

Abstract
Ethanol is a neuroteratogen and neurodegeneration is the most devastating consequence of developmental exposure to ethanol. A sublethal preconditioning has been proposed as a neuroprotective strategy against several central nervous system (CNS) neurodegenerative diseases. We have recently demonstrated that autophagy is a protective response to alleviate ethanol toxicity. A modest hypoxic preconditioning (1% oxygen) did not cause neurotoxicity but induced autophagy (Tzeng et al., 2010). We therefore hypothesize that the modest hypoxic preconditioning may offer a protection against ethanol-induced neurotoxicity. We showed here that the modest hypoxic preconditioning (1% oxygen) for 8 hours significantly alleviated ethanol-induced death of SH-SY5Y neuroblastoma cells. Under the normoxia condition, cell viability in ethanol-exposed cultures (316 mg/dl for 48 hrs) was 49 ± 6% of untreated controls; however, with hypoxic preconditioning, cell viability in the ethanol-exposed group increased to 78 ± 7% of the controls (p < 0.05; n = 3). Bafilomycin A1, an inhibitor of autophagosome and lysosome fusion, blocked hypoxic preconditioning-mediated protection. Similarly, inhibition of autophagic initiation by wortmannin also eliminated hypoxic preconditioning-mediated protection. In contrast, activation of autophagy by rapamycin further enhanced neuroprotection caused by hypoxic preconditioning. Taken together, the results confirm that autophagy is a protective response against ethanol neurotoxicity and the modest hypoxic preconditioning can offer neuroprotection by activating autophagic pathways.
Keywords: Alcohol, fetal alcohol spectrum disorders, protein degradation, neurodegeneration
Introduction
Ethanol is a neuroteratogen and fetal alcohol spectrum disorders (FASD) are caused by maternal ethanol exposure during pregnancy. FASD is the leading cause of mental retardation. Ethanol exposure causes neuroapoptosis in the developing brain, and the loss of neurons in the central nervous system (CNS) underlies many of the behavioral deficits observed in FASD (Luo 2009; Riley et al. 2011). It is important to understand the mechanisms of ethanol-induced neurodegeneration and develop approaches that can ameliorate ethanol neurotoxicity. It has been suggested that a prior sublethal conditioning, including hypoxia, ischemia and chemicals, can be cytoprotective against deadly insults. Intermittent hypoxia conditioning, the repeated exposure to moderately hypoxic atmospheres, protected neurons against ethanol withdrawal-induced mitochondrial damage (Ju et al. 2012). A modest hypoxic preconditioning (1% oxygen) alleviated MPP+ (a Parkinson’s disease mimetic)-induced neuronal death (Tzeng et al. 2010).
There are two major pathways that accomplish regulated protein catabolism: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal system (ALS). Autophagy is a lysosomal degradation pathway involved in the turnover of cellular macromolecules and organelles and is essential for survival, differentiation, development and homeostasis (Kroemer and Levine 2008). Although some of these functions overlap with those of the UPS, autophagy is primarily responsible for degrading long-lived proteins and maintaining amino acid pools in the setting of chronic starvation. The autophagy pathway is uniquely capable of degrading entire organelles such as mitochondria, peroxisomes and endoplasmic reticulum (ER), as well as intact intracellular microorganisms (Kroemer and Levine 2008). Autophagic degradation of cellular constituents can efficiently recycle essential nutrients to sustain basic biological processes. Autophagy is also used as a defense mechanism to clear intracellular microbes, misfolded proteins and damaged organelles. Constitutive clearance of cytosolic proteins by low-level basal autophagy is an additional important cytoprotective function, particularly to neurons. We have recently demonstrated that autophagy was a protective response to ethanol neurotoxicity (Chen et al. 2012). Since modest hypoxic preconditioning can stimulate autophagy (Tzeng et al. 2010), we hypothesized that hypoxic preconditioning may protect neuronal cells against ethanol-induced cell death through the autophagic pathway. We used SH-SY5Y neuroblastoma cells to test this hypothesis. SH-SY5Y cells were selected based the following reasons: (1) ethanol causes apoptotic death of SH-SY5Y cells, and is a widely used in vitro model for studying ethanol neurotoxicity (Johnson et al. 2007; Chen et al. 2008; Ramlochansingh et al. 2011; Yadav S et al. 2011); (2) in these cells, we have demonstrated that autophagy is a protective response to ethanol neurotoxicity (Chen et al. 2012); and (3) it was confirmed that hypoxic preconditioning activated autophagy in these cells (Tzeng et al. 2010). In this study, we show that hypoxic preconditioning offers protection against ethanol-induced death of SH-SY5Y neuroblastoma cells. Inhibition of autophagy blocks hypoxic preconditioning-mediated protection, whereas activation of autophagy enhances neuroprotection caused by hypoxic preconditioning.
Materials and Methods
Materials
The following materials were used: ethanol (Sigma-Aldrich, E7023), bafilomycin A1 (Sigma-Aldrich, B1793), wortmannin (Sigma-Aldrich, W1628), rapamycin (Sigma-Aldrich, R0395), anti-p62 antibody (Sigma-Aldrich, P0067), anti-tubulin antibody (Sigma-Aldrich, T0067), anti-LC3 antibody (Medical and Biological Laboratories, PM036), anti-beclin 1 antibody (Abcam, ab55878), anti-GAPDH (AbD Serotec, AHP1064) and MTT kit (Fisher, 507203844).
Cell culture and hypoxic preconditioning
Human neuroblastoma SH-SY5Y cells obtained from ATCC were grown in Eagle’s MEM containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, 25 μg/ml gentamicin, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C with 5% CO2 in a humidified incubator (Symphony 5.3A, Thermo Scientific). For hypoxic preconditioning, cells received an 8-hour hypoxic treatment (1% oxygen) and a subsequent ethanol exposure in a normoxia environment. To induce a hypoxic atmosphere, the oxygen concentration in the Galaxy 48R incubator (CO48R-120, New Brunswick, Enfield, CT) was preset at 1% and then flushed with a gas mixture of 5% CO2–95% N2 until the oxygen concentration in the chamber reached 1%. SH-SY5Y cells were placed in the hypoxic incubator for the indicated times.
Ethanol exposure protocol
A method utilizing sealed containers was used to maintain ethanol concentrations in the culture medium. With this method, ethanol concentrations in the culture medium can be accurately maintained (Luo et al. 2001). A pharmacologically relevant concentration of 0.4% (316 mg/dl or 69 mM) was used in this study. In general, the concentration for in vitro studies is higher than that required to produce a similar affect in vivo (Luo et al. 2001). FASD may be caused by binge drinking which could result in high blood alcohol concentrations (BAC > 300 mg/dl) (Adachi et al., 1991; Kuehn et al. 2012). Using animal models, we and others showed that a BAC of 300 mg/dl, which was achieved binge-like ethanol exposure paradigm, caused significant neuronal damage to the developing or mature brain (Kumar et al. 2011; Wang et al. 2012; Hayes et al. 2013).
Determination of cell viability
Cell viability was determined by MTT assay as previously described (Chen et al. 2008). The MTT assay is based on the cleavage of yellow tetrazolium salt MTT [3-(4,5-dimethylthiazol-2yl)-2,5-di-phenyl tetrazolium bromide] to purple formazan crystals by metabolically active cells. Briefly, the cells were cultured in 96-well microtiter plates and exposed to ethanol for 48 hours. After ethanol exposure, 10 μl of MTT labeling reagent was added to each well, and the plates were incubated at 37°C for 4 hours. The cultures were then solubilized, and spectrophotometric absorbance of the samples was detected by a microtiter plate reader. The wavelength to measure absorbance of formazan products is 570 nm, with a reference wavelength of 750 nm.
Immunoblotting
Cells were washed with phosphate-buffered saline (PBS; pH 7.4) and lysed with RIPA buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 1% Nonidet P-40 (NP-40), 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholic acid sodium, 0.1 mg/ml phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and 3% aprotinin] on ice for 10 min; solubilized cells were centrifuged and the supernatant was collected. The immunoblotting procedure has been previously described (Chen et al. 2012). Briefly, after the protein concentrations were determined, aliquots of the protein samples (20–40 μg) were loaded into the lanes of an SDS-polyacrylamide gel. The protein samples were separated by electrophoresis, and the separated proteins were transferred to nitrocellulose membranes. The membranes were blocked with either 5% BSA or 5% nonfat milk in 0.010 M PBS (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 hour. Subsequently, the membranes were probed with primary antibodies directed against target proteins for 2 hours at room temperature or overnight at 4°C. After three quick washes in TPBS, the membranes were incubated with a secondary antibody conjugated to horseradish peroxidase (Amersham, Arlington Heights, IL) diluted at 1:2,000 in TPBS for 1 hour. The immune complexes were detected by the enhanced chemiluminescence method (PerkinElmer, NEL105001EA). The blots were stripped and re-probed with an anti-tubulin or anti-GAPDH antibody. The images of immunoblots were documented using Gel Logic 2200 Pro (Carestream Health, Rochester, NY). The intensity of specific proteins was quantified using Carestream Molecular Image Software as previously described (Xu et al. 2012).
Statistical Analysis
Differences among treatment groups were tested by analysis of variance (ANOVA). p < 0.05 was considered statistically significant. Cases in which significant differences were detected, specific post-hoc comparisons between treatment groups were examined by Student-Newman-Keuls tests.
Results
Hypoxia induces autophagy
It was reported that a modest hypoxic preconditioning (1% oxygen) caused autophagy, which was demonstrated by an increase in LC3 lipidation and the formation of autohagic vacuoles in SH-SY5Y cells (Tzeng et al. 2010). During autophagic activation, the cytoplasmic LC3-I is processed and recruited to the autophagosomes, converting to LC3-II. This characteristic conversion of LC3 (LC3 lipidation) is a common marker for monitoring autophagic activity. To confirm this finding in our system, we treated SH-SY5Y cells with 1% oxygen and examined the expression of LC3 and beclin1. Exposure to 1% oxygen induced a duration-dependent increase in LC3-II in SH-SY5Y cells (Fig. 1A and B). Beclin1, the homolog of yeast Vps30 (also known as Atg6), is a critical regulator of autophagosome maturation and endocytic trafficking. Beclin1 is also frequently used to monitor autophagy. Exposure to 1% oxygen increased the levels of Beclin 1 (Fig. 1A and B). p62 is a protein associated with autophagosomes and is degraded in lysosomes after autophagosomes fuse with lysosomes. Our results indicated that hypoxia caused a down-regulation of p62 (Fig. 1C and D). Since p62 levels fluctuate in culture, a time matched-control was used. These results indicated that exposure to 1% oxygen activated autophagy.
Figure 1.


Effect of hypoxic preconditioning on autophagy. A: SHSY5Y cells maintained in media containing 10% serum were switched to serum-free media for 24 hours. Cells were then exposed to a hypoxic (5% CO2 and 1% O2) or controlled (5% CO2 and 20.9% O2) environment for 1 to 24 hours and cell lysates were collected. Protein was extracted and the levels of LC3 and beclin1 were analyzed by immunoblotting. B: Relative levels of LC3-II and beclin1 were determined by densitometry and normalized to tubulin levels as previously described (Chen et al. 2012). The hypoxia-induced changes were quantified relative to control groups. C: The expression of p62 was determined by immunoblotting. C: control; H: hypoxia. D: Relative levels of p62 were determined by densitometry and normalized to tubulin levels. The hypoxia-induced changes were quantified relative to time-matched control groups. The data represent the mean and SEM of three replications.
Hypoxia protects cells against ethanol-induced death
Exposure to 1% oxygen up to 24 hours did not affect the viability of SH-SY5Y cells (Tzeng et al. 2010). We confirmed this finding. In this study, we used 8 hours of hypoxic preconditioning to determine the protective effect of hypoxia because the duration of hypoxia sufficiently activated autophagy but did not affect cell viability. As previously shown (Chen et al. 2008), ethanol exposure (0.4% or 316 mg/dl) for 24 or 48 hours decreased the viability of SH-SY5Y cells (Fig. 3). However, in the groups that received hypoxic preconditioning, SH-SY5Y cells were more resistant to ethanol-induced cell death, indicating that hypoxia was protective. To establish the role of autophagy in hypoxia-induced protection, we either stimulated or inhibited autophagy during the hypoxic preconditioning and then examined the impact of these manipulations on hypoxia-induced protection. We have previously used rapamycin to activate autophagy, and bafilomycin A1 (an inhibitor of autophagosome and lysosome fusion) and wortmannin (an inhibitor of autophagosome initiation) to inhibit autophagy in SH-SY5Y cells (Chen et al. 2012). Rapamycin increased LC3 lipidation while bafilomycin A1 blocked the fusion of autophagosome and lysosome, resulting in increased LC3-II (Fig. 3). More importantly, stimulation of autophagy by rapamycin enhanced hypoxia-mediated protection whereas inhibition of autophagy by bafilomycin A1 completely blocked hypoxia-mediated protection (Fig. 4). Inhibition of the initiation of autophagy by wortmannin also eliminated hypoxia-mediated protection (Fig. 4).
Figure 3.
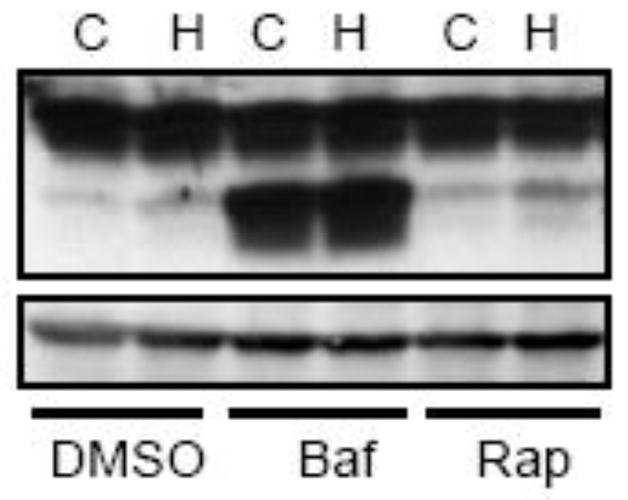
Manipulation of hypoxia-induced autophagy. SH-SY5Y cells were exposed to a hypoxic or controlled environment in the presence or absence of rapamycin (Rap, 10 μM) or bafilomycin (Baf, 10 nM) for 8 hours. The expression of LC3 was analyzed by immunoblotting as described in Fig. 1. C: control; H: hypoxia.
Figure 4.
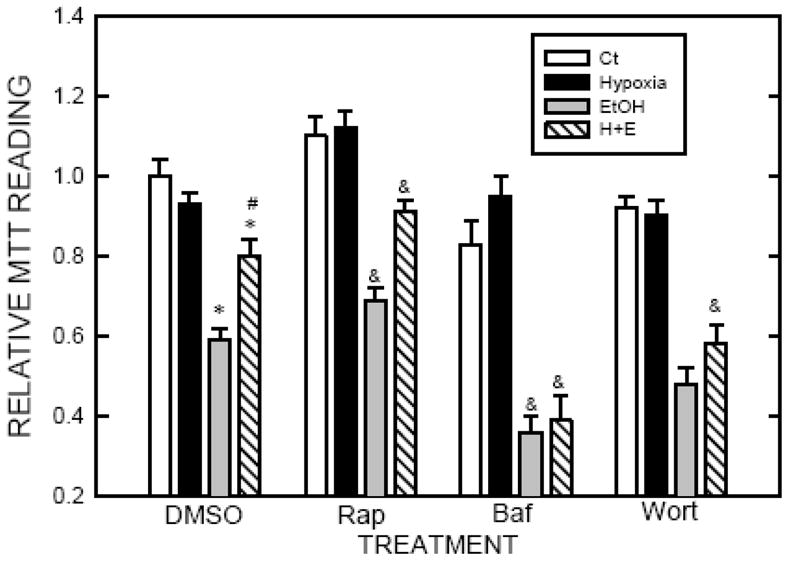
Effect of autophagy activators or inhibitors on hypoxic preconditioned-mediated protection. SH-SY5Y cells were exposed to a hypoxic or controlled environment in the presence or absence of rapamycin (Rap, 10 μM), bafilomycin (Baf, 10 nM) or wortmannin (Wort, 500 nM) for 8 hours. After that, cells were treated with ethanol (0.4% or 316 mg/dl) for 24 hours. Cell viability was determined by MTT assay as described under the Materials and Methods. The experiment was replicated three times. * denotes a statistically significant difference from no-ethanol-treated controls. # denotes a significant difference from ethanol-treated cells under normoxia condition. & denotes a statistically significant difference from corresponding groups in DMSO-treated cultures.
Discussion
Autophagy is a tightly regulated pathway involving the lysosomal degradation of cytoplasmic organelles or cytosolic components. This pathway can be stimulated by multiple forms of cellular stress, including nutrient or growth factor deprivation, hypoxia, oxidative stress, endoplasmic reticulum stress, DNA damage, protein aggregates, damaged organelles or intracellular pathogens (Kroemer et al. 2010).
The role of autophagy in the regulation of cell death has been controversial. Since the formation of autophagosomes is frequently associated with cellular stress and cell death, some believe that autophagy may promote cell death (Galluzzi et al. 2008). On the other hand, many view autophagy as a cellular self-defense response (Shen and Codogno 2011). Particularly, in neuropathies (Huntington’s, Alzheimer’s and Parkinson’s diseases) and ischemic heart disease, autophagy is more widely accepted as beneficial due to its role in eliminating ‘toxic assets’ and promoting cell viability (Glick et al. 2010). We have recently demonstrated that ethanol can activate autophagy in SH-SY5Y cells and the developing brain (Chen et al. 2012). Furthermore, enhancement of autophagic flux alleviates ethanol-induced neuronal death; contrarily, inhibition of autophagy exacerbates ethanol neurotoxicity.
A modest hypoxic preconditioning (1% oxygen) caused autophagy, which is demonstrated by an increase in LC3 lipidation and the formation of autohagic vacuoles in SH-SY5Y cells (Tzeng et al. 2010; Fig. 1). Our results indicate that hypoxic preconditioning (1% oxygen) significantly reduces ethanol-induced death of SH-SY5Y cells (Fig. 2). Bafilomycin A1, an inhibitor of autophagosome and lysosome fusion, blocks hypoxic preconditioning-mediated protection. Similarly, inhibition of autophagic initiation by wortmannin, also eliminates hypoxic preconditioned-mediated protection. In contrast, activation of autophagy by rapamycin further enhances the neuroprotection caused by hypoxic preconditioning (Fig. 4). Taken together, the results confirm that autophagy is a protective mechanism in response to ethanol neurotoxicity and hypoxic preconditioning-induced protection is likely mediated by activating the autophagic pathway.
Figure 2.

Effect of hypoxic preconditioning on ethanol-induced cell death. SH-SY5Y cells were cultured and exposed to hypoxic or controlled environment (normoxia) for 8 hours as described in Fig. 1. After that, cells were treated with ethanol (0.4% or 316 mg/dl) for 12–48 hours. Cell viability was determined by MTT assay as described under the Materials and Methods. Ct: normoxia; Hypoxia: 1% oxygen; EtOH: 0.4% (316 mg/dl) ethanol; H+E: hypoxia plus ethanol. The experiment was replicated three times. * denotes a statistically significant difference from no-ethanol-treated controls. # denotes a significant difference from ethanol-treated cells under normoxia condition.
The role of hypoxia in ethanol neurotoxicity has been controversial. Intermittent hypoxia conditioning minimizes neurocognitive impairment and stabilizes brain mitochondrial integrity during ethanol withdrawal (EW) in rats (Ju et al. 2012). However, an in vitro study shows that when administered simultaneously, hypoxia and low levels of ethanol exposure, can act synergistically producing a higher degree of neuronal injury in cultured cortical neurons (Genetta et al. 2007). The discrepancy may result from the difference in experimental paradigm and models being used. In the study by Genetta et al. (2007), cortical neurons were simultaneously exposed to 0.1% oxygen which is toxic to these neurons together with sub-toxic levels of ethanol. In our study, SH-SY5Y cells were pretreated by non-toxic hypoxia (1% oxygen) followed by ethanol exposure.
Hypoxic pretreatment has been reported to be neuroprotective in other models. For example, hypoxic preconditioning offers protection against kainate-induced neurotoxicity and brain ischemia by transient middle cerebral artery occlusion (Lin et al. 2003; Wang 2005). The mechanisms of hypoxic preconditioned-mediated neuroprotection are complex. The current findings and a recent study by Tzeng et al. (2010) suggests that the activation of autophagy may be a potential mechanism. However, other protective mechanisms may be operating. For example, it has been proposed that hypoxic preconditioning may increase the expression of antioxidant defense enzymes which in turn alleviate oxidative stress (Arthur et al., 2004; Wang et al. 2005). In addition, it is possible that hypoxic preconditioning may induce the expression of enzymes for ethanol metabolisms such as cytochrome P450 2E1, catalase, peroxidases, or alcohol dehydrogenase (ADH), resulting in altered ethanol metabolism. For example, hypoxic preconditioning has been shown to induce cytochrome P450 2C11 expression and increase catalase activity in the brain (Liu and Alkayed, 2005; Bigdeli et al., 2009). Therefore, further study is needed to explore these possibilities.
In summary, hypoxic preconditioning is protective against ethanol neurotoxicity in vitro. This protection appears to be mediated by enhanced autophagy. This result confirms our previous findings showing that autophagy alleviates ethanol-induced neurotoxicity. Enhancing autophagy may be a potential therapeutic strategy for treating ethanol neurotoxicity.
Acknowledgments
This research is supported by grants from National Institutes of Health (NIH) (AA019693 and AA015407).
References
- Adachi J, Mizoi Y, Fukunaga T, Ogawa Y, Ueno Y, Imamichi H. Degrees of alcohol intoxication in 117 hospitalized cases. J Stud Alcohol. 1991;52(5):448–53. doi: 10.15288/jsa.1991.52.448. [DOI] [PubMed] [Google Scholar]
- Arthur PG, Lim SC, Meloni BP, Munns SE, Chan A, Knuckey NW. The protective effect of hypoxic preconditioning on cortical neuronal cultures is associated with increases in the activity of several antioxidant enzymes. Brain Res. 2004;1017(1–2):146–54. doi: 10.1016/j.brainres.2004.05.031. [DOI] [PubMed] [Google Scholar]
- Bigdeli MR, Rasoulian B, Meratan AA. In vivo normobaric hyperoxia preconditioning induces different degrees of antioxidant enzymes activities in rat brain tissue. Eur J Pharmacol. 2009;611(1–3):22–9. doi: 10.1016/j.ejphar.2009.03.034. [DOI] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res. 2008;86(4):937–46. doi: 10.1002/jnr.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, Luo J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8(11):1577–89. doi: 10.4161/auto.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. 2008;8(2):78–91. doi: 10.2174/156652408783769616. [DOI] [PubMed] [Google Scholar]
- Genetta T, Lee BH, Sola A. Low doses of ethanol and hypoxia administered together act synergistically to promote the death of cortical neurons. J Neurosci Res. 2007;85(1):131–8. doi: 10.1002/jnr.21067. [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DM, Deeny MA, Shaner CA, Nixon K. Determining the threshold for alcohol-induced brain damage: new evidence with gliosis markers. Alcohol Clin Exp Res. 2013;37(3):425–34. doi: 10.1111/j.1530-0277.2012.01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Williams AN, Johnson C, Ou XM. The effects of antidepressant drug on ethanol-induced cell death. Drug Discov Ther. 2007;1(2):130–5. [PubMed] [Google Scholar]
- Ju X, Mallet RT, Downey HF, Metzger DB, Jung ME. Intermittent hypoxia conditioning protects mitochondrial cytochrome c oxidase of rat cerebellum from ethanol withdrawal stress. J Appl Physiol. 2012;112(10):1706–14. doi: 10.1152/japplphysiol.01428.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AMY, Dung SW, Chen CF, Hsu ML, Ho LT. Hypoxic preconditioning prevents cortical infarction by transient focal ischemia–reperfusion. Ann NY Acad Sci. 2003;993:168–178. doi: 10.1111/j.1749-6632.2003.tb07527.x. [DOI] [PubMed] [Google Scholar]
- Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab. 2005;25(8):939–48. doi: 10.1038/sj.jcbfm.9600085. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–10. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn D, Aros S, Cassorla F, Avaria M, Unanue N, Henriquez C, Kleinsteuber K, Conca B, Avila A, Carter TC, Conley MR, Troendle J, Mills JL. A prospective cohort study of the prevalence of growth, facial, and central nervous system abnormalities in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res. 2012;36(10):1811–9. doi: 10.1111/j.1530-0277.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Singh CK, Lavoie HA, Dipette DJ, Singh US. Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol Pharmacol. 2011;80(3):446–57. doi: 10.1124/mol.111.071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Lindström CL, Donahue A, Miller MW. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J Neurochem. 2001;76(5):1354–63. doi: 10.1046/j.1471-4159.2001.00129.x. [DOI] [PubMed] [Google Scholar]
- Luo J. GSK3beta in ethanol neurotoxicity. Mol Neurobiol. 2009;40(2):108–21. doi: 10.1007/s12035-009-8075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlochansingh C, Taylor RE, Tizabi Y. Toxic effects of low alcohol and nicotine combinations in SH-SY5Y cells are apoptotically mediated. Neurotox Res. 2011;20(3):263–9. doi: 10.1007/s12640-011-9239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev. 2011;21(2):73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7(5):457–65. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- Tzeng YW, Lee LY, Chao PL, Lee HC, Wu RT, Lin AM. Role of autophagy in protection afforded by hypoxic preconditioning against MPP+-induced neurotoxicity in SH-SY5Y cells. Free Radic Biol Med 2010. 2010;49(5):839–46. doi: 10.1016/j.freeradbiomed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Wang CH, Chang AY, Tsai MJ, Cheng HC, Liao LP, Lin AMY. Kainic acid-induced oxidative injury is attenuated by hypoxic preconditioning. Ann NY Acad Sci. 2005;1042:314–324. doi: 10.1196/annals.1338.054. [DOI] [PubMed] [Google Scholar]
- Wang X, Ke Z, Chen G, Xu M, Bower KA, Frank JA, Zhang Z, Shi X, Luo J. Cdc42-dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS One. 2012;7(5):e38075. doi: 10.1371/journal.pone.0038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Chen G, Fu W, Liao M, Frank JA, Bower KA, Fang S, Zhang Z, Shi X, Luo J. Ethanol disrupts vascular endothelial barrier: implication in cancer metastasis. Toxicol Sci. 2012;127(1):42–53. doi: 10.1093/toxsci/kfs087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Pandey A, Shukla A, Talwelkar SS, Kumar A, Pant AB, Parmar D. miR-497 and miR-302b regulate ethanol-induced neuronal cell death through BCL2 protein and cyclin D2. J Biol Chem. 2011;286(43):37347–57. doi: 10.1074/jbc.M111.235531. [DOI] [PMC free article] [PubMed] [Google Scholar]