

Abstract
Alpha-synuclein (SNCA) is crucial in the pathogenesis of Parkinson's disease (PD), yet mutations in the SNCA gene are rare. Evidence for somatic genetic variation in normal humans, also involving the brain, is increasing, but its role in disease is unknown. Somatic SNCA mutations, arising in early development and leading to mosaicism, could contribute to PD pathogenesis and yet be absent or undetectable in DNA derived from peripheral lymphocytes. Such mutations could underlie the widespread pathology in PD, with the precise clinical outcome dependent on their type and the timing and location of their occurrence. We recently reported a novel SNCA mutation (c.150T>G, p.H50Q) in PD brain-derived DNA. To determine if there was mosaicism for this, a PCR and cloning strategy was used to take advantage of a nearby heterozygous intronic polymorphism. No evidence of mosaicism was found. High-resolution melting curve analysis of SNCA coding exons, which was shown to be sensitive enough to detect low proportions of 2 known mutations, did not reveal any further mutations in DNA from 28 PD brain-derived samples. We outline the grounds that make the somatic SNCA mutation hypothesis consistent with genetic, embryological, and pathological data. Further studies of brain-derived DNA are warranted and should include DNA from multiple regions and methods for detecting other types of genomic variation. © 2013 Movement Disorder Society
Keywords: SNCA, alpha-synuclein, somatic mutation, mosaicism, etiology of Parkinson's disease
Alpha-synuclein (SNCA), encoded by the SNCA (PARK1) gene, is central to the pathogenesis of Parkinson's disease (PD).1,2 It is the major component of Lewy bodies. Misfolding into oligomers and fibrils is believed to underlie its toxicity, although the precise nature of the toxic species remains unclear. Three SNCA missense mutations have been reported in pedigrees with autosomal dominant inheritance.3–5 We recently identified the c.150T>G/p.H50Q mutation in DNA derived from the brain of a single apparently sporadic case of late-onset PD.6 Copy number variations (CNVs, duplications and triplications) have also been described,7,8 and noncoding variation in SNCA is a risk factor for sporadic PD.9 Several large studies analyzing DNA from blood lymphocytes have not found additional mutations, including 1 of more than 1900 mostly sporadic patients.10
Mutations occurring postzygotically are termed somatic and can lead to mosaicism (the presence of more than 1 genetically distinct cell in a single organism).11 The number of cell divisions in normal development has led to the suggestion that each gene may mutate several times postzygotically, with the term somatic evolutionary genomics used to refer to the accumulation of genetic change within the cell lineage of a single individual.12 Somatic mutations occurring in early embryogenesis in a dividing cell whose progeny will include neurons, derived from the ectoderm, could contribute to PD, but they could be missed when mesoderm-derived lymphocyte DNA is analyzed, in which they might be absent or present at a level below the 15%–30% resolution limit of Sanger sequencing.13–15 Low-level mosaicism could have been missed even in the few small studies in which SNCA was analyzed in PD brain-derived DNA, as the methods were not sensitive enough.16–18 Somatic mutation has previously been suggested as a cause of sporadic neurodegenerative disorders19,20 and was very recently hypothesized as a possible explanation of phenotypically discordant LRRK2 monozygotic twins.21,22 In Alzheimer's disease, a case with mosaicism from somatic mutation of presenilin-1 was described, with 14% mutant DNA in the cortex.23 Hereditary spastic paraplegia caused by mosaicism for a spastin mutation has been reported.24 Very recently, a novel form of neurodegeneration with brain iron accumulation has been found to be a result of mutations in WDR45, with a somatic origin in some cases.25 Mosaicism for triplet-repeat neurodegenerative disorders from somatic mutation of the expanded repeat has been described,26,27 including fragile X premutation syndrome, in which somatic instability in brain appears more pronounced than in blood,28 and c9orf72 in amyotrophic lateral sclerosis.29 Mosaicism for the expanded repeat may be the cause of intrafamilial variation in Friedreich's ataxia30 and is associated with onset age in Huntington's disease.31 In the special case of the mitochondrial genome, heteroplasmic somatic DNA deletions and point mutations in the substantia nigra (SN) are associated with PD.32–34
We therefore decided to test the hypothesis that SNCA somatic coding mutations present in the brain may contribute to PD by investigating the possibility of mosaicism for the H50Q mutation, and by using high-resolution melting curve (HRM) analysis for detection of possible low-proportion mosaicism in PD brain-derived DNA.
Patients and Methods
DNA from the brains of 28 patients with idiopathic PD from the Queen Square Brain Bank was analyzed. Patients had given informed consent for use of their brains in research, and the study was approved by the local ethics committee. The demographics and clinical detail of this cohort are summarized in Supporting Table 1. DNA was available from the SN and cerebellum in 5 cases, from the cerebellum in 7 cases, and from the caudate nucleus in 16 cases. The SN DNA had been previously sequenced in all 5 of the cases in which it was available, leading to the detection of the c.150T>G/p.H50Q mutation in 1 case.6 HRM analysis of PCR amplicons for all coding exons was performed using Idaho Technology HRM mastermix, amplicon melting on a Lightscanner (Idaho Technology, Salt Lake City, UT) and melt curves analysis with Call-IT 2.0 software. Further details and additional primers used for subcloning are shown in Supporting Table S1. HRM analysis is a robust and efficient method for screening small PCR amplicons for unknown mutations that relies on altered melting of heteroduplexes. Sensitivity is >99% for heterozygous point mutations and small insertions/deletions. In the case of mosaicism, a 5%–10% proportion of mutant DNA is detectable by HRM analysis,15 although detection of mutation proportion as low as 0.5% has been reported,35 making it highly suitable for screening for low-level mutations; the main limitation is the ability to analyze only small PCR amplicons, ideally 150–250 base pairs long.15
Results
Because of the a priori very low likelihood of an apparently sporadic late-onset PD case harboring a novel heterozygous missense mutation, we considered the possibility that the patient in whom we recently detected the novel c.150T>G (p.H50Q) mutation in the SN and cerebellum6 might have been a mosaic for a somatic mutation. Direct testing for the mutation in other tissues or in brain regions other than the SN or cerebellum was not possible, as no material was available. Inheritance could not be investigated, as relatives could not be traced. Mosaicism can be indirectly confirmed by determining the phase of a mutation in relation to a nearby heterozygous single-nucleotide polymorphism if a third allelic combination is present.23 Sequencing a PCR product including exon 3 but extending 430 bp upstream into intron 2 revealed heterozygosity for a known 5-base polymorphic insertion duplication (c.122–133_122–129dupTTTTT, rs72240586). A person heterozygous for both the c.150T>G exonic mutation and the 5T intronic duplication should have only 2 allele combinations, with the T and G at position c.150 each in cis with 1 of the rs72240586 variants; the presence of a third allele combination would indicate mosaicism and prove a somatic event (Fig. 1A). SN and cerebellar DNA was therefore amplified and subcloned in 2 independent experiments each; restriction digestion using BsgI (for which c.150T>G generates a restriction site) of direct colony PCR products was used to differentiate wild-type (wt) and mutant colonies. Sequencing of mutant colony PCR products from the SN and cerebellum revealed c.150G to be in cis with the rs72240586 duplication allele (Fig. 1B), whereas sequencing of all wt colony PCR products from 1 experiment of each DNA source demonstrated c.150T to be in cis with the reference (nonduplicated) rs72240586 allele (Fig. 1C), with no colony PCR products showing c.150T in cis with the rs72240586 duplication allele (Fig. 1D). Therefore, there was no evidence of mosaicism.
FIG. 1.

Determination of allele combinations at the rs72240586 polymorphic locus and c.150 (exon shown as light gray). A: Possible allele combinations in heterozygosity and mosaicism. The third combination indicates somatic origin of the c.150T>G mutation on the chromosome carrying rs72240586 allele B, with mosaicism evident, as both T and G are found in cis with allele B. B: Allele combination observed in chromosome without c.150T>G mutation. C: Allele combination observed in chromosome with c.150T>G mutation. D: Hypothetical third combination (not observed), which would have proven mosaicism by indicating that the mutation had arisen on the chromosome with the duplicated rs72240586 allele, but the original wild-type allele in cis with the rs72240586 duplication allele was also present.
To detect any SNCA somatic mutations that might be present at levels below the sensitivity of Sanger sequencing, we developed an HRM analysis protocol for the coding exons (2–6). We first verified that all known exon 3 mutations could be detected in the heterozygous state by performing HRM analysis on DNA samples carrying known mutations (SN with c.150T>G/p.H50Q and lymphocytes with c.157G>A/p.A53T and c.136G>A/p.E46K), all of which were differentiated from control DNA (Fig. 2). As our aim was to detect somatic mutations at levels below 50%, the lowest proportion detectable was determined by HRM analysis of serial dilutions of genomic DNA carrying the H50Q and A53T mutations (for which adequate DNA was available) with wt DNA. HRM analysis against controls in triplicate revealed that mutant DNA proportions of 12.5% and 2.5%, respectively, were detectable (Fig. 3). All coding exons were amplified and analyzed by HRM in all 28 samples, but no shifts in melting curves signifying additional mutations were detected.
FIG. 2.

HRM analysis of all known exon 3 mutations. A: Heterozygote c.157G>A (p.A53T) and c.150T>G (p.H50Q) in duplicate are differentiated from 3 controls in triplicate. B: c.136G>A (p.E46K) heterozygote in duplicate is differentiated from 2 controls in duplicate.
FIG. 3.

HRM analysis of DNA-carrying mutations, undiluted and diluted with control DNA, against control DNA in triplicate. The percentage of mutant DNA in each PCR template is shown. Left, c.157G>A (p.A53T), undiluted mutant percentage 50%, differentiated at 2.5% (1 in 20 dilution). Right, c.150T>G (p.H50Q), undiluted mutant percentage 50%, differentiated at 12.5% (1 in 4 dilution), not clearly differentiated at 5%.
Discussion
DNA sources for Somatic Mutation Studies
We found no evidence of mosaicism for H50Q within the cerebellum and SN, but the possibility of a somatic mutation arising very early in embryogenesis and therefore being present in all brain cells, precluding detection of mosaicism within a single tissue, could not be excluded.36,37 Detection of this mutation in additional cases would help to prove an inherited rather than a somatic origin. While this article was under review, the same mutation was reported in a Canadian familial patient, further supporting the conclusion that this was an inherited variant.38 We analyzed samples derived from different brain regions using HRM, which has proven sensitivity for low-level somatic mutations that could be missed by Sanger sequencing, but did not find any to support our hypothesis. The possibility that somatic mutations were present in other regions could not be excluded. Careful consideration needs to be given to the choice of brain region for the detection of hypothesized somatic mutations, although this may be limited by availability, as in our case. The pathology of PD extends well beyond the mesencephalon-derived SN, and even regions affected early in its course are anatomically and embryologically distant (the olfactory bulb arising from the prosencephalon, the dorsal motor nucleus of the vagus from the rhombencephalon, and enteric neurons from the neural crest39). Although the orderly progression described by Braak40 may explain this, we propose that the widespread early distribution of PD pathology could be partly explained by somatic mutations arising by the third week of embryogenesis; this would lead to neurons carrying the mutations found in all 3 primary brain vesicles (prosencephalon, mesencephalon, rhombencephalon) and the neural crest (Fig. 4). In this case, use of any neuroectodermal tissue could allow detection of relevant somatic mutations, and brain regions in which neurons are resistant to the disease pathology (eg cerebellum in PD) would have the highest chance of detection of surviving neurons carrying these mutations. It is important to emphasize that, although it may seem counterintuitive, mutations occurring before the 3 germ layers split could lead to mosaicism not restricted to the ectoderm, as already demonstrated in the case of the Alzheimer's mosaic, with an 8% mutation proportion in the mesoderm-derived blood, which was initially not detected23; such very early somatic mutations could therefore be detected in blood if sufficiently sensitive methods were used.
FIG. 4.
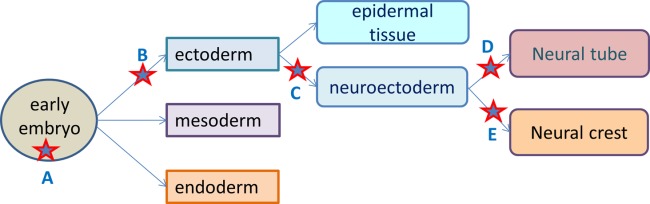
Schematic view of nervous system embryological derivation, with examples of somatic mutations affecting different cell populations (A–E). A: Mutation present in a proportion of cells from all germ layers. B: Mutation absent in nonectodermal tissues (eg, blood). C: Mutation present in all neuroectodermal tissue, including autonomic nervous system, but absent in skin. D: Mutation present throughout CNS. E: Mutation restricted to neural crest–derived structures (eg, adrenal medulla). As progenitor cell specification occurs very early, the diagram indicates the spatial distributions but not necessarily the precise timing of mutations.
Pathology Variants, Mosaicism Pattern, and Spread
As variations from the Braak model are well established in a proportion of PD cases,41 we suggest that the precise pathology pattern in Lewy body diseases could depend on the distribution of neurons derived from the precursor that acquired the mutation. This is consistent with reports of apparently multicentric early SNCA pathology in some incidental Lewy body disease cases.42 The unexplained asymmetry in PD could relate to an asymmetric, stochastic somatic mutation burden. Lewy body diseases with distinct profiles, such as pure autonomic failure that is accompanied by hyposmia as severe as PD,43 would be a result of a mosaic pattern that differs from PD. In an analogous way, somatic mutations have been proposed as the cause of patient-specific variations in light-chain deposition in systemic light chain-amyloidosis.44 A continuum of risk of PD and age of onset would be dependent on the relative mutation load of an individual's neural tissue, as suggested for neurodegenerative disease in general.12 Inherited mutations would lead to the highest risk and earliest onset; whereas somatic mutations very early in neuroectodermal development could lead to a similar situation, those arising later and affecting fewer neurons would result in later-onset disease, and those with the least neuronal involvement could result in clinically silent incidental Lewy body disease. This is consistent with the observation that, in general, age of onset is earlier and severity greater in those who have inherited a SNCA mutation than in sporadic cases and would lead us to expect a higher prevalence of somatic mutations and higher mutation load in younger-onset cases. It is worth noting that many structures derived from the neural crest (which forms as the dorsal neural tube closes and whose derivatives include the sensory ganglia, sympathetic chain and preaortic ganglia, parasympathetic gastrointestinal ganglia, Schwann cells, adrenal medulla, and salivary glands) demonstrate SNCA pathology45; in the adrenal gland, pathology is seen in the medulla and related nerve bundles, but not in the mesoderm-derived cortex. Somatic mutations occurring late in the neural crest lineage could underlie the occasional reports of isolated Lewy body–like pathology in peripheral structures such as the adrenal medulla46 and in heart and stellate ganglia.47
Somatic mutations occurring later in neurodevelopment and therefore restricted to a small region could modify protein conformation locally, and this could still lead to spread by permissive templating48 and prion-like propagation of pathology.49 The concept of spread of neurodegenerative disease from a small focus of cells with a somatic mutation has already been proposed, with particular emphasis on amyotrophic lateral sclerosis.12,50 In this scenario, a negative result in brain-derived DNA would not exclude an initiating somatic mutation, which would be extremely difficult to detect, as neurons carrying it would be among the first to die after triggering the pathogenic process; a similar issue was discussed in relation to mtDNA mutations, with SN neurons with high mtDNA mutation levels likely to be lost early in the disease course.34 It is clear that harboring a SNCA mutation is not enough to lead to death of any type of neuron, as even in cases with inherited mutations, which all neurons carry, the distribution of pathology is very specific.51 Selective vulnerability of neurons in PD is determined by a number of properties,52 and the eventual pathological effect of a somatic mutation would depend on the biological effect of the mutation per se, the pattern of mosaicism and mutation load, selective neuronal vulnerability, and patterns of interneuronal propagation that may occur.
Mosaicism for Other Types of Genomic Variants
We have focused our pilot work on attempting to detect small SNCA coding mutations, yet mosaicism for other types of genomic variation also merits consideration. CNV mosaicism is widespread,53 established very early in development,54 and present in normal brain.55 It is notable that a study of monozygotic twins discordant for PD or related phenotypes detected several copy number variations of somatic origin between cotwins of each pair, but as the DNA was derived from blood, no conclusion could be made on their pathogenicity.56 Somatic CNVs arise more frequently in chromosomal fragile sites,57,58 which include the regions where SNCA59 and PARK260 reside. CNVs can be induced by DNA “replication stress,”57 which is likely in the rapidly dividing neuronal precursors.61 Mosaicism for aneuploidy (gains or losses of whole chromosomes) is very common in humans.62 Aneuploidy is common in neural progenitor cells, from which the cerebral cortex is derived, and a significant proportion appear to survive into adulthood as postmitotic neurons,63 with aneuploidy estimated in 10% of human brain cells.64 Aneuploidy for chromosome 21 is more common in Alzheimer's disease brains than in controls,64 and hyperploid neurons in general are selectively vulnerable.65 Finally, the LINE-1 retrotransposon constitutes around 20% of the human genome, and abundant LINE-1 somatic rearrangements by a “copy-and-paste” mechanism arising specifically in neuronal precursors in embryonic development already indicate significant acquired variability in normal human neuronal genomes.66,67 No data on relevance to disease are available, but occasional somatic LINE-1 insertions were reported in healthy control brains in both SNCA and PARK2,66 confirming that these genes are susceptible to such disruption.
A Developmental and Evolutionary Perspective
Early somatic mutations leading to later neurodegeneration would be consistent with the “high initial load hypothesis” of the aging process, which proposes that initial damage of living organisms in fetal or early life may be responsible for later-onset degenerative disease.68 Even mtDNA mutations, generally considered to be acquired later in life, could arise in early development.69 Early somatic mutations may not, however, simply be accidents predisposing to later disease but could provide the basis for selection within an organism.11,70 This could be particularly relevant to selection of neuronal precursors and neurons that survive the massive apoptotic programmed cell death in early central nervous system development,71 which affects most neuronal populations, including dopaminergic neurons in the SN at the time of maximal competition for synaptic contact.72 A role for genetic variation (“chromosomal programming”) in the development of the nervous system, in a manner analogous to the immune system, was first suggested in 1967.73 Very recent data demonstrate that extreme aneuploidy is selected against in neurodevelopment.74 Multiple lines of evidence point to an important developmental role for SNCA, first in neuronal differentiation and later in synaptogenesis,75,76 with prominent perikaryal expression of SNCA in early development in the same neuronal groups later affected in PD.77 Therefore, we can speculate that certain somatic SNCA variants could be beneficial to neurons or their precursors in early development, undergoing positive selection within the organism, with PD a much later adverse consequence. As PD commonly develops past childbearing age, the mechanism that allows these early somatic changes would not be selected against and could indeed be favored by evolution if it allowed more genomic variation to provide a wider pool for selection of robust developing neurons. A similar explanation has already been put forward to account for LINE-1 mosaicism, suggesting that it favors generation of neuronal diversity, despite the possible risk of inducing neurological disease.78
Conclusions
The investigation of the potential contribution of somatic mutation to sporadic neurodegenerative disorders is in its infancy. Further large-scale analysis of brain DNA is warranted to test the hypothesis that somatic mutations of SNCA, or indeed other genes, may contribute to sporadic PD. The HRM protocol we have developed could be used for detecting low-proportion somatic coding SNCA mutations, but although HRM sensitivity can be improved even further to 0.1%–1% by the use of a PCR modification that preferentially amplifies low-level mutants,79 the falling cost and increasing versatility of next-generation sequencing should also allow its use for detection of low-proportion somatic mutations by using very high depth of coverage for targeted genomic regions; this was very recently demonstrated for selected mitochondrial80 and nuclear81,82 genes. Additional techniques need to be considered for detection of larger variants, such as FISH for aneuploidy; custom CGH arrays can improve sensitivity for mosaic CNVs,83 but the improving detection of CNVs in next-generation sequencing data and droplet digital PCR sensitivity of 0.1%53 is likely to revolutionize CNV mosaicism studies. Ideally, multiple brain regions should be sampled, and other neuroectoderm-derived tissues could be analyzed. Demonstration of mosaicism for any mutations detected in brain would preferably be confirmed by analysis of other tissues, and their collection by research facilities should be considered; conversely, clinicians should bear in mind that the absence of a detectable mutation in blood cannot exclude a somatic mutation.
Although we have not detected any evidence of somatic mutations in our pilot work, we believe that we have provided strong grounds for considering a contribution of somatic mutations in SNCA or other genes to PD in at least a proportion of cases. If SNCA and other PD genes are particularly susceptible to somatic mutation and/or if certain deleterious somatic variants paradoxically confer a selective developmental advantage to neuronal precursors or neurons, somatic mutation could underlie a substantial proportion of PD and other Lewy body-type pathology.
Acknowledgments
We are grateful to Dr. D. Mackay for technical help with HRM, to Professors M. de Pancorbo and L. Stefanis for providing genomic DNA with mutations as positive controls for HRM analysis, and to the Queen Square Brain Bank for brain samples.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Cookson MR, Bandmann O. Parkinson's disease: insights from pathways. Hum Mol Genet. 2010;19:R21–R27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- 3.Kruger R, Kuhn W, Muller T, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Zarranz JJ, Alegre J, Gomez-Esteban JC, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 6.Proukakis CBT, Mackay D, Houlden H, Schapira AH. A novel alpha-synuclein missense mutation in Parkinson's disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibanez P, Lesage S, Janin S, et al. α-Synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. 2009;66:102–108. doi: 10.1001/archneurol.2008.555. [DOI] [PubMed] [Google Scholar]
- 8.Mutez E, Leprêtre F, Le Rhun E, et al. SNCA locus duplication carriers: from genetics to Parkinson disease phenotypes. Hum Mutat. 2011;32:E2079–E2090. doi: 10.1002/humu.21459. [DOI] [PubMed] [Google Scholar]
- 9.Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg D, Niwar M, Maass S, et al. Alpha-synuclein and Parkinson's disease: implications from the screening of more than 1,900 patients. Mov Disord. 2005;20:1191–1194. doi: 10.1002/mds.20504. [DOI] [PubMed] [Google Scholar]
- 11.Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 12.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohlin A, Wernersson J, Engwall Y, Wiklund L, Bjork J, Nordling M. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat. 2009;30:1012–1020. doi: 10.1002/humu.20980. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vossen RH, Aten E, Roos A, den Dunnen JT. High-resolution melting analysis (HRMA): more than just sequence variant screening. Hum Mutat. 2009;30:860–866. doi: 10.1002/humu.21019. [DOI] [PubMed] [Google Scholar]
- 16.Chan P, Jiang X, Forno LS, Di Monte DA, Tanner CM, Langston JW. Absence of mutations in the coding region of the alpha-synuclein gene in pathologically proven Parkinson's disease. Neurology. 1998;50:1136–1137. doi: 10.1212/wnl.50.4.1136. [DOI] [PubMed] [Google Scholar]
- 17.El-Agnaf OM, Curran MD, Wallace A, et al. Mutation screening in exons 3 and 4 of alpha-synuclein in sporadic Parkinson's and sporadic and familial dementia with Lewy bodies cases. Neuroreport. 1998;9:3925–3927. doi: 10.1097/00001756-199812010-00029. [DOI] [PubMed] [Google Scholar]
- 18.Neystat M, Lynch T, Przedborski S, Kholodilov N, Rzhetskaya M, Burke RE. Alpha-synuclein expression in substantia nigra and cortex in Parkinson's disease. Mov Disord. 1999;14:417–422. doi: 10.1002/1531-8257(199905)14:3<417::aid-mds1005>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Pamphlett R. Somatic mutation: a cause of sporadic neurodegenerative diseases? Med Hypotheses. 2004;62:679–682. doi: 10.1016/j.mehy.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 20.Van Broeckhoven C. The future of genetic research on neurodegeneration. Nat Med. 2010;16:1215–1217. doi: 10.1038/nm.2225. [DOI] [PubMed] [Google Scholar]
- 21.Schneider SA, Johnson MR. Monozygotic twins with LRRK2 mutations: genetically identical but phenotypically discordant. Mov Disord. 2012;27:1203–1204. doi: 10.1002/mds.24991. [DOI] [PubMed] [Google Scholar]
- 22.Xiromerisiou G, Houlden H, Sailer A, Silveira-Moriyama L, Hardy J, Lees AJ. Identical twins with Leucine rich repeat kinase type 2 mutations discordant for Parkinson's disease. Mov Disord. 2012;27:1323–1323. doi: 10.1002/mds.24924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck JA, Poulter M, Campbell TA, et al. Somatic and germline mosaicism in sporadic early-onset Alzheimer's disease. Hum Mol Genet. 2004;13:1219–1224. doi: 10.1093/hmg/ddh134. [DOI] [PubMed] [Google Scholar]
- 24.Depienne C, Fedirko E, Faucheux J-M, et al. A de novo SPAST mutation leading to somatic mosaicism is associated with a later age at onset in HSP. Neurogenetics. 2007;8:231–233. doi: 10.1007/s10048-007-0090-4. [DOI] [PubMed] [Google Scholar]
- 25.Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancel G, Gourfinkel-An I, Stevanin G, et al. Somatic mosaicism of the CAG repeat expansion in spinocerebellar ataxia type 3/Machado-Joseph disease. Hum Mutat. 1998;11:23–27. doi: 10.1002/(SICI)1098-1004(1998)11:1<23::AID-HUMU4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 27.Chong SS, McCall AE, Cota J, et al. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1995;10:344–350. doi: 10.1038/ng0795-344. [DOI] [PubMed] [Google Scholar]
- 28.Lokanga RA, Entezam A, Kumari D, et al. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum Mutat. 2013;34:157–166. doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck J, Poulter M, Hensman D, et al. Large C9orf72 Hexanucleotide Repeat Expansions Are Seen in Multiple Neurodegenerative Syndromes and Are More Frequent Than Expected in the UK Population. Am J Hum Genet. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montermini L, Kish SJ, Jiralerspong S, Lamarche JB, Pandolfo M. Somatic mosaicism for Friedreich's ataxia GAA triplet repeat expansions in the central nervous system. Neurology. 1997;49:606–610. doi: 10.1212/wnl.49.2.606. [DOI] [PubMed] [Google Scholar]
- 31.Swami M, Hendricks AE, Gillis T, et al. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039–3047. doi: 10.1093/hmg/ddp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 33.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 34.Lin MT, Cantuti-Castelvetri I, Zheng K, et al. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol. 2012;71:850–854. doi: 10.1002/ana.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bastien R, Lewis TB, Hawkes JE, et al. High-throughput amplicon scanning of the TP53 gene in breast cancer using high-resolution fluorescent melting curve analyses and automatic mutation calling. Hum Mutat. 2008;29:757–764. doi: 10.1002/humu.20726. [DOI] [PubMed] [Google Scholar]
- 36.Kehrer-Sawatzki H, Cooper DN. Mosaicism in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes? J Med Genet. 2008;45:622–631. doi: 10.1136/jmg.2008.059329. [DOI] [PubMed] [Google Scholar]
- 37.Vadlamudi L, Dibbens LM, Lawrence KM, et al. Timing of de novo mutagenesis–a twin study of sodium-channel mutations. N Engl J Med. 2010;363:1335–1340. doi: 10.1056/NEJMoa0910752. [DOI] [PubMed] [Google Scholar]
- 38.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013 doi: 10.1002/mds.25421. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson's disease? Neuroscience. 2008;153:733–750. doi: 10.1016/j.neuroscience.2008.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braak H, Tredici KD, Udo R, Rob AIdV, Ernst NHJS, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 41.Jellinger KA. Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord. 2012;27:8–30. doi: 10.1002/mds.23795. [DOI] [PubMed] [Google Scholar]
- 42.Dickson D, Fujishiro H, DelleDonne A, et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acta Neuropathol. 2008;115:437–444. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein DS, Sewell L. Olfactory dysfunction in pure autonomic failure: Implications for the pathogenesis of Lewy body diseases. Parkinsonism Relat Disord. 2009;15:516–520. doi: 10.1016/j.parkreldis.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enqvist S, Sletten K, Stevens FJ, Hellman U, Westermark P. Germ line origin and somatic mutations determine the target tissues in systemic AL-amyloidosis. PLoS One. 2007;2:e981. doi: 10.1371/journal.pone.0000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beach T, Adler C, Sue L, et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fumimura Y, Ikemura M, Saito Y, et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J Neuropathol Exp Neurol. 2007;66:354–362. doi: 10.1097/nen.0b013e3180517454. [DOI] [PubMed] [Google Scholar]
- 47.Miki Y, Mori F, Wakabayashi K, Kuroda N, Orimo S. Incidental Lewy body disease restricted to the heart and stellate ganglia. Mov Disord. 2009;24:2299–2301. doi: 10.1002/mds.22775. [DOI] [PubMed] [Google Scholar]
- 48.Hardy J. Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ‘permissive templating’ as a general mechanism underlying neurodegeneration. Biochem Soc Trans. 2005;33:578–581. doi: 10.1042/BST0330578. [DOI] [PubMed] [Google Scholar]
- 49.Olanow CW, Prusiner SB. Is Parkinson's disease a prion disorder? Proc Natl Acad Sci U S A. 2009;106:12571–12572. doi: 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pamphlett R. The “somatic-spread” hypothesis for sporadic neurodegenerative diseases. Med Hypotheses. 2011;77:544–547. doi: 10.1016/j.mehy.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 51.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson's disease. Mov Disord. 2012;27:831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord. 2013;28:41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abyzov A, Mariani J, Palejev D, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mkrtchyan H, Gross M, Hinreiner S, et al. Early embryonic chromosome instability results in stable mosaic pattern in human tissues. PLoS One. 2010;5:e9591. doi: 10.1371/journal.pone.0009591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piotrowski A, Bruder CE, Andersson R, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 56.Bruder CE, Piotrowski A, Gijsbers AA, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet. 2008;82:763–771. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arlt MF, Wilson TE, Glover TW. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev. 2012;22:204–210. doi: 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 59.Rozier L, El-Achkar E, Apiou F, Debatisse M. Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer. Oncogene. 2004;23:6872–6880. doi: 10.1038/sj.onc.1207809. [DOI] [PubMed] [Google Scholar]
- 60.Mitsui J, Takahashi Y, Goto J, et al. Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am J Hum Genet. 2010;87:75–89. doi: 10.1016/j.ajhg.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez-Santiago B, Malats N, Rothman N, et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet. 2010;87:129–138. doi: 10.1016/j.ajhg.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A. 2001;98:13361–13366. doi: 10.1073/pnas.231487398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iourov IY, Vorsanova SG, Liehr T, Yurov YB. Aneuploidy in the normal, Alzheimer's disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis. 2009;34:212–220. doi: 10.1016/j.nbd.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 65.Arendt T, Bruckner MK, Mosch B, Losche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010;177:15–20. doi: 10.2353/ajpath.2010.090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baillie JK, Barnett MW, Upton KR, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479:534–537. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coufal NG, Garcia-Perez JL, Peng GE, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gavrilov LA, Gavrilova NS. Early-life programming of aging and longevity: the idea of high initial damage load (the HIDL hypothesis) Ann N Y Acad Sci. 2004;1019:496–501. doi: 10.1196/annals.1297.091. [DOI] [PubMed] [Google Scholar]
- 69.Khrapko K, Ebralidse K, Kraytsberg Y. Where and when do somatic mtDNA mutations occur? Ann N Y Acad Sci. 2004;1019:240–244. doi: 10.1196/annals.1297.040. [DOI] [PubMed] [Google Scholar]
- 70.Macosko EZ, McCarroll SA. Exploring the variation within. Nat Genet. 2012;44:614–616. doi: 10.1038/ng.2311. [DOI] [PubMed] [Google Scholar]
- 71.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 72.Oo TF, Burke RE. The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Brain Res Dev Brain Res. 1997;98:191–196. doi: 10.1016/s0165-3806(96)00173-3. [DOI] [PubMed] [Google Scholar]
- 73.Dreyer WJGW, Hood L. The Genetic, Molecular, and Cellular Basis of Antibody Formation: Some Facts and a Unifying Hypothesis. Cold Spring Harb Symp Quant Biol. 1967;32:353–367. [Google Scholar]
- 74.Peterson SE, Yang AH, Bushman DM, et al. Aneuploid cells are differentially susceptible to caspase-mediated death during embryonic cerebral cortical development. J Neurosci. 2012;32:16213–16222. doi: 10.1523/JNEUROSCI.3706-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bayer TA, Jakala P, Hartmann T, et al. Neural expression profile of alpha-synuclein in developing human cortex. Neuroreport. 1999;10:2799–2803. doi: 10.1097/00001756-199909090-00019. [DOI] [PubMed] [Google Scholar]
- 76.Galvin JE, Schuck TM, Lee VMY, Trojanowski JQ. Differential expression and distribution of α-, β-, and γ-synuclein in the developing human substantia nigra. Exp Neurol. 2001;168:347–355. doi: 10.1006/exnr.2000.7615. [DOI] [PubMed] [Google Scholar]
- 77.Raghavan R, Kruijff L, Sterrenburg MD, Rogers BB, Hladik CL, White CL., 3rd Alpha-synuclein expression in the developing human brain. Pediatric Develop Pathol. 2004;7:506–516. doi: 10.1007/s10024-003-7080-9. [DOI] [PubMed] [Google Scholar]
- 78.Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010;33:345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Milbury CA, Li J, Makrigiorgos GM. COLD-PCR-enhanced high-resolution melting enables rapid and selective identification of low-level unknown mutations. Clin Chem. 2009;55:2130–2143. doi: 10.1373/clinchem.2009.131029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Payne BA, Wilson IJ, Yu-Wai-Man P, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gerstung M, Beisel C, Rechsteiner M, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 82.Izawa K, Hijikata A, Tanaka N, et al. Detection of base substitution-type somatic mosaicism of the NLRP3 gene with >99.9% statistical confidence by massively parallel sequencing. DNA Res. 2012;19:143–152. doi: 10.1093/dnares/dsr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Valli R, Marletta C, Pressato B, et al. Comparative genomic hybridization on microarray (a-CGH) in constitutional and acquired mosaicism may detect as low as 8% abnormal cells. Molec Cytogenet. 2011;4:13. doi: 10.1186/1755-8166-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.