

Summary
Kaposi’s sarcoma—associated herpesvirus (KSHV) is the etiological agent of Kaposi’s sarcoma (KS), the most common cancer in AIDS patients. All herpesviruses express a conserved dimeric serine protease that is required for generating infectious virions, and is therefore of pharmaceutical interest. Given the past challenges of developing drug-like active-site inhibitors to this class of proteases, small-molecules targeting allosteric sites are of great value. In light of evidence supporting a strong structural linkage between the dimer interface and the protease active-site, we have focused our efforts on the dimer interface for identifying dimer disrupting inhibitors. Here, we describe a high throughput screening approach for identifying small molecule dimerization inhibitors of KSHV protease. The helical mimetic, small molecule library used, as well as general strategies for selecting compound libraries for this application will also be discussed. This methodology can be applicable to other systems where an alpha helical moiety plays a dominant role at the interaction site of interest, and in vitro assays to monitor function are in place.
Keywords: Human herpesvirus protease, Kaposi’s sarcoma-associated herpesvirus (KSHV), dimer disruptor, allosteric inhibitor, fluorogenic enzyme assay, alpha helical mimetic molecules, high throughput screening
1. Introduction
Herpesviruses make up one of the most prevalent viral families including eight human types that cause a variety of devastating illnesses. The standard course of treatment for common herpesviral infections is a class of broad—acting viral DNA replication inhibitors, which exhibit undesirable toxicity, poor oral bioavailability, and in some cases inadequate efficacy. Efforts by pharmaceutical companies to target the active-site of the essential dimeric serine protease of human herpesviruses (HHV) have yet to yield a drug-like candidate(1–6). Given the evidence supporting a conformational linkage between protease dimerization and activation, we have focused our efforts on identifying molecules that target the dimer interface(7–12). In the case of KSHV protease (KSHV Pr) the dimer interface covers approximately 2500 Å2, and includes the α-helix 5 of each monomer as the major constituent (Figure 1)(13). In vitro studies with KSHV Pr and other HHV proteases have shown the dimer interface to be very sensitive to genetic perturbation, where single point mutations often lead to a loss of dimerization and activity(11). Furthermore the dimerization affinity is weak; with a reported KD of 1.7µM for KSHV Pr(10). These characteristics define the dimer interface as a suitable candidate for dimerization inhibitors. We first tested this approach by inserting the key interfacial α—helix 5 residues on the internally—stabilized α—helix of a mini—protein(14). The resulting macromolecule disrupted the KSHV Pr dimer and inhibited enzyme activity, proving that targeting the dimer interface is a viable route for identifying novel inhibitors. In order to identify small molecule dimer disruptors of KSHV Pr, a workflow was developed that begins with high throughput screening (focus of this chapter), and continues with experiments that assess dimerization, mode of binding, and broad specificity against other HHV proteases (Figure 2) (15,16). Since two α-helices are the major component of the KSHV Pr dimer interface, screened a library of helical mimetic small molecules. This library was comprised of roughly 200 compounds that were originally developed by computational design to disrupt the interacting α—helix of p53 tumor suppressor protein with oncoprotein MDM2(17). Screening was performed in a 96—well plate format using a fluorogenic activity assay. The substrate used is an optimized hexa—peptide attached to 7—amino—4—carbamoylmethyl coumarin (ACC), where cleavage at the scissile bond releases the ACC group resulting in increased fluorescence(7,12). We have successfully used this approach to identify the small molecule inhibitor DD2 (Figure 3), which binds a novel allosteric pocket at the dimer interface of KSHV Pr, and traps it in an inactive monomeric state(16). Here we describe the important considerations in library selection as well as the detailed steps in the high throughput fluorogenic screening assay.
Figure 1.


KSHV Pr dimer. The top and side views of the dimeric crystal structure are shown. The active—site catalytic residues are in orange. The interfacial α—helix 5 moieties and the two independent catalytic triads are highlighted in black.
Figure 2.

The workflow for identifying dimer disruptors of HHV proteases. Inhibitors of KSHV Pr activity are identified by high throughput screening (HTS) of small molecule libraries. Hits are further characterized for mode of inhibition and dimer disruption through 2D—NMR assays that have been described previously. Lead compounds are then tested for efficacy against other HHV proteases. This method will focus on the HTS assay (dotted box).
Figure 3.
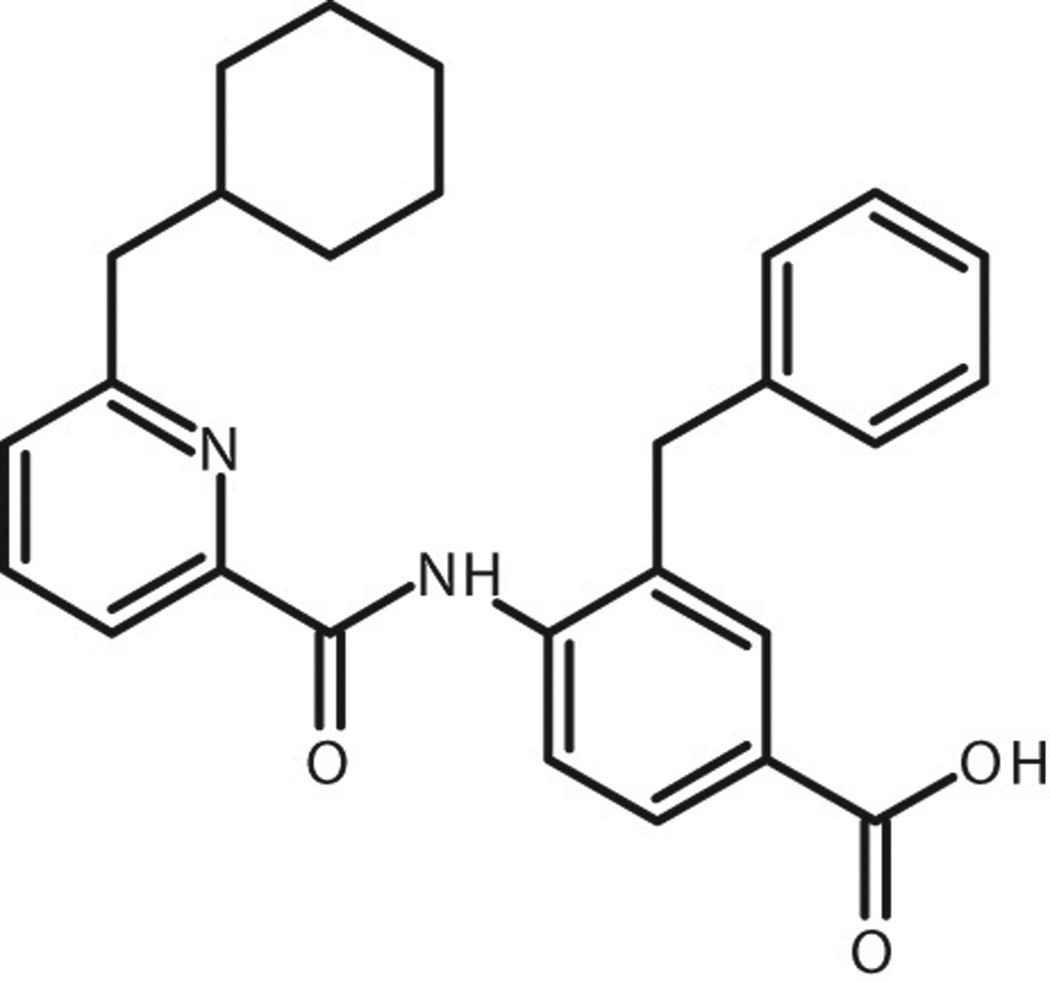
The structure of DD2.
2. Materials
2.1 Consumables and equipment
dimethyl sulfoxide (DMSO), 2—mercaptoethanol, and ethylenediaminetetraacetic acid (EDTA).
Round—bottom polypropylene 96—well assay plate, and plate sealing tape.
A 25 ml reagent reservoir, multichannel pipets (12—channel), and disposable tips for 10 and 200 µl volumes.
Fluorescence microplate reader, table-top centrifuge with microplate rotors, and a small incubator.
2.2 Buffer stocks
Prepare all aqueous solutions in MilliQ water followed by gentle shaking or mixing to ensure all solids have dissolved and all components have thoroughly mixed. Filter all 1 L stocks into sterile 1 L bottles. Store all reagent stocks at room temperature.
Prepare a 10 ml stock of 0.5M EDTA by dissolving 2.9 g EDTA into 10 ml of water.
Prepare a 1L stock of 1 M K2HPO4 by dissolving 141.9 g K2HPO4 into 1 L of water.
Prepare a 1L stock of 1 M KH2PO4 by dissolving 136.09 g KH2PO4 into 1 L of water.
Prepare 1 L stock of 3 M KCl by dissolving 223.6 g KCl into 1 L of water.
2.3 Assay buffer
The assay buffer consists of 25 mM potassium phosphate pH 8, 150 mM KCl, 0.1 mM EDTA, and 1 mM 2—mercaptoethanol.
Prepare a 100 ml stock of assay buffer by mixing 2.4 ml of 1M K2HPO4, 0.15 ml of 1 M KH2PO4, 5 ml of 3 M KCl, 20 µl of 0.5 M EDTA, and 92.48 ml water. Adjust pH to 8.0 using HCl and NaOH. Before use add 7 µl of 2–mercaptoethanol.
2.4 Protease expression and purification
Recombinant KSHV Pr is expressed in Escherichia coli and purified as reported previously(16). 80 µM aliquots of purified protease are flash frozen in storage buffer (same as assay buffer described in section 3.2) and stored at −20 °C. The total amount of protein required will vary depending on the assay and the number of plates being screened.
2.5 Protease substrate synthesis and purification
The protease substrate is an optimized hexa—peptide with the fluorogenic reporter group 7— amino-4—carbamoyl—methylcoumarin (ACC), which allows for monitoring enzyme activity spectroscopically. The peptide sequence is Ac—Pro—Val—Tyr—tBug—Gln—Ala—ACC with an observed KM of 8.5 ± 0.8 µM for KSHV Pr. Substrate is synthesized using standard FMOC chemistry and purified as reported previously(7,12). Substrate stocks of 10 mM in 100% DMSO are stored at −20 °C. The total amount of substrate required will vary depending on the assay and the number of plates being screened. (Ac = acetyl group, tBug = t-butyl glycine).
2.6 Test compounds
For general considerations on library selection see Note 1. Compound libraries are generally provided in ready-to-use format, dissolved in 100% DMSO and plated in either 96—well or 384—well plates. An additional dilution step with 100% DMSO or plate reformatting may be necessary depending on the screening assay conditions. In order to avoid screening artifacts resulting from compound aggregation or precipitation, a final screening concentration between 10—30 µM is recommended. Our compound library was in a ready-to-use format at a concentration of 1mM in a 96—well plate. Compound libraries are stored at −20 °C.
2.7 Control compound
Positive and negative controls are an important measure of assay performance and should ideally be included in every row (or column) of the assay plate. In assays, where timing is of particular importance, such as a protease assay, these controls allow for accurate calculation of % inhibition in each row (or column), as well as comparison of data across all rows (or columns). DMSO serves as the negative control and is used in the place of test compound. An ideal positive control may be a known active compound, such as a protease inhibitor in our case. The total amount of control compound required will vary depending on the assay and the number of plates being screened. At the time we performed our screen potent reversible KSHV Pr inhibitor did not exist, therefore substrate alone, which mimics the absence of protease activity, served as the positive control.
3. Methods
Prior to performing a high-throughput screen, it is critical to evaluate the quality of the screening assay by calculating a Z’—factor (see Note 2). High-throughput screening is typically performed in small assay volumes, therefore small fluctuations in liquid handling may affect the final readout. For this reason working with professionally calibrated pipettes is highly recommended. The following is a step—by—step protocol for manually performing a screen in one 96—well plate. The ability to screen multiple plates at once depends on the assay conditions and access to liquid handlers.
3.1 Summary of assay conditions
The protease assay is carried out in a final volume of 100 µl per well. It is composed of 98 µl of KSHV Pr in assay buffer, 1ul of test compound or DMSO, and 1ul of the protease substrate. These proportions ensure that the final concentration of protease is minimally changed upon substrate addition (see Methods).
In order to account for volume loss due to pipeting, add an extra 20% to all calculated reagent volumes. For example, to calculate the total volume of protease substrate required for 40 wells use the equation: [40 wells + (0.2 × 40 wells)] × 1 µl = 9.5 ml.
KSHV Pr is used at a final concentration of 2 µM. We selected a concentration close to the reported in vitro KD of 1.7 µM, in order to identify compounds that effectively compete with protease dimerization.
Test compounds are used at a final concentration of 10 µM. This concentration is typical for high throughput screening and helps avoid precipitation and aggregation.
Protease substrate is used at a final concentration of 100 µM, which is roughly 10 fold above its reported KM of 8.5 ± 0.8 µM for KSHV Pr. This concentration ensures that substrate binding is not the rate-limiting step in the screening assay.
Both the protease substrate and test compounds are dissolved in 100% DMSO. Therefore the final assay concentration of DMSO is 1% (v/v), which is well below the previously determined 5% DMSO tolerance of the assay (see Note 3).
3.2 Setup of 96-well plate
Column 1 is designated for the positive control. The components are 98 µl of assay buffer, 1 µl of DMSO, and 1ul of protease substrate (Figure 4).
Column 2 is designated for the negative control. The components are 98 µl of protease in assay buffer, 1 µl of DMSO, and 1ul of protease substrate.
Columns 3–12 are designated for the test compounds. The components are 98 µl of protease in assay buffer, 1 µl of test compound, and 1 µl of protease substrate.
Figure 4.

The 96—well plate arrangement. Positive (+) and negative (−) controls are located in columns 1 and 2 respectively. Test compounds are screened in columns 3—12. The wells in each row receive substrate at the same time. The controls in each row may be used to accurately calculate % inhibition.
3.3 Dispensing KSHV Pr to assay plate
Retrieve a frozen 80 µM vial of KSHV Pr from the freezer and thaw quickly by placing inside a 37 °C water bath. Once the protease is thawed, store the vial on ice.
Transfer 10.5 ml of assay buffer into clean 50—ml conical tube.
Add 275 µl of KSHV Pr from the thawed stock and mix. Label this conical tube “KSHV Pr”.
Carefully pour the entire mixture into a clean reagent reservoir labeled “KSHV Pr”.
Retrieve a clean 96—well plate, label it “Assay Plate”, and place on work bench at room temperature.
Load a 200 µl-volume multichannel pipette with eight clean tips, place inside “KSHV Pr” reservoir, and load 98 µl. Inspect all eight channels by eye to make sure the pipette is functioning properly. If large air bubbles are trapped inside the tip, empty all channels back into the “KSHV Pr” reservoir and try again.
Dispense contents into column 3, and then repeat until all remaining columns 4–12 receive KSHV Pr.
3.4 Dispensing test compounds to assay plate
Retrieve compound library from the −20 °C freezer and thaw at room temperature. Do not remove the plate seal.
Spin down plate in a table—top centrifuge at 3000 × g for 1 minute. After centrifugation remove the seal, label the plate “Compound Plate”, and place it on the work bench at room temperature.
Load a 10 µl-volume multichannel pipette with eight clean tips, place inside column 1 of “Compound Plate”, and load 1µl.
Dispense contents into column 3 of “Assay Plate”, such that compounds from wells A1—H1 of “Compound Plate” are transferred to wells A3—H3 of the “Assay Plate”.
Repeat steps 3—4 to transfer from columns 2—10 of “Compound Plate” to the corresponding columns 4—12 on the “Assay Plate”.
Columns 11—12 of the “Compound Plate” may be screened in a separate 96—well assay plate.
3.5 Dispensing the negative control to assay plate
Load a 200 µl—volume multichannel pipette with eight clean tips, place inside “KSHV Pr” reservoir, and load 98 µl.
Dispense contents into column 2 of “Assay Plate”.
In a fresh reagent reservoir, labeled “DMSO”, add 20 ml of DMSO.
Load a 10 µl—volume multichannel pipette with eight clean tips, place inside the “DMSO” reservoir and load 1 µl.
Dispense contents into column 2 of “Assay Plate”.
3.6 Dispensing the positive control to assay plate
Add 20 ml of assay buffer into a clean reagent reservoir labeled “Assay Buffer”.
Load a 200 µl—volume multichannel pipette with eight clean tips, place inside the “Assay Buffer” reagent reservoir, and load 98ul.
Dispense contents into column 1 of “Assay Plate”.
Load a 10 µl—volume multichannel pipette with eight clean tips, place inside “DMSO” reservoir and load 1 µl.
Dispense contents into column 1 of “Assay Plate”.
3.7 Incubation of assay plate
Load a 200 µl—volume multichannel pipet, with eight clean tips and set to 50 µl.
Place tips midway inside column 1 of “Assay Plate”, load and empty three times to mix contents.
Repeat steps 1—2 for columns 2—12.
Cover the plate using a sealing tape and spin down in a table—top centrifuge at 3000 × g for 1 minute.
Place plate inside 30 °C incubator for 30 minutes.
3.8 Addition of protease substrate to assay plate
Spin down “Assay Plate” in a table-top centrifuge at 3000 × g for 1 minute. Gently remove the plate seal.
Retrieve 10 mM stock of protease substrate from −20 °C freezer and thaw at room temperature.
In a fresh 96—well plate, labeled “Substrate Plate”, aliquot 15 µl of substrate into each well of row A.
Load a 10 µl—volume multichannel pipette with twelve clean tips, place inside row A of “Substrate Plate” and load 1 µl.
Dispense contents into row A of “Assay Plate”.
Load a 200 µl—volume multichannel pipette with twelve clean tips and set to 50 µl.
Place tips midway inside row A of “Assay Plate”, load and empty three times to mix contents.
Repeat steps 4—7 for rows B-H of “Assay Plate”.
3.9 Incubation of assay plate
Cover the plate using a new sealing tape and spin down in a table-top centrifuge at 3000 × g for 1 minute.
Place plate inside 30 °C incubator for 60 minutes. We have confirmed experimentally that the observed rate of substrate hydrolysis is linear during this time period.
3.10 Stopping the enzyme reaction
Spin down “Assay Plate” in a table-top centrifuge at 3000 × g for 1 minute.
Place “Assay Plate” on the work bench at room temperature. Carefully remove the sealing tape.
Load a 200 µl—volume multichannel pipette with twelve clean tips, place inside the “DMSO” reservoir, and load 100 µl.
Dispense contents into row A of “Assay Plate”.
Load a 200 µl—volume multichannel pipette with twelve clean tips and set to 100 µl.
Place tips midway inside row A of “Assay Plate”, load and empty three times to mix contents.
Repeat steps 2—5 for rows B-H of “Assay Plate”.
Cover the plate with a new sealing tape.
3.11 Reading the assay plate in fluorescence plate reader
Spin down “Assay Plate” in a table-top centrifuge at 3000 × g for 1 minute. Gently remove the plate seal.
Place in microplate reader and measure the endpoint fluorescence with the excitation and emission wavelengths set to 380 nm and 460 nm, respectively.
3.12 Data processing
-
For each row corrected endpoint fluorescence using the equation: EF—CP
EF is the endpoint fluorescence of the experimental and negative control wells. CP is the endpoint fluorescence of the positive control well.
-
For each row calculate the % inhibition using the equation: 1 – (TEF / NEF)
TEF is the corrected endpoint fluorescence of the experimental well with test compound. NEF is the corrected endpoint fluorescence of the negative control well.
Compounds that showed inhibition over 50% were considered true hits in our screen.
If possible repeat the screen and determine the average % inhibition from triplicate data set.
Follow up experiments will rule out false positives and determine if hits act as dimer disruptors (see Notes 4–7).
Acknowledgement
This work was supported by NIH grants T32 GMO7810, AIO67423 (C.S.C.), P50 GM 082250 and by the American Lebanese and Syrian Associated Charities and St Jude Children’s Research Hospital (R.K.G.).
Footnotes
KSHV Pr activity is regulated by a dimerization-driven conformational switch. The two α- helices located at the dimer interface became our rationale for screening a helical mimetic small-molecule library for inhibitors. α-helices make up the largest class of protein secondary structure and are commonly found at the interface of protein-protein interactions(18,19). Therefore significant effort has been aimed at developing non-peptide small molecule mimetics of α-helices. The key characteristic of such molecules is a rigid scaffold with functionalities presented in the same orientation as the i, i + 3 or i + 4, and i + 7 residue positions on an α—helix. The chemotypes of commonly reported scaffolds include biphenyls, allenes, alkylidene cycloalkanes, spiranes, benzylideneacetophenones, trisubstituted imidazole, indanes, polycyclic ethers, benzodiazepines and teraryl units(18). We had access to a small focused library of helical mimetic small molecules synthesized to disrupt the interaction of p53 and MDM2, which also includes an α-helix(17). The scaffolds were made of two or three aryl rings connected by amide bonds, with side chains in the i, i + 4, and i + 7 positions. It is worth noting that commercial helical mimetic libraries, as well as general protein-protein interaction libraries, are now available for purchase through companies like BioFocus. In the absence of suitable small molecule test candidates, peptidic alternatives such as hydrocarbon-stapled helical peptides can also serve as a starting point(20–22).
The Z’—factor is the measure of assay quality and is vital to identifying true hits in a high throughput screen(23). The value is typically calculated from the positive and negative control experiments, which in our screen correspond to the “low” and “high” signals respectively. The resulting numerical values range between 0 and 1, where numbers closest to 1 are favorable. The method of calculating the Z’—factor has been reported previously. Z’—factor optimization is achieved by varying the assay conditions such that the separation between the “low” and “high” signal is increased. We optimized our KSHV Pr assay by increasing the assay incubation temperature, the reaction incubation time, and including centrifugations steps.
DMSO exhibits an inhibitory affect in most assays, therefore its final concentration should be kept to a minimum. However, library compounds and protease substrates are generally dissolved in 100% DMSO stocks, and often require some DMSO to remain dissolved in aqueous buffer. For this reason it is important to determine the DMSO tolerance of screening assays. This is done by performing the basic assay in the presence of increasing concentration of DMSO until an inhibitory condition is reached. The DMSO tolerance for the KSHV Pr assay is 5% (v/v).
Some compounds form aggregates that interact with proteins in a non-specific manner(24,25). Aggregate forming compounds may result in a false-positive inhibitory effect when screened as part of large libraries. Including a small amount of detergent, such as 0.01% Triton X—100 (v/v), will eliminate aggregation-based hits in most cases(25). In the case of assays like the KSHV Pr assay that don’t tolerate detergents, including 1mg/ml BSA eliminates non-specific interactions. Ideally detergents are included in the initial screen. However, since the library we screened was small, we counter screened our hits in the presence of BSA.
Some compounds may exhibit fluorescent properties in the wavelengths monitored by the assay. For example a compound that absorbs light in the same region as our ACC substrate, may interfere with the final assay readout. Therefore, it’s important to monitor the abortions and emission properties of all screening hits at the wavelengths used to make read measurements.
A true inhibitor has a sigmoidal dose response cure with a hill slop value that is around 1. The concentration of inhibitor that achieves 50% inhibition is the IC50. Dose response curves are obtained by repeating the screening assay with increasing concentrations of compound, ranging from 0 to 100 µM. Generally a 3-fold dilution of compound is used (100 µM, 33.3 µM, 11.1 µM, etc.). Percent inhibition values (see Methods 3.11) are then plotted as a function of inhibitor concentration.
Since the goal of the screen is to identify dimer disruptors, secondary assays that monitor dimerization must be available. We used two independent but complementary methods to determine weather our hit DD2 was a dimer disruptor(7,16). The first was an FPLC assay and the second was a 2D—NMR approach using protease containing a reporter at the dimer interface. Both methods were carried out as described previously.
References
- 1.Borthwick AD, et al. Design and synthesis of pyrrolidine-5,5-trans-lactams (5-oxohexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 2. Potency and chirality. J Med Chem. 2002;45:1–18. doi: 10.1021/jm0102203. [DOI] [PubMed] [Google Scholar]
- 2.Borthwick AD, et al. Pyrrolidine-5,5-trans-lactams as novel mechanism-based inhibitors of human cytomegalovirus protease. Part 3: potency and plasma stability. Bioorg Med Chem Lett. 2002;12:1719–1722. doi: 10.1016/s0960-894x(02)00294-9. [DOI] [PubMed] [Google Scholar]
- 3.Borthwick AD, et al. Design and synthesis of monocyclic beta-lactams as mechanism-based inhibitors of human cytomegalovirus protease. Bioorg Med Chem Lett. 1998;8:365–370. doi: 10.1016/s0960-894x(98)00032-8. [DOI] [PubMed] [Google Scholar]
- 4.Gopalsamy A, et al. Design and syntheses of 1,6-naphthalene derivatives as selective HCMV protease inhibitors. J Med Chem. 2004;47:1893–1899. doi: 10.1021/jm030540h. [DOI] [PubMed] [Google Scholar]
- 5.Ogilvie W, et al. Peptidomimetic inhibitors of the human cytomegalovirus protease. J Med Chem. 1997;40:4113–4135. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]
- 6.Waxman L, Darke PL. The herpesvirus proteases as targets for antiviral chemotherapy. Antivir Chem Chemother. 2000;11:1–22. doi: 10.1177/095632020001100101. [DOI] [PubMed] [Google Scholar]
- 7.Marnett AB, Nomura AM, Shimba N, Ortiz de Montellano PR, Craik CS. Communication between the active sites and dimer interface of a herpesvirus protease revealed by a transition-state inhibitor. Proc Natl Acad Sci U S A. 2004;101:6870–6875. doi: 10.1073/pnas.0401613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. Induced structure of a helical switch as a mechanism to regulate enzymatic activity. Nat Struct Mol Biol. 2005;12:1019–1020. doi: 10.1038/nsmb1006. [DOI] [PubMed] [Google Scholar]
- 9.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. One functional switch mediates reversible and irreversible inactivation of a herpesvirus protease. Biochemistry. 2006;45:3572–3579. doi: 10.1021/bi0523658. [DOI] [PubMed] [Google Scholar]
- 10.Pray TR, Nomura AM, Pennington MW, Craik CS. Auto-inactivation by cleavage within the dimer interface of Kaposi's sarcoma-associated herpesvirus protease. J Mol Biol. 1999;289:197–203. doi: 10.1006/jmbi.1999.2791. [DOI] [PubMed] [Google Scholar]
- 11.Pray TR, Reiling KK, Demirjian BG, Craik CS. Conformational change coupling the dimerization and activation of KSHV protease. Biochemistry. 2002;41:1474–1482. doi: 10.1021/bi011753g. [DOI] [PubMed] [Google Scholar]
- 12.Lazic A, Goetz DH, Nomura AM, Marnett AB, Craik CS. Substrate modulation of enzyme activity in the herpesvirus protease family. J Mol Biol. 2007;373:913–923. doi: 10.1016/j.jmb.2007.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reiling KK, Pray TR, Craik CS, Stroud RM. Functional consequences of the Kaposi's sarcoma-associated herpesvirus protease structure: regulation of activity and dimerization by conserved structural elements. Biochemistry. 2000;39:12796–12803. doi: 10.1021/bi001019h. [DOI] [PubMed] [Google Scholar]
- 14.Shimba N, Nomura AM, Marnett AB, Craik CS. Herpesvirus protease inhibition by dimer disruption. J Virol. 2004;78:6657–6665. doi: 10.1128/JVI.78.12.6657-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahian T, et al. Inhibition of a Viral Enzyme by a Small Molecule Dimer Disruptor. Nature Chemical Biology. 2009;9:640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu F, et al. Proteomimetic libraries: design, synthesis, and evaluation of p53-MDM2 interaction inhibitors. J Comb Chem. 2006;8:315–325. doi: 10.1021/cc050142v. [DOI] [PubMed] [Google Scholar]
- 18.Cummings CG, Hamilton AD. Disrupting protein-protein interactions with non-peptidic, small molecule alpha-helix mimetics. Curr Opin Chem Biol. 14:341–346. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Davis JM, Tsou LK, Hamilton AD. Synthetic non-peptide mimetics of alpha-helices. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 20.Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moellering RE, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 24.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 25.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]