

Abstract
Radiotherapy is routinely used for the treatment of lung cancer. However, the mechanisms underlying ionizing radiation (IR)-induced senescence and its role in lung cancer treatment are poorly understood. Here, we show that IR suppresses the proliferation of human non-small cell lung cancer (NSCLC) cells via an apoptosis-independent mechanism. Further investigations reveal that the anticancer effect of irradiation correlates well with IR-induced premature senescence, as evidenced by increased senescence-associated β-glactosidase (SA-β-gal) staining, decreased BrdU incorporation and elevated expression of p16INK4a (p16) in irradiated NSCLC cells. Mechanistic studies indicate that the induction of senescence is associated with activation of the p53-p21 pathway, and that inhibition of p53 transcriptional activity by PFT-α attenuates IR-induced tumor cell killing and senescence. Gain-of-function assays demonstrate that restoration of p53 expression sensitizes H1299 cells to irradiation, whereas knockdown of p53 expression by siRNA inhibits IR-induced senescence in H460 cells. Furthermore, treatment with Nutlin-3a, a small molecule inhibitor of MDM2, enhances IR-induced tumor cell killing and senescence by stabilizing the activation of the p53-p21 signaling pathway. Taken together, these findings demonstrate for the first time that pharmacological activation of p53 by Nutlin-3a can sensitize lung cancer cells to radiation therapy via promoting IR-induced premature senescence.
Keywords: Non-small cell lung cancer, Radiotherapy, Senescence, p53, Nutilin-3a, siRNA
1. Introduction
Although anticancer agent-induced apoptosis was thought to be the primary mechanism whereby cytotoxic therapy suppresses tumor growth, treatment with chemo- or radiation-therapy is not invariably cytotoxic to all tumor cells [1–5]. Cellular senescence is characterized by an irreversible cell cycle arrest that can be triggered by many types of intrinsic and extrinsic stresses, including radiation [4–10]. More importantly, senescence has been recognized as an indispensable cellular response of tumor cells to anticancer therapy [2,4,6,9,11]. However, it is largely unknown if IR-induced premature senescence contributes to the treatment outcomes of cancer radiotherapy. Senescence limits the life span and proliferative capacity of cells, therefore the induction of senescence is regarded as an important mechanism for cancer prevention and treatment [10–13]. Consistent with this hypothesis, it has been shown that restoration of p53 in cancer cells leads to tumor suppression by the induction of senescence rather than apoptosis in sarcomas and liver cancer [14,15]. These findings strongly suggest that induction of senescence might be an important mechanism of action underlying chemotherapy and radiation-induced tumor suppression.
Lung cancer is the leading cause of cancer deaths [16]. Even with current advanced treatment, the 5-year overall survival rate is less than 16% and has not changed appreciably over many decades [16,17]. This poor prognosis emphasizes the urgent need for the development of novel therapeutic approaches to more effectively manage this deadly disease. Radiotherapy is used in over 50% of patients during the course of cancer treatment and is effective both as a curative modality and for palliation [18]. However, previous studies indicated that many epithelial-derived tumors including several lung cancer types show poor response to radiation-induced apoptosis [6,19], underscoring the importance for a better understanding of the molecular mechanisms whereby radiotherapy suppresses lung cancer growth. Therefore, the goal of this study was to determine the role of IR-induced senescence in the tumor-suppressive effect of radiotherapy in NSCLC cells.
We and others have demonstrated that IR induces senescence in a variety of mouse and human cells [6,8,20,21]. However, it remains to be determined if and how IR induces senescence in lung cancer cells and whether targeting the senescence signaling pathway could sensitize lung cancer to radiotherapy. The present studies demonstrate for the first time that IR-induced tumor cell suppression is associated with the induction of senescence but not that of apoptosis in NSCLC cells, suggesting that induction of senescence may play a critical role in mediating the tumor suppressive effect of IR and that targeting of the senescence signaling pathway could be exploited as a novel strategy to improve the efficacy of lung cancer radiotherapy.
2. Materials and methods
2.1. Reagents
BrdU, Pifithrine-α (PFT-α) and mouse anti-BrdU monoclonal antibody were purchased from Sigma (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) and other culture media were obtained from Invitrogen (Carlsbad, CA). Phosphorylated p53 (Ser15), total p53 and p21 antibodies were purchased from Cell Signaling (Danvers, MA). SA-β-gal staining kit was purchased from Cell Signaling. Nutlin-3a was obtained from Cayman Chemical (Ann Arbor, MI).
2.2. Cell lines and culture
Human non-small cell lung cancer (NSCLC) cell lines A549 and H460 were purchased from American Type Culture Collection. A549 cells were cultured in DMEM medium containing 10% FBS, 2mM L-glutamine and 100 microgram/ml of penicillin-streptomycin (Invitrogen). H460 cells were grown in RPMI-1640 medium containing 10% FBS, 2mM L-glutamine and 100μg/ml of penicillin-streptomycin.
2.3. Radiation and clonogenic survival assay
The NSCLC cells were irradiated with a 137Cs irradiator. Irradiated or non-irradiated control cells were cultured in 60 mm dishes for 10–12 days to allow formation of cell colonies. The colonies were fixed and stained with 0.5% crystal violet (Sigma) in methanol for 30min. The number of colonies (≥50 cells) was scored using a microscope. The surviving fraction was calculated as the ratio of the plating efficiency of the treated cells to that of control cells.
2.4. Propidium iodide staining and flow cytometric analysis
The effects of IR on cell cycle progression and apoptosis (detected by sub G1 cells) were measured using propidium iodide (PI) staining and flow cytometric analysis as previously described [16].
2.5. Senescence-associated β-galactosidase (SA-β-gal) staining
In situ staining of SA-β-gal was performed using a senescence β-galactosidase staining kit (Cell Signaling) as we reported previously [21,22].
2.6. BrdU incorporation assay
BrdU incorporation assay was used to determine cellular senescence as we reported previously [21,22].
2.7. Western blotting
Western blotting was performed as previously described [22], using antibodies specific for phospho-p53 and p21, and p16 (BD Biosciences). All blots were stripped and reprobed with β-actin antibody (Sigma) as loading control.
2.8. Statistical analysis
All experiments were repeated independently at least three times. Comparisons between two experimental groups were performed by Student's t-test. Multiple group comparisons were performed using analysis of variance (ANOVA). Differences were considered significant if p < 0.05.
3. Results
3.1. IR suppresses the clonogenic growth of NSCLC cells via an apoptosis-independent mechanism
Clonogenic cell survival assays were performed to investigate the ability of IR to suppress lung cancer cell growth. The results show that IR causes a dose-dependent inhibition of the clonogenic growth of A549 and H460 lung cancer cells (Fig. 1A and B). Moreover, the data also demonstrate that A549 cells are more resistant to IR-induced cell killing than H460 cells (Fig. 1B). Consistent with our observations, it was reported that the radioresistance property of A549 cells is likely mediated through an epithelial growth factor receptor (EGFR)-dependent mechanism [23].
Fig. 1.

IR suppresses the growth of NSCLC cells via an apoptosis-independent mechanism. (A) Clonogenic survival assays show that the number of cancer cell-derived colonies decreases with IR doses. (B) The results of clonogenic assays were normalized to the clonogenic survival of non-irradiated control cells and the survival fractions of A549 and H460 cells were plotted. (C–F) PI staining and flow cytometric analyses were employed to examine apoptosis (Sub G1 cells) and cell cycle distribution in H460 and A549 cells at 7 days after 6Gy of irradiation. (C) Representative flow cytometry graphs of cell cycle analysis of irradiated and control H460 cells. (D) The results of cell cycle analysis of H460 cells are presented as mean ± SEM (n = 3). (E) Representative flow cytometry graphs of cell cycle analysis of irradiated and control A549 cells. (F) The results of cell cycle analysis of A549 cells are presented as mean ± SEM (n = 3). (G and H) Activation of caspase-3 was determined by Western blotting at 24 h after IR or camptothecin (CPT, 5 μM) treatment. *p < 0.05 vs. control; **p <0.01 vs. control.
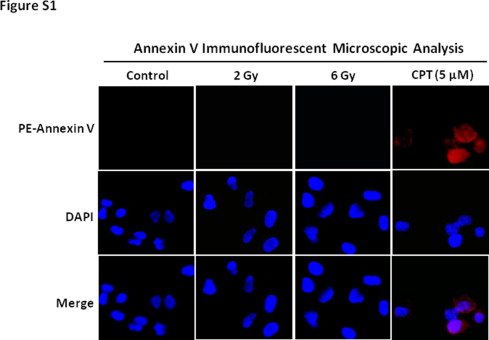
Next, we investigated whether apoptosis is involved in IR-induced clonogenic growth suppression of NSCLC cells. Flow cytometric sub-G1 assays show that IR does not induce any significant changes in apoptosis in both A549 and H460 cells even after 6 Gy of irradiation (Fig. 1C–F). In contrast, we observed a significant increase of G1 arrest in irradiated NSCLC cells (Fig. 1C–F). These results suggest that IR-induced cell killing in NSCLC cells is likely apoptosis-independent. To confirm the induction of apoptosis in lung cancer cells, caspase-3 activation was assessed by Western blotting. The data indicate that neither 2 Gy nor 6 Gy of irradiation induces significant changes in caspase-3 activation in A549 and H460 cells. In contrast, camptothecin (CPT) treatment causes a substantial increase in activated caspase-3 expression (Fig. 1G and H). Moreover, we also show that CPT treatment but not 2 or 6 Gy of irradiation increases Annexin V staining in H460 cells (Supplementary Fig. S1). Together, these data demonstrate that induction of apoptosis is not a primary mechanism underlying IR-induced cell killing in NSCLC cells, suggesting that IR suppresses the growth of NSCLC cells via an apoptosis-independent mechanism.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.lungcan.2013.04.017.
3.2. IR induces premature senescence in NSCLC cells in a dose-dependent manner
To determine the role of senescence in IR-induced tumor cell killing, we exposed H460 cells to different doses of IR (0-6 Gy) and examined senescence in irradiated lung cancer cells using SA-β-gal staining, a widely used biomarker of cellular senescence [24]. The results reveal a substantial increase in SA-β-gal positive senescent cells in irradiated lung cancer cells (Fig. 2A and B). Similar results were also observed in A549 cells (Supplementary Fig. S2). Moreover, high levels of p16 expression, another important biomarker of senescence [25], were detected in irradiated H460 cells (Fig. 3A). In addition, BrdU incorporation assays show that the senescent lung cancer cells are unable to synthesize DNA and incorporate BrdU (Fig. 2A and C). Together, these findings demonstrate for the first time that IR induces senescence in NSCLC cells in a dose-dependent manner.
Fig. 2.

IR induces premature senescence in NSCLC cells in a dose-dependent manner. (A) SA-β-gal staining increased (upper panel) and BrdU incorporation decreased (lower panel) with radiation dose in irradiated H460 cells. (B) The percentage of SA-β-gal positive senescent cells is presented as mean ± SEM (n = 3). (C) The percentage of BrdU positive proliferating cells is presented as mean±SEM (n = 3). **p < 0.001 vs. control.
Fig. 3.

Activation of the p53-p21 pathway is involved in IR-induced senescence in NSCLC cells. (A) The activation of p53 in irradiated H460 cells was determined by Western blotting using a phosphorylated p53 (P-p53, Ser15) specific antibody. The expression levels of p21 and p16 were examined by Western blotting. Relative density of protein bands was quantified using Image J software (NIH) and normalized to β-actin. (B) The specificity of PFT-α to inhibit the transcriptional activity of p53 in NSCLC cells was determined by Western blotting. (C) H460 cells were pre-incubated with p53 specific inhibitor PFT-α (10 μM) prior to radiation exposure and clonogenic survival assays were performed to determine whether inhibition of p53 by PFT-α affects IR-induced tumor cell killings. (D and E) SA-β-gal staining and BrdU incorporation assays were performed to determine senescent cells in irradiated H460 cells. (F) Knockdown of p53 expression by siRNA was confirmed by Western blotting. (G) The effect of p53 knockdown on IR-induced senescence was determined by SA-β-gal assays. a, p < 0.01 vs. control; b, p < 0.05 vs. IR; #, p < 0.001 vs. control; $, p < 0.01 vs. IR; ***, p < 0.001 vs. NC-siRNA + IR.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.lungcan2013.04.017.
3.3. IR induces senescence in NSCLC cells via activation of the p53-p21 pathway
Next, we sought to investigate the mechanisms underlying IR-induced senescence in NSCLC cells. Previous studies from our group and those of others have shown that p53 is a key modulator of stress-induced senescence [8,10,26]. However, it has yet to be determined if p53 is required for IR-induced senescence in NSCLC cells. Our findings here demonstrate that IR activates the p53–p21 pathway in a dose-dependent manner as evidenced by increased expression of phosphorylated p53 and p21 in irradiated H460 cells (Fig. 3A), suggesting a role of p53 in IR-induced senescence in NSCLC cells.
To further examine the role of p53 in IR-induced tumor cell killing and senescence in NSCLC cells, we investigated if inhibition of p53 with a specific inhibitor, Pifithrine-alpha (PFT-α) [27], had any impact on IR-induced tumor cell killing and senescence. The specificity of PFT-α to inhibit the transcriptional activity of p53 in NSCLC cells was verified by Western blotting analyses (Fig. 3B). Clonogenic assays demonstrate that preincubation with PFT-α significantly attenuates IR-induced tumor cell killing (Fig. 3C). Interestingly, the impaired tumor cell killing by PFT-α is associated with a decrease of senescence induction in irradiated H460 cells (Fig. 3D and E), suggesting that inhibition of p53 transcriptional activity diminishes IR-induced tumor cell killing via suppressing senescence induction in irradiated lung cancer cells. Moreover, we also show that knockdown of p53 expression by siRNA inhibits IR-induced senescence in H460 cells (Fig. 3F and G; Supplementary Fig. S3). These results demonstrate that the p53–p21 pathway plays an import role in regulating IR-induced premature senescence in NSCLC cells.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.lungcan.2013.04.017.
3.4. Restoration ofp53 expression sensitizes lung cancer cells to radiation
Because many tumors exhibit defects in p53 signaling which are associated with therapy-resistance [28–31], we decided to investigate if restoration of p53 expression can sensitize H1299 cells to radiation and thus improve the efficacy of radiotherapy. H1299 cells were transfected with a p53 expression vector (pCEP4-p53) as previously described [32] and expression of p53 in transfected cells was confirmed by Western blotting (Fig. 4A). Clonogenic assays were performed to examine the radiosensitivity of H1299 cells transfected with pCEP4-p53. As shown in Fig. 4B, the results indicate that restoration of p53 expression sensitizes H1299 cells to IR-induced cell killing, demonstrating that p53 plays a critical role in mediating the anticancer effects of radiation therapy.
Fig. 4.

Restoration of p53 expression sensitizes lung cancer cells to IR-induced cell killing. (A) H1299 cells were transfected with pCEP4-p53 or pCEP4 empty vectors as control to restore the expression of p53 as described previously [32]. The expression of p53 in transfected cells was detected by Western blotting. (B) Clonogenic assays were performed to examine IR-induced tumor cell killing. *p < 0.01 vs. H1299/pCEP4 cells.
3.5. Pharmacological activation of the p53 pathway enhances IR-induced tumor cell killing in NSCLC cells
Since previous studies from our group and those of others have shown that p53 is a key regulator of senescence induction [8,10,26], we investigated if pharmacological activation of p53 could enhance IR-induced cell killing and senescence in NSCLC cells. Nutlin-3a (Nut) is a small molecule inhibitor of MDM2 that can selectively stabilize p53 protein and protect it from degradation [33]. As shown in Fig. 5A, our data demonstrate that Nut treatment substantially increases IR-induced p53 activation and p21 expression in H460 cells. Moreover, clonogenic assays show that a combination of Nut and IR combined treatment leads to a synergistic inhibition of the clonogenic growth of H460 cells (Fig. 5B). Interestingly, the increased tumor cell killing by IR and Nut combined treatment correlates with the increased expression of senescence biomarkers (increased SA-β-gal staining and decreased BrdU incorporation) in irradiated lung cancer cells (Fig. 5C and D). These results suggest that pharmacological activation of the p53–p21 pathway can enhance IR-induced tumor cell killing by promoting senescence induction in irradiated lung cancer cells. In addition, clonogenic assays also revealed that Nut does not increase IR-induced cell killing in H1299 cells (p53 null), suggesting that the radiosensitizing activity of Nut may require the presence of functional p53 in NSCLC cells (Supplementary Fig. S4).
Fig. 5.

Activation of p53 with Nutlin-3a enhances IR-induced tumor cell killing in NSCLC cells. (A) Lung cancer cells were pre-incubated with 3 μM of Nutlin-3a (Nut) for 30min prior to irradiation. The status of p53 activation was determined by p-p53 (Ser15) Western blotting analyses at 24 h after 4Gy of IR. The expression of total p53 and p21 were also determined by Western blotting. (B) Clonogenic assay was employed to detect IR-induced tumor cell killing. (C) SA-β-gal staining was performed to determine senescent cells in irradiated H460 cells. (D) BrdU incorporation assay was used to detect the proliferating cells. a, p < 0.05 vs. control (Ctl); b, p < 0.001 vs. Ctl; c, p < 0.01 vs. IR.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.lungcan.2013.04.017.
4. Discussion
Radiation is widely used as a therapeutic approach for the management of a variety of human cancers, including clinically inoperable lung cancers [34-36]. However, even with high dose escalation, the treatment outcome of lung cancer radiotherapy is very disappointing and has not improved for many decades [16,36]. Clearly, there is a critical need for the development of novel therapeutic strategies to improve the efficacy of lung cancer radiotherapy. Although we and others have shown that IR can induce premature senescence in mouse bone marrow, human fibroblasts and breast cancer cells [6,8,20,26], the exact contributions of apoptosis and senescence to lung cancer radiotherapy remains to be defined. Here, we show for the first time that the tumor suppressive effect of IR is associated with the induction of senescence but not apoptosis in irradiated NSCLC cells. These findings suggest that IR-induced senescence may play an important role in mediating the anticancer effects of radiotherapy. In agreement with this suggestion, it was found that the induction of senescence correlates with treatment outcomes in previous animal studies [10,11]. These findings strongly support the hypothesis that induction of senescence is a central mechanism of action underlying IR-induced anticancer effects.
Our previous studies and those of others have demonstrated that the p53–p21 pathway plays a critical role in regulating IR-induced premature senescence [8,10,26]. However, the role of p53 in IR-induced senescence in lung cancer cells is largely unknown. The present study demonstrates that IR-induced senescence in NSCLC cells is associated with p53 activation and increased p21 expression, implicating the p53–p21 pathway in IR-induced senescence in NSCLC cells. MDM2 is a transcriptional target of p53, which negatively regulates the p53 pathway by increasing p53 degradation via its function as an E3 ubiquitin ligase [37]. Overexpression of MDM2 has been observed in several human cancers and is believed to account for therapy-resistance [38,39]. Our studies show that Nutlin-3a enhances IR-induced tumor cell killing and senescence in NSCLC cells, suggesting that pharmacological activation of the p53-p21 pathway can sensitize lung cancer to radiotherapy by promoting IR-induced senescence in irradiated cancer cells. It is worth mentioning that the induction of senescence by Nutlin-3a treatment alone was not stronger than IR (Fig. 5C), suggesting that a single dose of Nutlin-3a and brief treatment may only cause a transient activation of p53 and increase in p21 expression, thus repeated administration of Nutlin-3a might be needed to achieve the optimal senescence induction effects in this regard. Consistent with our studies, there is evidence that Nutlin-3a promotes senescence and radiosensitizes laryngeal carcinoma cells harboring wild-type p53 [40].
In summary, our present studies demonstrate for the first time that the tumor suppressive effect of IR is associated with the induction of senescence but not apoptosis in NSCLC cells. These findings strongly support the hypothesis that induction of senescence is a central mechanism of action underlying IR-induced anticancer effects. In agreement with this hypothesis, it has been shown that restoration of p53 in cancer cells leads to tumor suppression by the induction of senescence rather than apoptosis in sarcomas and liver cancer [14,15]. Collectively, these findings suggest that targeting the senescence signaling pathway may represent a novel therapeutic strategy to sensitize lung cancer to radiotherapy. Further preclinical and clinical studies are warranted to determine whether pharmacological activation of p53 with Nutlin-3a can sensitize lung cancer to radiotherapy by promoting IR-induced senescence in tumor tissues.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors want to thank Dr. Yi-Ching Wang for providing the pCEP4-p53 plasmids. This study was supported in part by NIH grants CA138313, DC00422, DC012058 and HL106451.
Footnotes
Conflict of interest statement: None.
References
- 1.Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–15. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 2.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 3.Rebbaa A, Zheng X, Chou PM, Mirkin BL. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene. 2003;22:2805–11. doi: 10.1038/sj.onc.1206366. [DOI] [PubMed] [Google Scholar]
- 4.Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA. 2002;99:389–94. doi: 10.1073/pnas.012602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin DX, Schimke RT. BCL-2 expression delays drug-induced apoptosis but does not increase clonogenic survival after drug treatment in HeLa cells. Cancer Res. 1995;55:4922–8. [PubMed] [Google Scholar]
- 6.Suzuki M, Boothman DA. Stress-induced premature senescence (SIPS) – influence of SIPS on radiotherapy. J Radiat Res. 2008;49(2):105–12. doi: 10.1269/jrr.07081. [DOI] [PubMed] [Google Scholar]
- 7.Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. 2008;76:947–57. doi: 10.1016/j.bcp.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 8.Jones KR, Elmore LW, Jackson-Cook C, Demasters G, Povirk LF, Holt SE, et al. p53-dependent accelerated senescence induced by ionizing radiation in breast tumor cells. Int J Radiat Biol. 2005;81:445–58. doi: 10.1080/09553000500168549. [DOI] [PubMed] [Google Scholar]
- 9.Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–46. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 11.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102(20):1536–46. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 13.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–5. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumor clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 16.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics 2009. CA Cancer J Clin. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 17.Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 18.Hall EJ, Wu CS. Radiation-induced second cancers: the impact of 3D-CRT and IMRT. Int J Radiat Oncol Biol Phys. 2003;56(1):83–8. doi: 10.1016/s0360-3016(03)00073-7. [DOI] [PubMed] [Google Scholar]
- 19.Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61(10):3986–97. [PubMed] [Google Scholar]
- 20.Wang Y, Schulte BA, Larue AC, Ogawa M, Zhou D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood. 2006;107:358–66. doi: 10.1182/blood-2005-04-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Scheiber MN, Neumann C, Calin GA, Zhou D. MicroRNA regulation of ionizing radiation-induced premature senescence. Int J Radiat Oncol Biol Phys. 2011;81:839–48. doi: 10.1016/j.ijrobp.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Meng A, Zhou D. Inhibition of phosphatidylinostol 3-kinase uncouples H2 O2-induced senescent phenotype and cell cycle arrest in normal human diploid fibroblasts. Exp Cell Res. 2004;298:188–96. doi: 10.1016/j.yexcr.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005;76:157–61. doi: 10.1016/j.radonc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 24.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng A, Wang Y, Van Zant G, Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 2003;63:5414–9. [PubMed] [Google Scholar]
- 27.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 28.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 29.Mori S, Ito G, Usami N, Yoshioka H, Ueda Y, Kodama Y, et al. p53 apoptotic pathway molecules are frequently and simultaneously altered in nonsmall cell lung carcinoma. Cancer. 2004;100:1673–82. doi: 10.1002/cncr.20164. [DOI] [PubMed] [Google Scholar]
- 30.Lai SL, Perng RP, Hwang J. p53 gene status modulates the chemosensitivity of non-small cell lung cancer cells. J Biomed Sci. 2000;7(1):64–70. doi: 10.1007/BF02255920. [DOI] [PubMed] [Google Scholar]
- 31.Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–30. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 32.Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, et al. Dysregulation of p53/Sp1 control leads to DNAmethyltransferase-1 overexpression in lung cancer. Cancer Res. 2010;70(14):5807–17. doi: 10.1158/0008-5472.CAN-09-4161. [DOI] [PubMed] [Google Scholar]
- 33.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 34.Sibley GS, Jamieson TA, Marks LB, Anscher MS, Prosnitz LR. Radiotherapy alone for medically inoperable stage I non-small-cell lung cancer: the Duke experience. Int J Radiat Oncol Biol Phys. 1998;40(1):149–54. doi: 10.1016/s0360-3016(97)00589-0. [DOI] [PubMed] [Google Scholar]
- 35.Park SY, Kim YM, Pyo H. Gefitinib radiosensitizes non-small cell lung cancer cells through inhibition of ataxia telangiectasia mutated. Mol Cancer. 2010;9:222. doi: 10.1186/1476-4598-9-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sura S, Yorke E, Jackson A, Rosenzweig KE. High-dose radiotherapy for the treatment of inoperable non-small cell lung cancer. Cancer J. 2007;13(4):238–42. doi: 10.1097/PPO.0b013e31813ffd7b. [DOI] [PubMed] [Google Scholar]
- 37.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 38.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 39.Muthusamy V, Hobbs C, Nogueira C, Cordon-Cardo C, McKee PH, Chin L, et al. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 2006;5(5):447–54. doi: 10.1002/gcc.20310. [DOI] [PubMed] [Google Scholar]
- 40.Arya AK, El-Fert A, Devling T, Eccles RM, Aslam MA, Rubbi CP, et al. Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br J Cancer. 2010;103(2):186–95. doi: 10.1038/sj.bjc.6605739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.