1) is not better than that in Ω(n, k) for some k < m.
The optimality scoring is done using the MUL-tree technique discussed above, and we now describe the topological operations that we employ to search the phylogenetic network space. For every phylogenetic network N, we define two disjoint neighborhoods: Δ(N), which contains networks with the same number of reticulation nodes as that in N, and Δ+1(N), which contains networks with one more reticulation node than that in N. Given a phylogenetic network N, a neighbor N′ ∈ Δ(N) is obtained by either relocating the source of one edge in N or relocating the destination of one reticulation edge in N. Relocating the source of one edge in N follows three steps:
Choose two distinct edges (u1, v1) and (u2, v2) in N such that u1 is neither a reticulation node nor a predecessor of v2.
Delete node u1 and the four edges (u1, v1), (u2, v2), (w, u1), and (u1, z), where w is the parent node of u1 and z is a child node of u1 other than v1.
Add a new node x and four new edges (u2, x), (x, v2), (x, v1), and (w, z) to the network.
Relocating the destination of one reticulation edge in N follows three steps:
Choose two distinct edges (u1, v1) and (u2, v2) in N such that v1 is a reticulation node and v2 is not a predecessor of u1.
Delete node v1 and the four edges (u1, v1), (u2, v2), (w, v1), and (v1, z), where w is a parent node of v1 other than u1 and z is the child node of v1.
Add new node x and four new edges (u2, x), (x, v2), (u1, x), and (w, z) to the network.
Given a phylogenetic network N, a neighbor N′ ∈ Δ+1(N) is obtained from N by adding a single edge to form a new reticulation node using the following three steps:
Choose two distinct edges (u1, v1) and (u2, v2) in N such that v2 is not a predecessor of u1.
Delete both (u1, v1) and (u2, v2).
Add two new nodes x1 and x2 and five new edges (u1, x1), (x1, v1), (u2, x2), (x2, v2), and (x1, x2).
Given a collection 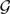 of gene trees, we search in Ω(n, k) as follows. Assume the current optimal network in the search is N ∈ Ω(n, k) and we search for the next optimal network in Ω(n, k). We compute minN′∈ Δ(N)XL(N′,
of gene trees, we search in Ω(n, k) as follows. Assume the current optimal network in the search is N ∈ Ω(n, k) and we search for the next optimal network in Ω(n, k). We compute minN′∈ Δ(N)XL(N′, 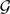 ) and compare this value to XL(N,
) and compare this value to XL(N, 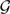 ). If the latter is larger, we replace the current network N by the new optimal one and continue the search in Ω(n, k) from the new network; otherwise, we stop the search in Ω(n, k) since the local optimum has been reached. If the search has stopped and k has reached a pre-specified upper bound of the number of reticulation nodes, the entire search terminates and the current network is returned as the inferred optimal network. If the pre-specified upper bound is not reached, the search moves up to Ω(n, k + 1) by computing minN′Δ+1(N)
XL(N′,
). If the latter is larger, we replace the current network N by the new optimal one and continue the search in Ω(n, k) from the new network; otherwise, we stop the search in Ω(n, k) since the local optimum has been reached. If the search has stopped and k has reached a pre-specified upper bound of the number of reticulation nodes, the entire search terminates and the current network is returned as the inferred optimal network. If the pre-specified upper bound is not reached, the search moves up to Ω(n, k + 1) by computing minN′Δ+1(N)
XL(N′, 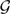 ) and compare this value to XL(N,
) and compare this value to XL(N, 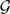 ). If the latter value is larger, we replace the current network N by the new optimal one and continue the search in Ω(n, k +1) from the new network N using Δ(N); otherwise, the search terminates and N is returned as the optimal phylogenetic network inferred. It is important to note that since the optimal network in Ω(n, 0) is the optimal species tree under the MDC criterion, the globally optimal network in this subspace can be found efficiently (without search) using the method of Than and Nakhleh (2009).
). If the latter value is larger, we replace the current network N by the new optimal one and continue the search in Ω(n, k +1) from the new network N using Δ(N); otherwise, the search terminates and N is returned as the optimal phylogenetic network inferred. It is important to note that since the optimal network in Ω(n, 0) is the optimal species tree under the MDC criterion, the globally optimal network in this subspace can be found efficiently (without search) using the method of Than and Nakhleh (2009).
Handling gene tree uncertainty.—When analysing biological data sets, gene-tree topologies are estimated from sequence data. Consequently, these gene-tree estimates may have uncertainty associated with them. We handle this uncertainty in two different ways. First, consider a case where for each gene, a non-binary tree is obtained, such as in an analysis involving bootstrapping followed by contraction of all branches with low support or in an analysis that considers the strict consensus of all optimal trees under a maximum parsimony analysis. In this case, for each gene we have a tree g that is not necessarily binary, and we replace Equation (1) by
| (2) |
where b(g) is the set of all binary refinements of g. Of course, if g contains nodes of very high degrees, this approach is computationally infeasible if done in a brute-force fashion (explicitly considering all possible refinements). However, using our MUL-tree conversion technique, the efficient algorithms of (Yu et al. 2011b, 2011c) apply directly and achieve this computation in polynomial time (in the size of the MUL-tree), as opposed to the exponential time (in the size of the MUL-tree) of the brute-force approach.
The second way of dealing with gene-tree uncertainty is by incorporating the posterior probabilities computed by a Bayesian inference of the gene-tree topologies. For each locus i, let g1i ,...,gqi be the set of gene trees along with their posterior probabilities p1i,...,pqi For a gene-tree topology g, let pg be the sum of posterior probabilities associated with all gene trees that have the same topology as g over all loci. Then, we replace Equation (1) by
| (3) |
where 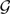 is the set of all distinct gene-tree topologies computed over all loci.
is the set of all distinct gene-tree topologies computed over all loci.
Results
Evaluating Inference on Simulated Data
To study the performance of our criterion and method in terms of the phylogenetic network they infer and the inheritance probabilities they estimate, we first used simulated data. We considered four phylogenetic networks (Fig. 4) depicting evolutionary scenarios that present different challenges. The phylogenetic network in Scenario I includes speciation after hybridization. Scenario II presents two independent hybridization events involving terminal taxa (leaves). Scenario III includes a hybrid species that further speciates, and then the two sister taxa hybridize again. Scenario IV includes two hybridization events the more recent of which involves a descendant and a descendant of a parent of the earlier hybrid. These different phylogenetic networks allow us to examine how combinations of speciation and hybridization affect the detectability of hybridization in particular, and the inference of phylogenetic networks in general. Further, we varied the inheritance probabilities associated with the hybridization events in the phylogenetic networks. For Scenario I, we considered α ∈ {0.0, 0.3, 0.5}, and for Scenario II and III we considered (α, β) ∈ {(0.0, 0.5), (0.3, 0.3), (0.5, 0.5)}. Since the hybridization events in Scenario IV are overlapping, we considered (α, β) ∈ {(0.0, 0.5), (0.3, 0.3), (0.5, 0.0), (0.5, 0.5), (0.5, 1.0)} in this case. The rationale for selecting the three values 0.0, 0.3, and 0.5 is that they represent no hybridization, “skewed” hybridization (different genetic contributions of the two parents to the hybrid), and perfect hybridization (equal genetic contributions of the two parents to the hybrid). Finally, to vary the extent of deep coalescence within each of the four evolutionary histories, we considered two settings for the branch lengths t1, ..., t4 (as measured in coalescent units): setting 1, in which t1 = t2 = t3 = t4 = 1.0, and setting 2, in which t1 = t2 = t3 = t4 = 2.0. As the extent of ILS increases as branches become shorter, we expect setting 1 to provide more challenging data for the method.
Figure 4.

Phylogenetic networks depicting different hybridization/divergence/extinction scenarios. The α and β parameters denote the proportions (or, probabilities) of alleles that are inherited from the “left” parents of the reticulation nodes (1 − α and 1 − β denote the proportions of the alleles that are inherited from the “right” parents of the nodes).
Using each combination of phylogenetic network, inheritance probabilities, and branch length setting, we used the ms program (Hudson 2002) to generate 10, 25, 50, 100, 500, 1000, and 2000 gene trees within the branches of the phylogenetic networks. To obtain statistically significant results, we generated 100 data sets per parameter setting and evaluated the performance as averaged over these 100 data sets, for each point in the parameter space. In these experiments, a single allele per species per gene was sampled.
Using the input sets of gene-tree topologies, we inferred phylogenetic networks along with inheritance probabilities. In this experiments, we started our search from the optimal species tree under MDC by the exact method of Than and Nakhleh (2009). In this section, we assume knowledge of the true number of hybridization events and made inference with these (known) numbers of hybridization events. More specifically, for data sets corresponding to Scenario I, we inferred phylogenetic networks with single hybridization events, and for the other three scenarios, we inferred phylogenetic networks with two hybridization events. We discuss later the issues arising when we do not control for the number of hybridization events. We compared each inferred phylogenetic network against the (known) true phylogenetic network in terms of the topology and estimated inheritance probability. For comparing the topologies of two phylogenetic networks, we used the dissimilarity measure of (Nakhleh et al. 2004; Than et al. 2008a) which computes the symmetric difference between the two sets of taxa clusters induced by the two networks. Results of the application of our methods to gene trees under Scenarios I, II, and III are given in Figure 5.
Figure 5.

Accuracy of the inferred phylogenetic networks and inheritance probabilities. The three columns from left to right correspond to Scenarios I, II, and III in Figure 4, respectively. One allele per gene per species is sampled.
In terms of the accuracy of the inferred phylogenetic network topology, we observe that as the number of gene trees used increases, the error in the estimated network decreases. For all three evolutionary scenarios, using about 50 gene trees under time setting 2 for branch lengths results in phylogenetic network inferences with 0 error. However, the performance is different under time setting 1, which incorporates larger extents of ILS. Here, we see that using about 50 gene trees results in correct network inference only under Scenario II, which is the least challenging for all scenarios considered. When we consider Scenario I, which adds to Scenario II the complexity of divergence after hybridization, we observe that the number of genes required to obtain accurate phylogenetic networks increases significantly (by an order of magnitude). For Scenario III, we observe that even with 2000 gene trees, the search heuristic fails to identify the true phylogenetic network. It is important to note here that we must distinguish between the performance of the optimality criterion and that of the search heuristic employed for inference. In this case, our search heuristic begins with a species tree that minimizes the number of extra lineages (or, deep coalescences) over all possible tree candidates, given the set of gene trees. Using this tree, the search proceeds in a hill descent fashion, each time exploring all neighboring topologies of the current optimal network, and continuing with the best found. An artifact of this search heuristic is that if the true network cannot be obtained from the starting tree in any possible way, then this search heuristic would not converge to the true network. Of course, this problem could be ameliorated by random restarts of the search heuristic or by exhaustively starting from all possible trees. Although the former is also not guaranteed to result in convergence to the true network, the latter is prohibitive but for data sets with very small numbers of taxa, given the exponentially large size of the tree space. Nevertheless, we have inspected the cases pertaining to Scenario III and verified that the reason behind the lack of convergence to 0-error networks is the criterion: The number of extra lineages in the optimal network that the heuristic infers is smaller than that in the true network. This is not surprising, since parsimonious reconciliation and inference is known to have consistency issues, even when ILS is the only event at play (Than and Nakhleh 2009, 2010; Than and Rosenberg 2011). Finally, we observe that the performance is better for inheritance probabilities that are closer to 0.5. This is due to the fact that under these settings the contributions of the two parents to the genetic makeup of a hybrid species is more balanced, providing more phylogenetic signal for the method to infer the correct evolutionary history.
In terms of estimating the inheritance probabilities, the results show that our search heuristic makes very good estimates, regardless of the evolutionary scenario and branch length setting. Even though branch length setting 2 yields slightly more accurate estimates, which is expected, it is important to note that the method produces very good estimates even for the shorter branch lengths, where the extent of ILS is much larger. Further, it is worth emphasizing that these good estimates are obtained even with the smallest data sets (in terms of the number gene trees). This is a strength of the method.
More Loci or More Alleles?
Given the finite resources associated with any phylogenomic analysis, a natural question to ask is: In order to obtain more accurate inferences of phylogenetic networks and inheritance probabilities, should one sample more loci across the genomes or more alleles per locus? To explore this question, we used the above simulation procedure to generate gene trees under evolutionary Scenario IV, where 1, 2, 4, and 8 alleles per locus per species were sampled. The multi-allele gene trees were then used as input in the inference procedure. The results of this experiment are shown in Figure 6.
Figure 6.

The effect of the number of alleles. Accuracy of the phylogenetic networks and inheritance probabilities estimated from gene trees simulated under Scenario IV, with true inheritance probabilities α = β = 0.3, where the number of alleles sampled per species also varies. Top and bottom rows correspond to time settings 1 and 2, respectively.
Several observations are in order. First, in the case of this evolutionary scenario, the ability of the method to infer the correct topology of the phylogenetic network is not affected much by the branch length settings, unlike the performance on the other three scenarios. However, in this case, the method always overestimates the inheritance probability (by about 5% hybridization), more so in the case of time setting 1. Second, in this case, the estimates of the probability β of the lower (closer to the leaves) hybridization are more accurate than that of the estimates of α, which is unlike Scenarios II and III, where we did not observe any differences in the quality of the estimates of the two hybridization events. The reason for this is that in this scenario, some lineages, or alleles, from species D that trace different parents at the hybridization event undergo a further hybridization event, affecting the coalescence patterns toward the root. Regarding the benefit obtained by increasing the number of alleles, none are observed in terms of the inheritance probability, and some are observed in terms of the phylogenetic network accuracy under time setting 1. That is, if the branches are very short, sampling two alleles, instead of one, improves the quality of the inferred network significantly. However, adding alleles beyond that does not seem to add more power, or signal, to the method. Under the other three scenarios, a single allele was already sufficient to provide highly accurate estimates. In summary, given the experimental settings we used here, there does not seem to be much benefit in sampling many alleles per species. Rather, sampling more loci per genome, particularly when the number of loci afforded is smaller than 100, provides more benefit. It is worth mentioning that the probabilistic method of Yu et al. (2012) yields very accurate estimates of the inheritance probabilities under this evolutionary scenario, even when a single allele is sampled per species [see the supplementary material of Yu et al. (2012)].
Evaluating Inference on a Yeast Data Set
To study the performance of our framework on biological data, we reanalysed the yeast data set of Rokas et al. (2003). This data set consists of 106 loci, each present in exactly a single copy in each of seven Saccharomyces species, S. cerevisiae (Scer), S. paradoxus (Spar), S. mikatae (Smik), S. kudriavzevii (Skud), S. bayanus (Sbay), S. castellii (Scas), S. kluyveri (Sklu), and the outgroup fungus Candida albicans (Calb). We reconstructed gene trees from sequence data using maximum parsimony in PAUP* (Swofford 1996) and Bayesian inference in MrBayes (Huelsenbeck and Ronquist 2001). In each of 106 gene trees, the genes from the five species Scer, Spar, Smik, Skud, and Sbay formed a monophyletic group. From a parsimony perspective, all coalescent events involving genes from these five species occur at or below their most recent common ancestor. Therefore, in our analysis, we only focused on the evolutionary history of these five species.
It is important to note that the gene trees used in the analysis here are not all binary. In the case where the gene trees were inferred by maximum parsimony, we used the strict consensus of all optimal trees found for each gene, which resulted in non-binary trees. In the case of Bayesian inference, we used each gene tree with its posterior probability. See Methods section for how we accounted for uncertainty in gene trees using these two approaches.
Using our method, we inferred the optimal species networks containing 0, 1, and 2 reticulation nodes. The resulting species networks inferred from gene trees reconstructed by maximum parsimony are shown in Figure 7 along with inheritance probabilities and total number of extra lineages. The optimal species tree in Figure 7A has been reported by several studies (Rokas et al. 2003; Edwards et al. 2007; Than and Nakhleh 2009). The optimal species network containing one reticulation node in Figure 7B has also been proposed as an alternative evolutionary history under the stochastic framework of (Bloomquist and Suchard, 2010), the parsimony framework of Than and Nakhleh (2009) and the likelihood framework of Yu et al. (2012). It is worth mentioning that the inheritance probability inferred by our method is almost the same as that inferred by the probabilistic approach of Yu et al. (2012). The optimal species network with two reticulation nodes in Figure 7C was not reported in any of the aforementioned studies.
Figure 7.
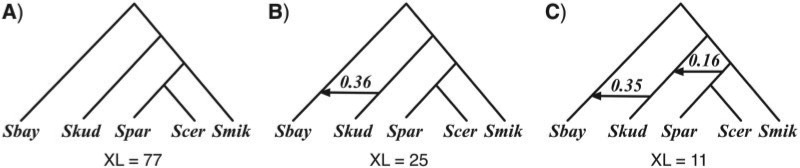
Analysis of the yeast data set, where gene trees are reconstructed using MP. Optimal species phylogenies, along with inheritance probabilities, inferred from gene trees reconstructed by maximum parsimony for the yeast data set of (Rokas et al. 2003). A) The optimal species tree (network with 0 reticulation nodes). B) The optimal species network containing one reticulation node. C) The optimal species network containing two reticulation nodes. For each species phylogeny, the total number of extra lineages (XL) is computed using Equation (2) and reported.
For gene trees reconstructed using MrBayes, the inferred species networks are shown in Figure 8. The optimal species tree in Figure 8A has been reported as a very close candidate (Edwards et al. 2007; Than and Nakhleh 2009). The optimal species network containing one reticulation node in Figure 8B has the same topology as the one inferred from gene trees reconstructed by maximum parsimony in Figure 7B, but with a slightly higher inheritance probability.
Figure 8.
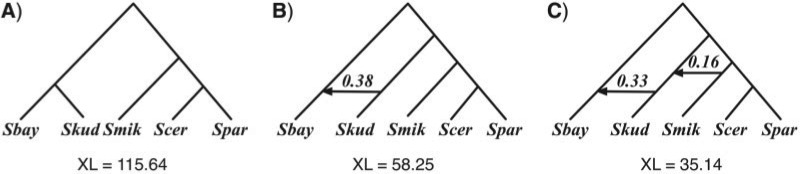
Analysis of the yeast data set, where gene trees are reconstructed using Bayesian inference. Optimal species phylogenies, along with inheritance probabilities, inferred from gene trees reconstructed by MrBayes for the yeast data set of (Rokas et al. 2003). A) The optimal species tree (network with 0 reticulation nodes). B) The optimal species network containing one reticulation node. C) The optimal species network containing two reticulation nodes. For each species phylogeny, the total number of extra lineages (XL) is computed using Equation (3) and reported.
The Model Selection Problem
A major confounding issue that arises when inferring phylogenetic network topologies is that of determining the correct number of reticulation events (Nakhleh 2010). As we observed in the yeast data set analysis, adding a single reticulation node to the optimal species tree reduces the number of extra lineages by about 70%. Further, adding an additional reticulation node to the optimal species network with a single reticulation node reduces the number of extra lineages by about a half. This is the classical model selection problem arising in the domain of phylogenetic networks: Increasing the complexity of the phylogenetic network topology by adding more reticulation nodes to it mostly improves the fit of the data. Simply minimizing the sum of the number of hybridization events and deep coalescence events does not solve the problem. Further, minimizing a weighted sum of these two numbers raises the questions of how to weight them and whether weights are data-dependent or not.
As we pointed out above, when analysing the simulated data, we assumed knowledge of the true number of reticulation nodes. To understand the performance of the method when this assumption is removed, we inferred phylogenetic networks with up to four reticulation nodes from the data we generated, and explored the number of extra lineages in these inferred networks as a function of the number of reticulation nodes. The results for Scenario III are shown in Figure 9; similar results were observed under the other scenarios.
Figure 9.

Network complexity and the number of extra lineages. The decrease in the number of extra lineages in the inferred phylogenetic network as a function of the increase in number of hybridization events inferred. The results were obtained from data pertaining to Scenario III under two different settings of the inheritance probabilities and two different settings of the branch lengths.
As the figure shows, the number of extra lineages of the optimal species networks keeps decreasing as more reticulation nodes are added. Thus, using the minimization of the number of extra lineages as the optimality criterion, without penalizing complexity, may result in gross overestimation of the amount of reticulation in the data.
Performance on Large Data Sets
We recently proposed another exact method for computing the number of extra lineages of a phylogenetic network and showed that it is much faster than the MUL-tree based one (Yu and Nakhleh 2012). Since both methods are exact, substituting one for the other does not affect the inference method. Still, the method based on MUL-trees has its advantages in that it is applicable efficiently to unrooted and non-binary gene trees, as discussed above. Since we seek to evaluate the performance of parsimonious inference of phylogenetic networks, we employ the method of Yu and Nakhleh (2012) for scoring phylogenetic networks.
We conducted experiments on simulated data sets that are much larger than the ones used above. We first generated 100 random species trees with 10, 20, and 40 taxa using PhyloGen Rambaut (2012) and set the total heights of those species trees to 8 Ne, 16 Ne, and 32 Ne, respectively. From each species tree, we then generated random species networks with 1, 2, 3, 4, and 5 reticulation nodes, respectively. When expanding a species network with k reticulation nodes to a species network with k +1 reticulation nodes, we randomly selected two existing edges in the species network and connected their midpoints from the higher one to the lower one and then the lower one becomes a new reticulation node. For every reticulation node, we assigned random values from 0 to 1 as its inheritance probability. Finally, we simulated 25, 50, 100, and 200 gene trees, respectively, within the branches of each species network using the ms program Hudson (2002).
Using the input sets of gene-tree topologies, we inferred phylogenetic networks using the search procedure described above, and assuming knowledge of the true number of reticulation nodes. The running times of the method are shown in Figure 10. It is not surprising that the running time increases with the increase in the numbers of taxa and reticulation nodes. But overall, our method is able to finish the computations on all data sets in a reasonable amount of time. For the largest data set which has 40 taxa and 5 reticulation nodes, 75% of the computations finished within 24 h. The outliers in the figure indicate that some data sets took much more time than others, especially for larger data sets. This occurs because the topology of the phylogenetic network and gene trees affect the running time, even when keeping the numbers of taxa and reticulation nodes fixed.
Figure 10.

Running time of phylogenetic network inference. The three columns from left to right correspond to data sets with 10, 20, and 40 taxa, respectively. The six rows from bottom to top correspond to data sets with 0, 1, 2, 3, 4, and 5 reticulation nodes, respectively. In each sub-figure, the x-axis is the number of gene trees sampled and the y-axis is the running time in seconds.
In addition to the running times, we also investigated the topological accuracy of the inferred phylogenetic network. Since for each run we know the true network Nm and the inferred network Ni, the two networks can be compared using the normalized symmetric difference of the two; that is, by calculating the number of clusters that appear in one but not both of the networks, and dividing the number by twice the number of clusters in Nm. This measure was first introduced in Nakhleh et al. (2003) and implemented in PhyloNet (Than et al. 2008a). The values of this measure for the pairs of phylogenetic networks we consider here range between 0, indicating identical networks, and 1, indicating the pair of networks disagree on every cluster. Results on the accuracy of the inferred phylogenetic networks are given in Figure 11. For a fixed number of taxa, the error of network inference increases with the number of reticulation nodes. It is expected because the addition of reticulation nodes increases the complexity of the phylogenetic networks. On the other hand, for a fixed number of reticulation nodes, the error of network inference decreases as the number of taxa increases. This happens because for a network with larger number of taxa, the randomly added reticulation nodes may have a higher chance to be independent of each other, which actually makes the inference easier. Last but not least, as the number of gene trees sampled increases, the accuracy improves, albeit slightly. This may be due to issues with the search strategy, issues with the MDC criterion, or both.
Figure 11.

Accuracy of inferred phylogenetic networks. The three columns from left to right correspond to data sets with 10, 20, and 40 taxa, respectively.
Discussion
In this study, we extended the MDC criterion (Maddison 1997; Than and Nakhleh 2009) in order to define a parsimonious reconciliation of a gene-tree topology within the branches of a phylogenetic network. By doing so, the resulting reconciliation accounts simultaneously for ILS and hybridization. Further, we devised a local search heuristic for searching the phylogenetic network space to identify optimal ones under the new criterion. We applied our criterion and search heuristic to simulated data and a biological data set, and demonstrated the quality of inferences.
A central technique that we use in our study entails converting a phylogenetic network into its corresponding multi-labeled tree, or MUL-tree. This technique enables applying existing, tree-based criteria and methods to phylogenetic networks by employing them on the tree representation of the network. Indeed, in Yu et al. (2012), we showed how to apply standard coalescent-based probabilistic computations to MUL-trees, and in this study we demonstrated how to extend parsimony-based tree reconciliations to phylogenetic networks by working indirectly on the MUL-tree representations. A further potential use of MUL-trees might be in facilitating phylogenetic network space search to enable efficient inference techniques.
When inferring phylogenetic networks, the classical model selection problem arises: More complex networks (i.e., ones with more hybridization) may be found to fit the data better than less complex ones. Even though there is a quadratic bound on the maximum number of reticulation events in terms of the number of leaves in the phylogenetic network, methods that do not account for this issue would result in gross overestimations of hybridization. This is one of the major problems with parsimonious reconciliations.
Although various biological events can cause gene trees to disagree with each other as well as with the species phylogeny, a major confounding factor that must be accounted for when conducting analyses is uncertainty in gene trees. As gene trees are estimated from sequence data using computational methods, not all branching patterns in these trees can be inferred with certainty. Therefore, it is very important that criteria and methods account for this issue. We showed two ways of doing so, by allowing for non-binary gene trees and by considering posterior distributions.
Just as parsimony approaches can have consistency issues when inferring trees from sequences (Felsenstein 1978), their counterparts for species tree inference from gene trees suffer from similar issues (Than and Rosenberg 2011). We expect that this issue would arise also in the case of parsimonious inference of phylogenetic networks. Nevertheless, we showed in this study that for many cases of hybridization and ILS, parsimony obtains the same results as a probabilistic framework, within a fraction of the time that the latter approach takes. We believe one of the best uses of parsimony would be to quickly obtain a good initial network that can be used to seed searches for phylogenetic networks under probabilistic approaches.
One major simplifying assumption that we made here is ignoring other discord factors, such as gene duplication and loss. We will explore ways of incorporating duplication and loss into our framework, potentially along the lines of combining the idea of MUL-tree with that of the locus tree (Rasmussen and Kellis 2012). Further, while we specifically address hybridization in this study, the framework is applicable in theory to HGT in general. However, when only a single gene or very few genes are transferred, a large extent of ILS might overwhelm the signal for HGT.
Supplementary Material
Supplementary material, including data files and/or online-only appendices, can be found at http://datadryad.org and in the Dryad data repository (DOI:10.5061/dryad.sr534).
Funding
This work was supported in part by the National Science Foundation [grant numbers DBI-1062463, CCF-1302179]; the National Library of Medicine [grant R01LM009494]; an Alfred P. Sloan Research Fellowship, and a Guggenheim Fellowship (to L.N.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NSF, National Library of Medicine, the National Institutes of Health, the Alfred P. Sloan Foundation, or the John Simon Guggenheim Memorial Foundation.
References
- Arnold M.L. Natural hybridization and evolution. Oxford: Oxford University Press; 1997. [Google Scholar]
- Bansal M., Alm E. Kellis M. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics. 2012;28:i283–i291. doi: 10.1093/bioinformatics/bts225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. The role of hybridization in evolution. Mol. Ecol. 2001;10:551–568. doi: 10.1046/j.1365-294x.2001.01216.x. [DOI] [PubMed] [Google Scholar]
- Beiko R., Hamilton N. Phylogenetic identification of lateral genetic transfer events. BMC Evol. Biol. 2006;6:15. doi: 10.1186/1471-2148-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist E., Suchard M. Unifying vertical and nonvertical evolution: a stochastic ARG-based framework. Syst. Biol. 2010;59:27–41. doi: 10.1093/sysbio/syp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordewich M., Semple C. On the computational complexity of the rooted subtree prune and regraft distance. Ann. Comb. 2004;8:409–423. [Google Scholar]
- Cranston K.A., Hurwitz B., Ware D., Stein L., Wing R.A. Species trees from highly incongruent gene trees in rice. Syst. Biol. 2009;58:489–500. doi: 10.1093/sysbio/syp054. [DOI] [PubMed] [Google Scholar]
- Degnan J., Rosenberg N. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009;24:332–340. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Edwards S.V., Liu L., Pearl D.K. High-resolution species trees without concatenation. Proc. Natl Acad. Sci. USA. 2007;104:5936–5941. doi: 10.1073/pnas.0607004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978;27:401–410. [Google Scholar]
- Hobolth A., Dutheil J., Hawks J., Schierup M., Mailund T. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 2011;21:349–356. doi: 10.1101/gr.114751.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Joly S., McLenachan P.A., Lockhart P.J. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 2009;174:E54–E70. doi: 10.1086/600082. [DOI] [PubMed] [Google Scholar]
- Kingman J.F.C. The coalescent. Stochast. Proc. Appl. 1982;13:235–248. [Google Scholar]
- Kubatko L.S. Identifying hybridization events in the presence of coalescence via model selection. Syst. Biol. 2009;58:478–488. doi: 10.1093/sysbio/syp055. [DOI] [PubMed] [Google Scholar]
- Kuo C.-H., Wares J.P., Kissinger J.C. The Apicomplexan whole-genome phylogeny: an analysis of incongurence among gene trees. Mol. Biol. Evol. 2008;25:2689–2698. doi: 10.1093/molbev/msn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Yu L.L., Kubatko L., Pearl D.K., Edwards S.V. Coalescent methods for estimating phylogenetic trees. Mol. Phylogenet. Evol. 2009;53:320–328. doi: 10.1016/j.ympev.2009.05.033. [DOI] [PubMed] [Google Scholar]
- MacLeod D., Charlebois R., Doolittle F., Bapteste E. Deduction of probable events of lateral gene transfer through comparison of phylogenetic trees by recursive consolidation and rearrangement. BMC Evol. Biol. 2005;5:27. doi: 10.1186/1471-2148-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison W. Gene trees in species trees. Syst. Biol. 1997;46:523–536. [Google Scholar]
- Maddison W.P., Knowles L.L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 2006;55:21–30. doi: 10.1080/10635150500354928. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005;20:229–237. doi: 10.1016/j.tree.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybrid speciation. Nature. 2007;446:279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- Meng C., Kubatko L.S. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene-tree incongruence: a model. Theor. Popul. Biol. 2009;75:35–45. doi: 10.1016/j.tpb.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Nakhleh L. Evolutionary phylogenetic networks: models and issues. In: Heath L., Ramakrishnan N., editors. The problem solving handbook for computational biology and bioinformatics. New York: Springer; 2010. pp. 125–158. [Google Scholar]
- Nakhleh L., Ruths D., Wang L. RIATA-HGT: a fast and accurate heuristic for reconstructing horizontal gene transfer. In: Wang L., editor. 2005. pp. 84–93. Proceedings of the Eleventh International Computing and Combinatorics Conference (COCOON 05) lNCS #3595. [Google Scholar]
- Nakhleh L., Sun J., Warnow T., Linder C., Moret B., Tholse A. Towards the development of computational tools for evaluating phylogenetic network reconstruction methods. Proceedings of the Eighth Pacific Symposium on Biocomputing. 2003;8:315–326. [PubMed] [Google Scholar]
- Nakhleh L., Warnow T., Linder C. Proceedings of the 8th Annual International Conference on Research in Computational Molecular Biology (RECOMB04) 2004. Reconstructing reticulate evolution in species–theory and practice; pp. 337–346. [DOI] [PubMed] [Google Scholar]
- Pollard D.A., Iyer V.N., Moses A.M., Eisen M.B. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2006;2:e173. doi: 10.1371/journal.pgen.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. 2012. Phylogen v1.1. Available from: URL http://treebioedacuk/software/phylogen/ (last accessed January 2013) [Google Scholar]
- Rannala B., Yang Z. Phylogenetic inference using whole genomes. Annu. Rev. Genomics Hum. Genet. 2008;9:217–231. doi: 10.1146/annurev.genom.9.081307.164407. [DOI] [PubMed] [Google Scholar]
- Rasmussen M.D., Kellis M. Unified modeling of gene duplication, loss, and coalescence using a locus tree. Genome Res. 2012;22:755–765. doi: 10.1101/gr.123901.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg L. Hybrid origins of plant species. Annu. Rev. Ecol. Syst. 1997;28:359–389. [Google Scholar]
- Rokas A., Williams B.L., King N., Carroll S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Swofford D.L. PAUP*: phylogenetic analysis using parsimony (*and other methods). Version 4.0. Sunderland (MA): Sinauer Associates; 1996. [Google Scholar]
- Syring J., Willyard A., Cronn R., Liston A. Evolutionary relationships among Pinus (Pinaceae) subsections inferred from multiple low-copy nuclear loci. Am. J. Bot. 2005;92:2086–2100. doi: 10.3732/ajb.92.12.2086. [DOI] [PubMed] [Google Scholar]
- Takuno S., Kado T., Sugino R.P., Nakhleh L., Innan H. Population genomics in bacteria: a case study of staphylococcus aureus. Mol. Biol. Evol. 2012;29:797–809. doi: 10.1093/molbev/msr249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C., Nakhleh L. Species tree inference by minimizing deep coalescences. PLoS Comput. Biol. 2009;5:e1000501. doi: 10.1371/journal.pcbi.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C., Nakhleh L. Inference of parsimonious species phylogenies from multi-locus data by minimizing deep coalescences. In: Knowles L., Kubatko L., editors. Estimating species trees: practical and theoretical aspects. Hoboken, New Jersey: Wiley-VCH; 2010. pp. 79–98. [Google Scholar]
- Than C., Rosenberg N. Consistency properties of species tree inference by minimizing deep coalescences. J. Comput. Biol. 2011;18:1–15. doi: 10.1089/cmb.2010.0102. [DOI] [PubMed] [Google Scholar]
- Than C., Ruths D., Innan H., Nakhleh L. Confounding factors in HGT detection: statistical error, coalescent effects, and multiple solutions. J. Comput. Biol. 2007;14:517–535. doi: 10.1089/cmb.2007.A010. [DOI] [PubMed] [Google Scholar]
- Than C., Ruths D., Nakhleh L. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics. 2008a;9:322. doi: 10.1186/1471-2105-9-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C., Sugino R., Innan H., Nakhleh L. Efficient inference of bacterial strain trees from genome-scale multi-locus data. Bioinformatics. 2008b;24:i123–i131. doi: 10.1093/bioinformatics/btn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M.A., Ane C., Dewey C.N., Larget B.R., Payseur B.A. Fine-scale phylogenetic discordance across the house mouse genome. PLoS Genet. 2009;5:e1000729. doi: 10.1371/journal.pgen.1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Degnan J., Nakhleh L. The probability of a gene-tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 2012;8:e1002660. doi: 10.1371/journal.pgen.1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Nakhleh L. 2012. Fast algorithms for reconciliation under hybridization and incomplete lineage sorting. arXiv 1212.1909. [Google Scholar]
- Yu Y., Than C., Degnan J., Nakhleh L. Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Syst. Biol. 2011a;60:138–149. doi: 10.1093/sysbio/syq084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Warnow T., Nakhleh L. Proceedings of the 15th Annual International Conference on Research in Computational Molecular Biology (RECOMB). Vol. 6577 of Lecture Notes in Bioinformatics. Berlin, Heidelberg: Springer; 2011b. Algorithms for MDC-based multi-locus phylogeny inference; pp. 531–545. [Google Scholar]
- Yu Y., Warnow T., Nakhleh L. Algorithms for MDC-based multi-locus phylogeny inference: beyond rooted binary gene trees on single alleles. J. Computa. Biol. 2011c;18:1543–1559. doi: 10.1089/cmb.2011.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]