

Abstract
The neural crest (NC) is first induced as an epithelial population of cells at the neural plate border requiring complex signaling between bone morphogenetic protein, Wnt, and fibroblast growth factors to differentiate the neural and NC fate from the epidermis. Remarkably, following induction, these cells undergo an epithelial-to-mesenchymal transition (EMT), delaminate from the neural tube, and migrate through various tissue types and microenvironments before reaching their final destination where they undergo terminal differentiation. This process is mirrored in cancer metastasis, where a primary tumor will undergo an EMT before migrating and invading other cell populations to create a secondary tumor site. In recent years, as our understanding of NC EMT and migration has deepened, important new insights into tumorigenesis and metastasis have also been achieved. These discoveries have been driven by the observation that many cancers misregulate developmental genes to reacquire proliferative and migratory states. In this review, we examine how the NC provides an excellent model for studying EMT and migration. These data are discussed from the perspective of the gene regulatory networks that control both NC and cancer cell EMT and migration. Deciphering these processes in a comparative manner will expand our knowledge of the underlying etiology and pathogenesis of cancer and promote the development of novel targeted therapeutic strategies for cancer patients. © 2013 Wiley Periodicals, Inc.
INTRODUCTION
The neural crest (NC) is a population of transient, multipotent cells that are specified at the border of the neural plate between the neural and non-neural ectoderm in vertebrate embryos. These cells undergo an epithelial-to-mesenchymal transition (EMT), delaminate, and migrate away from the neural tube to populate various tissues and contribute multiple cell fates to the developing embryo, including pigment cells, neurons and glia of the peripheral nervous system, and craniofacial cartilage.1,2 The genes that regulate these developmental processes have been extensively studied in many model systems, including Xenopus, zebrafish, chick, and mouse, and are highly conserved between these vertebrate species. The process of EMT involves downregulation of characteristic epithelial genes such as the adhesion genes E-cadherin, Claudins, and Occludins and the upregulation of mesenchymal markers such as fibronectin, vitronectin, and vimentin (Figures 1 and 2). After EMT, NC cell migration involves complex interactions between the cells and the environment in which they migrate including positive responses to attractive signals such as chemokines, as well as avoidance of repulsive signals such as ephrins and semaphorins (Figure 3). In combination, these signals direct NC cells along restricted migratory paths. In addition, NC cells use cell autonomous activation of matrix metalloproteinases (MMPs) and ADAMs (a disintegrin and metalloproteinase; adamlysins) to break down the extracellular matrix (ECM), including proteins such as fibronectin, and to facilitate migration to their final destinations.
FIGURE 1.

Comparison of transcriptional regulation in neural crest (NC) cells and cancer cells. The diagram depicts the role of growth factors and their signaling pathways in initiating epithelial-to-mesenchymal transition (EMT) and migration in both NC cells and cancer cells. The actions of the specific transcription factors discussed in the text are indicated.
FIGURE 2.
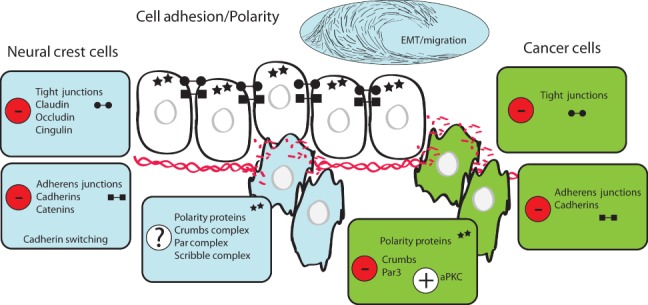
Comparison of cell adhesion and polarity changes in neural crest (NC) cells and cancer cells. The diagram depicts alterations in tight junction components (circle barbells), adherens junction components (square barbells), and polarity proteins (stars) during epithelial-to-mesenchymal transition (EMT) and migration in NC cells (light blue) and cancer cells (green) as they differentiate and migrate away from normal epithelial cells (white). Passage of cells through the basement membrane and disruption of the extracellular matrix (ECM; red intertwined lines and red broken fragments) is indicated.
FIGURE 3.

Comparison of migration and guidance in neural crest (NC) cells and cancer cells. The diagram depicts degradation of the extracellular matrix (ECM; red intertwined lines and red broken fragments) and the role of guidance cues (ligands shown as spirals and receptors shown as  ) during EMT and migration in NC cells (light blue) and cancer cells (green).
) during EMT and migration in NC cells (light blue) and cancer cells (green).
The processes described for NC development share many characteristics with the progression of cancer. For example, primary epithelial tumors display EMT characteristics similar to those displayed during NC EMT before they delaminate and begin to metastasize. Key features of cancer progression can involve a loss of junctional proteins, disruption of the basement membrane, and upregulation of mechanisms to escape cell death. While NC cell EMT and migration are tightly regulated and conserved processes, a key feature of cancer cell movement is a misregulation of these same processes. This includes the usurping of developmental programs including those that direct cell polarity, adhesion, and cell morphology. For instance, during cancer EMT, abnormal upregulation of key developmental transcriptional regulators such as the Snail, Twist, and ZEB transcription factors occurs.3–6 Similar to NC cells, as cancer cells migrate during metastasis, they can upregulate proteins such as MMPs to break down the ECM and invade tissues to form metastatic lesions. Additionally, recent studies suggest that, like NC cells, migrating tumor cells respond to chemoattractive signals that promote migration to their secondary invasion site. The parallels between the genes and proteins involved in EMT and migration in both NC development and cancer progression make NC cells an excellent model for investigating the genetic regulation of cancer progression and metastasis (summarized in Table 1). Several recent reviews have documented the similarities between NC development and cancer migration.7–9 Here, we directly compare at a systems level NC and cancer cell EMT and migration, and demonstrate how this information provides a unique perspective on cancer cell migration and metastasis.
TABLE 1.
Genes with Roles in Both Neural Crest Development and Cancer Progression
| Gene | Role in Neural Crest Development | Role in Cancer Progression |
|---|---|---|
| Transcription factors | ||
| Snail/Slug | Induction of EMT10–13 | Induction of EMT and metastasis3,14–19 |
| Zeb | Induction of EMT and NC migration20–24 | Induction of EMT and metastasis3,25 |
| Sox10 | NC differentiation (melanocytes) and survival26–30 | Melanoma formation31 |
| Twist | Induction of EMT and NC migration, differentiation32,33 | Induction of EMT and metastasis34–36 |
| Adhesion and polarity | ||
| Claudins and Occludins | Downregulated for migration37–40 | Downregulation correlates with migration41 |
| E-cadherin | Downregulated during NC specification42–44 | Downregulation correlates with migration45 |
| N-cadherin | Downregulated for NC delamination and migration10,42,46–48 | Downregulation correlates with migration49,50 |
| Crumbs complex | Polarity of NC cells51 | Downregulation correlates with EMT52–55 |
| Par complex | Polarity of NC cells51 | Misregulation correlates with EMT56–59 |
| Migration | ||
| MMPs and ADAMs | Degradation of ECM for NC migration60–65 | Degradation of ECM for metastasis66–76 |
| SDF1/CXCR4 | Directional NC migration77–79 | Metastasis and survival80,81 |
| Ephrins and semaphorins | Directional NC migration8,82–85 | Misregulated in cancer86,87 |
ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; MMPs, matrix metalloproteinases; NC, neural crest.
TRANSCRIPTION FACTORS IN NC EMT AND CANCER
EMT in both the NC and cancer is triggered by various signaling pathways including BMP, Wnt, and signals from the ECM.4,8 One of the hallmark targets of these signaling pathways are the Snail family of transcription factors including Snail1 and Snail2 (also known as Slug), which are known to have an important role in both NC specification and EMT as well as in cancer progression (Figures 1 and 4). Snail is expressed in the NC prior to EMT in both zebrafish and chick embryos, following initial specification of the NC.88,89 Snail is thought to be an initial early target of transcription factors that specify the NC or ‘NC specifiers’ such as Foxd3 in zebrafish.90 In chick, once Snail transcription factors are expressed in the NC, they initiate EMT by repressing N-cadherin and thereby mediating the switch to a mesenchymal phenotype.10 In chick and Xenopus, loss of Snail family members results in a failure of EMT, and failure of NC delamination and migration.11,12 Similarly, in zebrafish, loss of the two Snail transcription factors results in massive embryonic failure of EMT in multiple tissues.13 However, in the mouse, the requirement for Snail in NC formation and delamination may not be conserved.91 Snail proteins have also been found to be crucial for cancer cell EMT through similar mechanisms. Snail1 has been shown to transcriptionally repress E-cadherin in invasive carcinoma cells and epithelial tumors.14,15 Furthermore, upregulation of Snail proteins has been shown to correlate with an increased incidence of tumor metastasis, recurrence, and an overall poorer prognosis in human cancers including breast, ovarian, colon, liver, and squamous cell carcinomas, suggesting that Snail is involved in increased EMT and tumor cell migration in human patients.3 In addition, inhibition of Snail in tumor cell lines or in orthotopic mouse models can reverse EMT and invasiveness.16–18 Interestingly, in studies of transformed primary human melanocytes, which form metastatic melanomas in nude mice, the upregulation of Snail2 expression is required for metastatic spread.19 Furthermore, in patients with benign nevi that have not undergone malignant transformation, Snail2 is highly expressed and is significantly correlated with the expression of other NC migration-associated genes.19 These data suggest that while expression of Snail2 is not sufficient for malignant transformation and malignant spread, Snail2 and other NC developmental genes may play an important role in the biology of malignant melanoma and other invasive cancers.
FIGURE 4.

Pathway analysis showing integration between signaling pathways, transcription factors, and adhesion genes. Wnt and BMP act at the top of the hierarchy to initiate the induction of EMT cascade. The transcriptional network, including Twist1/2, Zeb1/2, Snail1/2, Sox10, and Mitf, is active both in neural crest development and cancer. The transcription factors then interact with adhesion genes on the cell surface including N-cadherin (N-cad), E-cadherin (E-cad), Occludins (Ocln), Claudins (Cldn), and extracellular matrix proteins such as fibronectin (FN) and intermediate filament proteins such as vimentin (Vim). (The network was built with Ingenuity Pathway Analysis, Ingenuity Systems, Inc., Redwood City, CA and modified in Adobe Illustrator).
Another key family of factors that regulate EMT in NC and cancer cells is the Zeb family of transcription factors, containing Zeb1 and Zeb2. Zeb transcription factors are expressed in the NC in Xenopus and mouse during early development, as well as in a subset of NC derivatives.92 Loss of Zeb factors leads to a defect in NC migration in the mouse embryo and a persistence of E-cadherin after differentiation of the neuroepithelium from the ectoderm and after EMT,20,21 correlating with the role of Zeb proteins as transcriptional repressors of E-cadherin.22 Furthermore, mutations in the human Zeb protein have also been linked to the neurocristopathy Hirschprung's disease, which is characterized by a failure of enteric NC cells to migrate into and populate the gut.23,24 Zeb factors also repress E-cadherin in tumor progression. Similar to Snail, high expression levels of Zeb1 or Zeb2 correlate with a decrease in E-cadherin expression in a multitude of human cancers including breast, endometrial, colon, uterine, pancreatic, and non-small cell lung cancers.3,25 This suggests that Zeb factors correlate with increased metastasis and poor prognosis.
The transcription factor Sox10 is also an important activator of NC fate and functions at many stages of NC cell development. The pattern of Sox10 expression in the NC is highly conserved across zebrafish, Xenopus, chick, and mice, and it is also expressed in human NC precursors.26 Initially expressed at the premigratory stage, Sox10 expression is maintained in most migratory NC progenitors. However, Sox10 functions predominantly in cell differentiation and survival, as NC cells lacking Sox10 form and migrate normally but undergo apoptosis prior to terminal differentiation.26,27 For example, Sox10 regulates differentiation of the melanocyte lineage through direct transcriptional regulation of the microphthalmia-associated transcription factor (MITF).28 Accordingly, mutations in the Sox10 gene disrupt differentiation of the melanocyte lineage, such as in the murine Dominant megacolon (Dom) and zebrafish colorless (cls) mutants.29,30 In addition to a role in NC development, recent work suggests a critical role for Sox10 in the development and persistence of human cancer. In human patients, virtually all congenital nevi and melanomas have upregulated SOX10 expression. Furthermore, in a mouse model of melanoma, loss of one allele of Sox10 or knockdown with shRNA in human melanoma cells completely abolishes in vivo melanoma formation.31 These results suggest that targeting of Sox10 expression may suppress the formation of giant congenital nevi and melanomas in human patients.
Additional transcription factors such as the helix-loop-helix (HLH) family including Twist1, E proteins, and Id HLH proteins also have a demonstrated role in EMT. Some of these proteins are known to repress E-cadherin expression, similar to Snail and Zeb, but also may have a role in cell cycle and proliferation control.93 Twist1 is required in the developing mouse NC for proper migration and differentiation.32,33 In cancer, Twist is a repressor of E-cadherin and also activates the expression of several mesenchymal genes such as vimentin and fibronectin.34 It is thought that Twist1 induces EMT by activating Snail2.35 Moreover, increased Twist expression is associated with later-stage progression of tumors and correlates with increased invasion and metastasis as well as poor survival in human cancer.36 Other HLH proteins such as Id proteins have also been shown to be deregulated in a number of human cancers, suggesting that their roles in developmental EMT could be recapitulated in cancer progression.94
CHANGES IN CELLULAR ADHESION AND POLARITY ARE REQUIRED FOR NC AND CANCER EMT
Both NC cell development and cancer metastasis rely on the dynamic reorganization of cellular adhesions during EMT and migration.95–97 The transition from an epithelial adhesive cellular phenotype to a migratory mesenchymal phenotype is a key feature of NC cell development. As NC cells arise from the neuroepithelium, they exhibit epithelial cell adhesion. Epithelial cell adhesion is maintained through two intercellular adhesion complexes: tight junctions and adherens junctions.
Tight junctions are comprised of families of transmembrane proteins, Claudins and Occludins, which localize to the apical zone in neuroepithelial cells and maintain adhesion with adjacent cells. Increasing evidence implicates the disruption of tight junctions as a critical step during NC cell EMT and migration (Figures 2 and 4). Claudins and Occludins are downregulated in the neural tube prior to NC cell migration,37 and Snail, a known transcriptional mediator of EMT, has been shown to directly repress Claudin and Occludin gene expression.38,39 Furthermore, the downregulation of the tight junction protein Claudin-1 promotes migration of chick cranial NC, whereas overexpression impedes crest migration.40 Additionally, inhibition of the tight junction-associated scaffolding protein Cingulin was recently shown to increase the size of the migratory NC cell domain.98 In cancer cell biology, disruption of tight junctions is thought to reduce cell adhesion allowing for cancer cell migration, as well as to increase vascular permeability and metastatic spread. Misregulation of tight junction proteins has been observed in a vast range of primary human cancers and model systems, including cancer of the breast, lung, brain and peripheral nerves, skin, oral cavity, endocrine organs, and genitourinary and gastrointestinal tracts.41
The disruption of adherens junctions has also been described in both NC cell development and cancer metastasis. Adherens junctions are comprised of several proteins, including classical cadherins and catenins. Classical cadherins form the core of adherens junctions, stabilizing adhesion to neighboring cells through the homophilic interaction of the extracellular domain, and anchoring adhesion to the actin or microtubule cytoskeleton through interaction with catenins.99 Differential regulation of cadherin expression plays a critical role in cell–cell interaction during NC cell EMT.42 The downregulation of E-cadherin, a type 1 cadherin associated with epithelial cell integrity, coincident with the upregulation of N-cadherin and cadherin-6b, initially defines the neuroepithelium and premigratory NC cells in zebrafish, mouse, and chick.42–44 Transitions in cadherin isoform expression also appear to be critical for cells to acquire a motile phenotype.100 The downregulation of N-cadherin and Cad6 occurs prior to NC cell migration, switching expression to a less adherent type 2 cadherin: Cad7 in chick and Cad11 in mouse and Xenopus.10,42,46,47 Conversely, continued expression of N-cadherin inhibits NC delamination by maintaining adherens junctions and sequestering β-catenin from functioning in cell signaling,48 highlighting the critical role of cadherin regulation in EMT. The involvement of cadherins in human cancer has also been well established. The disruption of junctional proteins, including E-cadherin, is required for cancer cell movement and thus is a prominent feature in most human carcinomas, leading to an invasive phenotype and poor prognosis.45 Deregulation of N-cadherin has also been implicated in cancer metastasis, promoting motility in human breast cancer cells regardless of their E-cadherin expression.49,50
Cellular polarity is established through the asymmetric distribution of cellular organelles and proteins, and is essential for a variety of cellular processes including directed cell migration. Disruption of cell polarity is one of the defining features of EMT in both NC cell development and tumor metastasis. Because they form initially as part of the neuroepithelium, NC cells exhibit an epithelial apical–basolateral polarity that is altered at the onset of EMT and migration. At the molecular level, apical–basolateral polarity is established by evolutionarily conserved polarity proteins that form multiprotein complexes. Polarity complexes are localized in specific cellular domains and maintain cellular polarity through mutually antagonistic interactions: the Crumbs complex [CRB(1-3)/Pals1/PatJ], localized at the apical cell cortex, stabilizes the localization of the Par complex (Par3/Par6/aPKC) at the tight junction. The Par complex is also excluded from the basolateral domain by the Scribble complex (Scribble/Dgl/Lgl).51 Although the processes of establishing and maintaining apical–basolateral cell polarity have been well studied, the mechanisms underlying the reorganization of cellular polarity during NC EMT remain unclear.
Nonetheless, despite the limited understanding of how polarity proteins are regulated during EMT, increasing evidence implicates disruption of polarity proteins in cancer progression and poor clinical prognosis. For example, the downregulation of Crumbs protein CRB3 is required for tumor formation in mouse epithelial cells and correlates with high levels of vimentin and reduced expression of E-cadherin, both hallmarks of EMT, suggesting that CRB3 may normally function to maintain tight junctions, apicobasal polarity, contact inhibited growth, and suppress migration and metastasis.52 Snail and Zeb1 have been shown to directly bind to the promoter and repress the transcription of the Crumbs gene, highlighting Crumbs regulation as a potential mediator of EMT.53–55 Par3 expression and subsequent stability of the PAR complex are regulated by transforming growth factor β (TGFβ) signaling,56 a key regulator of EMT in tumor formation.57,58 This suggests that TGFβ pathways may mediate loss of apicobasal cell polarity and drive EMT associated with cancer progression.56 In addition, the overexpression of aPKC, a component of the PAR complex, has also been observed in cancer progression and correlates with a poor clinical prognosis in ovarian cancer, breast cancer, and non-small cell lung cancer.59 The precise mechanisms by which polarity proteins function in NC EMT and in tumor progression remain unclear; however, accumulating evidence of their involvement highlights the potential importance of polarity proteins in both NC cell and cancer cell biology.
NC CELLS AND METASTATIC CANCER CELLS USE SIMILAR MIGRATION STRATEGIES
Following delamination, NC cells migrate from the dorsal neural tube to disparate tissues where they will differentiate. Depending on the environment through which they transit, NC cells exhibit different cellular strategies during migration. Cranial NC cells, for example, migrate as sheets of cells, consisting of both leader and follower cells. The leading cells respond to chemoattractants, whereas the following cells utilize contact inhibition to maintain polarity and directionality in their migration to the ventral pharyngeal arches.4 Trunk and enteric NC cells exhibit a very different mechanism of migration whereby they move in streams or single-cell chains.101,102 Similarly, cancer cells can exhibit one or more of these strategies to migrate from the primary tumor, either in single cells or in sheets, typically along blood vessels to new tissues.103
There are several processes that are common between NC cell and cancer cell migration (Figure 3). First, the cells must break down the ECM through which they migrate using MMPs that degrade components of the ECM. MMP-2 and MMP-8 are expressed as NC cells exit the neural tube and begin migrating60,61 and are required for NC cell migration.62,63 Cancer cells also use MMPs for degradation of ECM during EMT and migration.66 As with NC cells, MMP-2 enhances cancer cell migration in vitro,67 in an orthotopic mouse model of breast cancer,68 and is associated with a decrease in disease-free survival in human prostate69 and non-small cell lung cancer patients.70 There is also evidence that MMP-2, along with MMP-9 and MMP-14, have a role in promoting invasion and angiogenesis in mice.71
ADAMs are another family of metalloproteinases involved in cell migration. In Xenopus, ADAM9 and ADAM13 are required for breakdown of the ECM by cranial NC cells64 and the cleavage of cadherin-11.65 Several of the ADAM family members have also been implicated in the progression of cancer through the inhibition of apoptosis and the promotion of cell proliferation and angiogenesis.72 Additional studies have shown a correlation between many of the ADAM proteins and high metastatic rate and poor prognosis in human patients,73–76 although the multiple signaling functions of ADAMs could also contribute to these results.
Once NC cell migration begins, both attractive and inhibitory signals from the environment guide their movement. A positive cue for NC migration is the chemokine SDF1/CXCL12 from the microenvironment, which activates the CXCR4 receptor within migrating NC cells. Expression of the SDF1 ligand in the pharyngeal arches, along with expression of the CXCR4 receptor in the anterior migratory stream of NC cells, is necessary for proper migration of the cranial NC to populate the craniofacial region in zebrafish77 and Xenopus.78 Similarly, SDF-1/CXCR4 signaling is also required in the mouse trunk in order for NC cells to migrate and populate dorsal root ganglia.79 The role of chemokines and their receptors in cancer cell biology and metastatic spread was first documented in metastatic human breast cancer.80 In the past decade, this signaling pathway has been implicated in the metastatic spread of a multitude of human tumors. The expression of chemokine receptors by tumor cells is thought to provide them with access to the normal migratory pathways utilized in development during organogenesis, as well as access to pathways that direct the migration of discrete populations of immune cells to specific target organs for the establishment of regional immunity.81 Furthermore, expression of chemokines by tumor cells has also been proposed to promote tumor cell growth, angiogenesis, and the formation of immunotolerant microenvironments.81
Directed migration of NC cells also relies on inhibitory signals to keep the migrating cells spatially organized. The key signals involved in this process are the ligands, ephrin and semaphorin. They are expressed in regions where the NC cells do not normally migrate, such as the scleratome in the trunk or the tissues in between streams of migrating cells.8,82 The receptors for these ligands, Eph and neuropilin, are expressed by cephalic NC cells. Activation of these signaling pathways prevents migration of NC cells into specific zones by inducing collapse of cellular projections.8 Loss of normal Eph/ephrin or neuropilin/semaphorin signaling results in ectopic migration of NC into other tissues and mixing of NC streams, ultimately preventing proper patterning of the embryo.83–85 There is evidence that guidance molecules including ephrin and semaphorin are misregulated in human cancers including lung and breast cancer, suggesting that these signals may have a role in tumor progression and metastasis; however, the complexity of these signaling interactions complicates our understanding of how these molecules function in a metastatic environment.86,87 Further understanding of the role of pathfinding in tumor cell invasion will likely yield a wealth of information on how tumor cells metastasize.
CONCLUSIONS
NC cells and tumor cells both undergo dynamic processes to transition from an epithelial layer to migratory cells. The molecular interactions that govern these developmental processes in NC cells are echoed by the misregulation of these same processes throughout tumorigenesis and cancer cell migration, making NC EMT and migration an excellent model for understanding cancer formation, progression, and metastasis. Interestingly, the deregulation of several early developmental genes is necessary and/or sufficient for cancer progression, suggesting that these genes regulate key steps in cell proliferation, cell death avoidance, and invasiveness. Identifying how this reprogramming occurs, either within a stem-like cell residing in a mature tissue or within a mature fully differentiated cell, will be critical in raising our understanding of the progression of cancer. A vast array of molecular tools and model systems are available and have been used repeatedly in the study of the NC, which can provide great insight into the intricacies of human cancer. Many complex and intriguing questions still remain in the processes of EMT and migration, such as how cell adhesion molecules are regulated temporally and spatially, how migrating cells communicate with each other and the environment, how components of the ECM are regulated during migration, how cells at the end of migration begin to colonize within their target tissue, whether there is a mesenchymal-to-epithelial transition of migrating cells within the target tissue, and how this is regulated. Exploring these questions through the development of the NC may provide key insights into the regulation of tumor cell adhesion, communication, and migration through tissues during cancer metastasis, which will lead to the identification of disease predictors and potential therapeutic targets.
Acknowledgments
We thank Mary Reyland and Jenean O'Brien for critically reviewing this manuscript. Davalyn R. Powell is supported by National Institutes of Health (F31-DE022237). Work in the Britt lab is supported by the National Institutes of Health (R01-EY018376). Work in the Artinger lab is supported by the National Institutes of Health (R01-DE017699). This publication is in partial fulfillment of the requirements of the course CSDV 7850, Independent Study.
REFERENCES
- 1.Bronner ME, LeDouarin NM. Development and evolution of the neural crest: an overview. Dev Biol. 2012;366:2–9. doi: 10.1016/j.ydbio.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- 3.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 4.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 5.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Strobl-Mazzulla PH, Bronner ME. Epithelial to mesenchymal transition: new and old insights from the classical neural crest model. Semin Cancer Biol. 2012;22:411–416. doi: 10.1016/j.semcancer.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012;366:34–54. doi: 10.1016/j.ydbio.2011.12.041. [DOI] [PubMed] [Google Scholar]
- 9.Kulesa PM, Gammill LS. Neural crest migration: patterns, phases and signals. Dev Biol. 2010;344:566–568. doi: 10.1016/j.ydbio.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell. 2005;8:179–192. doi: 10.1016/j.devcel.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 11.LaBonne C, Bronner-Fraser M. Snail-related transcriptional repressors are required in Xenopus for both the induction of the neural crest and its subsequent migration. Dev Biol. 2000;221:195–205. doi: 10.1006/dbio.2000.9609. [DOI] [PubMed] [Google Scholar]
- 12.Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264:835–839. doi: 10.1126/science.7513443. [DOI] [PubMed] [Google Scholar]
- 13.Blanco MJ, Barrallo-Gimeno A, Acloque H, Reyes AE, Tada M, Allende ML, Mayor R, Nieto MA. Snail1a and Snail1b cooperate in the anterior migration of the axial mesendoderm in the zebrafish embryo. Development. 2007;134:4073–4081. doi: 10.1242/dev.006858. [DOI] [PubMed] [Google Scholar]
- 14.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De HerrerosA. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 15.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 16.Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26:1862–1874. doi: 10.1038/sj.onc.1209997. [DOI] [PubMed] [Google Scholar]
- 17.Tang P, Yu Z, Zhang K, Wang Y, Ma Z, Zhang S, Chen D, Zhou Y. Slug down-regulation by RNA interference inhibits invasion growth in human esophageal squamous cell carcinoma. BMC Gastroenterol. 2011;11:60. doi: 10.1186/1471-230X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Wang X, Zhang K, Sun L, Liu J, Lu H. Short interfering RNA directed against Slug blocks tumor growth, metastasis formation, and vascular leakage in bladder cancer. Med Oncol. 2011;28(suppl 1):S413–S422. doi: 10.1007/s12032-010-9728-4. [DOI] [PubMed] [Google Scholar]
- 19.Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, Weinberg RA. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. 2005;37:1047–1054. doi: 10.1038/ng1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van de Putte T, Francis A, Nelles L, van Grunsven LA, Huylebroeck D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum Mol Genet. 2007;16:1423–1436. doi: 10.1093/hmg/ddm093. [DOI] [PubMed] [Google Scholar]
- 21.Van de Putte T, Maruhashi M, Francis A, Nelles L, Kondoh H, Huylebroeck D, Higashi Y. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am J Hum Genet. 2003;72:465–470. doi: 10.1086/346092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 23.Cacheux V, Dastot-Le Moal F, Kaariainen H, Bondurand N, Rintala R, Boissier B, Wilson M, Mowat D, Goossens M. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet. 2001;10:1503–1510. doi: 10.1093/hmg/10.14.1503. [DOI] [PubMed] [Google Scholar]
- 24.Wakamatsu N, Yamada Y, Yamada K, Ono T, Nomura N, Taniguchi H, Kitoh H, Mutoh N, Yamanaka T, Mushiake K, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet. 2001;27:369–370. doi: 10.1038/86860. [DOI] [PubMed] [Google Scholar]
- 25.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 26.Hong CS, Saint-Jeannet JP. Sox proteins and neural crest development. Semin Cell Dev Biol. 2005;16:694–703. doi: 10.1016/j.semcdb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 28.Harris ML, Baxter LL, Loftus SK, Pavan WJ. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:496–513. doi: 10.1111/j.1755-148X.2010.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herbarth B, Pingault V, Bondurand N, Kuhlbrodt K, Hermans-Borgmeyer I, Puliti A, Lemort N, Goossens M, Wegner M. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc Natl Acad Sci U S A. 1998;95:5161–5165. doi: 10.1073/pnas.95.9.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelsh RN, Eisen JS. The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development. 2000;127:515–525. doi: 10.1242/dev.127.3.515. [DOI] [PubMed] [Google Scholar]
- 31.Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, Claudinot S, Okoniewski M, Beermann F, Mihic-Probst D, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol. 2012;14:882–890. doi: 10.1038/ncb2535. [DOI] [PubMed] [Google Scholar]
- 32.Soo K, O'Rourke MP, Khoo PL, Steiner KA, Wong N, Behringer RR, Tam PP. Twist function is required for the morphogenesis of the cephalic neural tube and the differentiation of the cranial neural crest cells in the mouse embryo. Dev Biol. 2002;247:251–270. doi: 10.1006/dbio.2002.0699. [DOI] [PubMed] [Google Scholar]
- 33.Vincentz JW, Barnes RM, Rodgers R, Firulli BA, Conway SJ, Firulli AB. An absence of Twist1 results in aberrant cardiac neural crest morphogenesis. Dev Biol. 2008;320:131–139. doi: 10.1016/j.ydbio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aaku-Saraste E, Hellwig A, Huttner WB. Loss of occludin and functional tight junctions, but not ZO-1, during neural tube closure–remodeling of the neuroepithelium prior to neurogenesis. Dev Biol. 1996;180:664–679. doi: 10.1006/dbio.1996.0336. [DOI] [PubMed] [Google Scholar]
- 38.Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116((Pt 10)):1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Estrada OM, Culleres A, Soriano FX, Peinado H, Bolos V, Martinez FO, Reina M, Cano A, Fabre M, Vilaro S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem J. 2006;394((Pt 2)):449–457. doi: 10.1042/BJ20050591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fishwick KJ, Neiderer TE, Jhingory S, Bronner ME, Taneyhill LA. The tight junction protein claudin-1 influences cranial neural crest cell emigration. Mech Dev. 2012;129:275–283. doi: 10.1016/j.mod.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788:872–891. doi: 10.1016/j.bbamem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development. 1995;121:1321–1332. doi: 10.1242/dev.121.5.1321. [DOI] [PubMed] [Google Scholar]
- 43.Inoue T, Chisaka O, Matsunami H, Takeichi M. Cadherin-6 expression transiently delineates specific rhombomeres, other neural tube subdivisions, and neural crest subpopulations in mouse embryos. Dev Biol. 1997;183:183–194. doi: 10.1006/dbio.1996.8501. [DOI] [PubMed] [Google Scholar]
- 44.Liu Q, Liu B, Wilson AL, Rostedt J. cadherin-6 message expression in the nervous system of developing zebrafish. Dev Dyn. 2006;235:272–278. doi: 10.1002/dvdy.20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1:a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development. 1998;125:2963–2971. doi: 10.1242/dev.125.15.2963. [DOI] [PubMed] [Google Scholar]
- 47.Vallin J, Girault JM, Thiery JP, Broders F. Xenopus cadherin-11 is expressed in different populations of migrating neural crest cells. Mech Dev. 1998;75:171–174. doi: 10.1016/s0925-4773(98)00099-9. [DOI] [PubMed] [Google Scholar]
- 48.Shoval I, Ludwig A, Kalcheim C. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development. 2007;134:491–501. doi: 10.1242/dev.02742. [DOI] [PubMed] [Google Scholar]
- 49.Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999;147:631–644. doi: 10.1083/jcb.147.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol. 2008;9:833–845. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karp CM, Tan TT, Mathew R, Nelson D, Mukherjee C, Degenhardt K, Karantza-Wadsworth V, White E. Role of the polarity determinant crumbs in suppressing mammalian epithelial tumor progression. Cancer Res. 2008;68:4105–4115. doi: 10.1158/0008-5472.CAN-07-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26:6979–6988. doi: 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537–544. doi: 10.1158/0008-5472.CAN-07-5682. [DOI] [PubMed] [Google Scholar]
- 55.Whiteman EL, Liu CJ, Fearon ER, Margolis B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene. 2008;27:3875–3879. doi: 10.1038/onc.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Nie J, Zhou Q, Liu W, Zhu F, Chen W, Mao H, Luo N, Dong X, Yu X. Downregulation of Par-3 expression and disruption of Par complex integrity by TGF-β during the process of epithelial to mesenchymal transition in rat proximal epithelial cells. Biochim Biophys Acta. 2008;1782:51–59. doi: 10.1016/j.bbadis.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 59.Huang L, Muthuswamy SK. Polarity protein alterations in carcinoma: a focus on emerging roles for polarity regulators. Curr Opin Genet Dev. 2010;20:41–50. doi: 10.1016/j.gde.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai DH, Vollberg TM, Sr, Hahn-Dantona E, Quigley JP, Brauer PR. MMP-2 expression during early avian cardiac and neural crest morphogenesis. Anat Rec. 2000;259:168–179. doi: 10.1002/(SICI)1097-0185(20000601)259:2<168::AID-AR7>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 61.Giambernardi TA, Sakaguchi AY, Gluhak J, Pavlin D, Troyer DA, Das G, Rodeck U, Klebe RJ. Neutrophil collagenase (MMP-8) is expressed during early development in neural crest cells as well as in adult melanoma cells. Matrix Biol. 2001;20:577–587. doi: 10.1016/s0945-053x(01)00166-4. [DOI] [PubMed] [Google Scholar]
- 62.Anderson RB. Matrix metalloproteinase-2 is involved in the migration and network formation of enteric neural crest-derived cells. Int J Dev Biol. 2010;54:63–69. doi: 10.1387/ijdb.082667ra. [DOI] [PubMed] [Google Scholar]
- 63.Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229:42–53. doi: 10.1002/dvdy.10465. [DOI] [PubMed] [Google Scholar]
- 64.Alfandari D, Cousin H, Gaultier A, Smith K, White JM, Darribere T, DeSimone DW. Xenopus ADAM 13 is a metalloprotease required for cranial neural crest-cell migration. Curr Biol. 2001;11:918–930. doi: 10.1016/s0960-9822(01)00263-9. [DOI] [PubMed] [Google Scholar]
- 65.McCusker C, Cousin H, Neuner R, Alfandari D. Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell. 2009;20:78–89. doi: 10.1091/mbc.E08-05-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 67.Xu X, Wang Y, Chen Z, Sternlicht MD, Hidalgo M, Steffensen B. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 2005;65:130–136. [PubMed] [Google Scholar]
- 68.Mendes O, Kim HT, Lungu G, Stoica G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin Exp Metastasis. 2007;24:341–351. doi: 10.1007/s10585-007-9071-0. [DOI] [PubMed] [Google Scholar]
- 69.Trudel D, Fradet Y, Meyer F, Harel F, Tetu B. Significance of MMP-2 expression in prostate cancer: an immunohistochemical study. Cancer Res. 2003;63:8511–8515. [PubMed] [Google Scholar]
- 70.Leinonen T, Pirinen R, Bohm J, Johansson R, Kosma VM. Increased expression of matrix metalloproteinase-2 (MMP-2) predicts tumour recurrence and unfavourable outcome in non-small cell lung cancer. Histol Histopathol. 2008;23:693–700. doi: 10.14670/HH-23.693. [DOI] [PubMed] [Google Scholar]
- 71.Masson V, de la Ballina LR, Munaut C, Wielockx B, Jost M, Maillard C, Blacher S, Bajou K, Itoh T, Itohara S, et al. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 2005;19:234–236. doi: 10.1096/fj.04-2140fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rocks N, Paulissen G, El Hour M, Quesada F, Crahay C, Gueders M, Foidart JM, Noel A, Cataldo D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008;90:369–379. doi: 10.1016/j.biochi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 73.Jiang C, Zhang Y, Yu HF, Yu XT, Zhou SJ, Tan YF. Expression of ADAM8 and its clinical values in diagnosis and prognosis of hepatocellular carcinoma. Tumour Biol. 2012;33:2167–2172. doi: 10.1007/s13277-012-0477-1. [DOI] [PubMed] [Google Scholar]
- 74.Roy R, Wewer UM, Zurakowski D, Pories SE, Moses MA. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem. 2004;279:51323–51330. doi: 10.1074/jbc.M409565200. [DOI] [PubMed] [Google Scholar]
- 75.Xiao LJ, Lin P, Lin F, Liu X, Qin W, Zou HF, Guo L, Liu W, Wang SJ, Yu XG. ADAM17 targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to promote prostate cancer cell invasion. Int J Oncol. 2012;40:1714–1724. doi: 10.3892/ijo.2011.1320. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Tan YF, Jiang C, Zhang K, Zha TZ, Zhang M. High ADAM8 expression is associated with poor prognosis in patients with hepatocellular carcinoma. Pathol Oncol Res. 2013;19:79–88. doi: 10.1007/s12253-012-9560-6. [DOI] [PubMed] [Google Scholar]
- 77.Olesnicky Killian EC, Birkholz DA, Artinger KB. A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev Biol. 2009;333:161–172. doi: 10.1016/j.ydbio.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, Mayor R. Collective chemotaxis requires contact-dependent cell polarity. Dev Cell. 2010;19:39–53. doi: 10.1016/j.devcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Belmadani A, Tran PB, Ren D, Assimacopoulos S, Grove EA, Miller RJ. The chemokine stromal cell-derived factor-1 regulates the migration of sensory neuron progenitors. J Neurosci. 2005;25:3995–4003. doi: 10.1523/JNEUROSCI.4631-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 81.Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597–606. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 82.Krull CE, Lansford R, Gale NW, Collazo A, Marcelle C, Yancopoulos GD, Fraser SE, Bronner-Fraser M. Interactions of Eph-related receptors and ligands confer rostrocaudal pattern to trunk neural crest migration. Curr Biol. 1997;7:571–580. doi: 10.1016/s0960-9822(06)00256-9. [DOI] [PubMed] [Google Scholar]
- 83.Gammill LS, Gonzalez C, Gu C, Bronner-Fraser M. Guidance of trunk neural crest migration requires neuropilin 2/semaphorin 3F signaling. Development. 2006;133:99–106. doi: 10.1242/dev.02187. [DOI] [PubMed] [Google Scholar]
- 84.Schwarz Q, Maden CH, Davidson K, Ruhrberg C. Neuropilin-mediated neural crest cell guidance is essential to organise sensory neurons into segmented dorsal root ganglia. Development. 2009;136:1785–1789. doi: 10.1242/dev.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith A, Robinson V, Patel K, Wilkinson DG. The EphA4 and EphB1 receptor tyrosine kinases and ephrin-B2 ligand regulate targeted migration of branchial neural crest cells. Curr Biol. 1997;7:561–570. doi: 10.1016/s0960-9822(06)00255-7. [DOI] [PubMed] [Google Scholar]
- 86.Harburg GC, Hinck L. Navigating breast cancer: axon guidance molecules as breast cancer tumor suppressors and oncogenes. J Mammary Gland Biol Neoplasia. 2011;16:257–270. doi: 10.1007/s10911-011-9225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nasarre P, Potiron V, Drabkin H, Roche J. Guidance molecules in lung cancer. Cell Adh Migr. 2010;4:130–145. doi: 10.4161/cam.4.1.10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sakai D, Suzuki T, Osumi N, Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. 2006;133:1323–1333. doi: 10.1242/dev.02297. [DOI] [PubMed] [Google Scholar]
- 89.Thisse C, Thisse B, Postlethwait JH. Expression of snail2, a second member of the zebrafish snail family, in cephalic mesendoderm and presumptive neural crest of wild-type and spadetail mutant embryos. Dev Biol. 1995;172:86–99. doi: 10.1006/dbio.1995.0007. [DOI] [PubMed] [Google Scholar]
- 90.Montero-Balaguer M, Lang MR, Sachdev SW, Knappmeyer C, Stewart RA, De La Guardia A, Hatzopoulos AK, Knapik EW. The mother superior mutation ablates foxd3 activity in neural crest progenitor cells and depletes neural crest derivatives in zebrafish. Dev Dyn. 2006;235:3199–3212. doi: 10.1002/dvdy.20959. [DOI] [PubMed] [Google Scholar]
- 91.Murray SA, Gridley T. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc Natl Acad Sci U S A. 2006;103:10300–10304. doi: 10.1073/pnas.0602234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66:773–787. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 94.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113((Pt 22)):3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 95.Dow LE, Humbert PO. Polarity regulators and the control of epithelial architecture, cell migration, and tumorigenesis. Int Rev Cytol. 2007;262:253–302. doi: 10.1016/S0074-7696(07)62006-3. [DOI] [PubMed] [Google Scholar]
- 96.Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2012;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 97.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 98.Wu CY, Jhingory S, Taneyhill LA. The tight junction scaffolding protein cingulin regulates neural crest cell migration. Dev Dyn. 2011;240:2309–2323. doi: 10.1002/dvdy.22735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Etienne-Manneville S. Control of polarized cell morphology and motility by adherens junctions. Semin Cell Dev Biol. 2011;22:850–857. doi: 10.1016/j.semcdb.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 100.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008;121((Pt 6)):727–735. doi: 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- 101.Anderson RB, Newgreen DF, Young HM. Neural crest and the development of the enteric nervous system. Adv Exp Med Biol. 2006;589:181–196. doi: 10.1007/978-0-387-46954-6_11. [DOI] [PubMed] [Google Scholar]
- 102.Zhang Y, Kim TH, Niswander L. Phactr4 regulates directional migration of enteric neural crest through PP1, integrin signaling, and cofilin activity. Genes Dev. 2012;26:69–81. doi: 10.1101/gad.179283.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]