

Abstract
Hereditary breast cancers stem from germline mutations in susceptibility genes such as BRCA1, BRCA2 and PALB2, whose products function in the DNA damage response and redox regulation. Autophagy is an intracellular waste disposal and stress mitigation mechanism important for alleviating oxidative stress and DNA damage response activation; it can either suppress or promote cancer, but its role in breast cancer is unknown. Here we show that, similar to Brca1 and Brca2, ablation of Palb2 in mouse mammary gland resulted in tumor development with long latency and the tumors harbored mutations in Trp53. Interestingly, impaired autophagy, due to monoallelic loss of the essential autophagy gene Becn1, reduced Palb2-associated mammary tumorigenesis in Trp53-wild type but not conditionally null background. These results indicate that, in the face of DNA damage and oxidative stress elicited by PALB2 loss, p53 is a barrier to cancer development, whereas autophagy facilitates cell survival and tumorigenesis.
Introduction
About 5–10% of breast cancer occurs in the form of inherited predisposition in certain high-risk families in which women tend to develop the disease at higher frequencies and at younger ages than the general population. Interestingly, nearly all of the known familial breast cancer genes function, at least in part, in the repair and/or signaling response to DNA damage, particularly double strand breaks (DSBs) (1). In addition, several of the susceptibility genes, e.g. BRCA1, PALB2, TP53 and ATM, etc., also share a function in reducing cellular levels of reactive oxygen species (ROS), which cause genome damage and promote tumorigenesis (2–6). Thus, a major fraction of hereditary breast cancer appears to result from a common root, namely genome instability.
BRCA1 and BRCA2 perform key functions in genome stability maintenance by promoting faithful repair of DSBs by homologous recombination (HR) and other relevant processes (7, 8). We discovered PALB2 as a major BRCA2 binding partner that controls its chromatin association and function in HR (9). Subsequent work established PALB2 as a BRCA2-like tumor suppressor that is mutated in familial breast and pancreatic cancers as well as Fanconi anemia (10–16). More recently, PALB2 was shown to directly bind BRCA1 as well and link BRCA1 and BRCA2 in HR repair (17, 18). Importantly, multiple patient-derived missense mutations that abrogate PALB2 binding have been identified in both BRCA1 and BRCA2 and shown to disable their HR-repair function (9, 18), indicating that the three proteins function together in a BRCA complex/pathway to promote HR and suppress tumor development.
Contrary to an expectation that mice lacking Brca1 or Brca2 may develop breast cancer, complete knockout of either gene was found to result in early embryonic lethality (19). It was then realized that these genes were indispensable for HR, which is essential not only for tumor suppression but also for mammalian development. Consistent with a role of PALB2 as a linker between BRCA1 and BRCA2 in HR, systemic knockout of Palb2 in mouse resulted in phenotypes similar to that of Brca1 and Brca2, including early embryonic lethality and induction of the cyclin-dependent kinase inhibitor p21 (20, 21).
The p53 transcription program plays essential roles in regulating many critical aspects of cell and tissue physiology that collectively prevent tumorigenesis. Virtually 100% of BRCA1-associated human breast cancers harbor mutations or deletions of the TP53 gene, and BRCA2 and PALB2 tumors also frequently contain TP53 mutations (11, 22, 23). Similarly, somatic mutations in Trp53 are frequently found in mammary tumors that develop in Brca1 and Brca2 conditional knockout (CKO) mouse models (24, 25), and Trp53 co-deletion or heterozygosity strongly accelerated mammary gland tumor development in all Brca1 and Brca2 models tested (26–30). Moreover, loss of p53 partially rescues the embryonic lethality and developmental defect caused by the knockout of each of the 3 genes (21, 31). The evidence indicates that inactivation of the p53 pathway may be a prerequisite for mammary epithelial cells (MECs) to survive the DNA damage and escape the resulting cell cycle checkpoint following BRCA1/2 loss and perhaps also that of PALB2.
Autophagy is an intracellular waste disposal and recycling process whereby damaged organelles and certain proteins are engulfed in double-membrane vesicles (autophagosomes) and delivered to lysosomes for degradation (32). By eliminating damaged mitochondria and toxic protein aggregates and perhaps through other unknown mechanisms, autophagy mitigates oxidative stress and promotes genome stability, thereby suppressing tumorigenesis (33–35). Indeed, monoallelic loss of the essential autophagy gene Beclin 1 (Becn1) in mice leads to increased tumor development at old ages (35–37). Interestingly, autophagy has been shown to be upregulated in RAS-driven cancers, and these cancer cells appear to be “addicted” to and rely on autophagy for survival (38, 39). Thus, autophagy can also facilitate tumor development, presumably by mitigating oxidative stress and promoting tumor cell fitness and nutrient recycling (40, 41).
In this study, we generated and characterized a model of PALB2-associated breast cancer. Moreover, using this model we explored the role of p53 and autophagy in breast cancer associated with oxidative stress and DNA damage. Our results demonstrate that an inactivation of p53 is critical for most, if not all, Palb2-associated tumorigenesis, that autophagy facilitates the development of such breast cancer by promoting tumor cell survival and that the effect of autophagy on mammary tumorigenesis is influenced by p53 status.
Results
Mammary Tumor Development in Palb2 Conditional Knockout Mice
To gain new insights into PALB2-mediated tumor suppression, we targeted the mouse Palb2 gene by inserting loxP sites into introns 1 and 3 (Fig. 1A). Cre-mediated excision of exons 2 and 3 would render exon 4 out of frame and result in a functionally null Palb2 gene (42). To inactivate Palb2 in the mammary gland, Palb2-floxed animals were crossed with mice bearing a Cre transgene driven by the mammary gland specific promoter of whey acidic protein (Wap-cre) (43). The resulting females were mated to undergo two rounds of pregnancy and lactation to induce maximal Cre expression in alveolar MECs, and then monitored for tumor development. As shown in Fig. 1B, 19 out of 29 (66%) of mice with MEC-specific knockout of Palb2 developed 20 mammary tumors (T50=607 days), directly demonstrating that Palb2 acts as a tumor suppressor in the mammary gland. None of the 18 control animals (with Wap-cre) developed mammary tumors.
Figure 1.

Mammary tumor development in mice with tissue-specific ablation of Palb2. A, Schematic representation of the generation of the Palb2-floxed and knockout alleles. The full gene structure of Palb2 is shown on top. B, Kaplan-Meier survival curves of mice with mammary gland-specific deletion of Palb2, Trp53 or both genes. C, Diverse histology of Palb2-associated mouse mammary tumors. I-IV, the 4 different types of histology observed; V-VIII, enlarged views of the center regions of I-IV, respectively; IX, a solid tumor with a well-formed pushing margin; X, a solid tumor invading into fat tissue; XI-XII, higher power views of tumor cell nuclei and mitotic figures.
Characteristics of Palb2-associated Mammary Tumors
Eighteen of the 20 mammary tumors that developed in the Palb2f/f;Wap-cre mice were analyzed for histology and immunophenotypes. Four characteristic histological types were observed- solid (poorly differentiated adenocarcinoma), tubular (well differentiated adenocarcinoma), sarcomatoid (post epithelial to mesenchymal transition (EMT)) and adenosquamous (adenocarcinoma with squamous differentiation) (Fig. 1C). Ten of the 18 tumors (56%) were mostly solid with varying degrees of tubule formation, one was largely tubular, 3 were mostly sarcomatoid, 2 had squamous differentiation and the remaining 2 were mixtures of solid and sarcomatoid with ongoing EMT (Table 1). Necrosis was a common feature in solid areas but rarely seen in other areas or tumors. Nuclear grades were generally high except in the tubular areas of a few tumors. Although well-defined pushing margins were observed for all of the tumors, at least 15 of them were found to have invasive borders in one or more areas (Fig. 1C and S1). Moreover, 10 out of the 18 tumors appeared to have invaded into skin or muscle at the time of collection. Additional views of histology are shown in Fig. S1.
Table 1. Characteristics of mammary tumors developed in Palb2 CKO mice with Becn1+/+ and Becn1+/− backgrounds.
See Figure 1 for definitions and examples of histology and ER, PR and p53 immuno grades. “±” indicates weak staining in only some areas of the tumors. For γH2A.X, “+++”, “++” and “+” denote tumors in which ≥30%, 10–30% and 1–10% of cells, respectively, show 3 strongly stained foci or virtually pan-nuclear staining; “SF” denotes tumors in which a single, large focus was observed per cell, which is possibly the inactivated X chromosome. Grade assignment for 8-oxo-dG staining is based on the relative staining intensity, as all positive tumors have greater than 60% of cells showing positive staining. Trp53 mutations are shown at both DNA and protein levels, separated by a forward slash. N.D., Trp53 cDNA sequence not determined due to poor quality of RNA isolated from frozen tissues.
| Mouse ID | Becn1 | Latency | Tumor | Histology (H&E) | Immunohistochemistry | Trp53 mutation | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| ER | PR | γH2A.X | 8-oxo-dG | p53 | ||||||
| 914 | +/+ | 383 | 1 | Solid+Tubular | ++ | ++ | +++ | +++ | +++ | G809A/R270H |
| 949 | +/+ | 398 | 1 | Solid+Tubular | + | − | ++ | +++ | +++ | G720A/M240I, G721T/G241W |
| 741 | +/+ | 422 | 1 | Sarcomatoid+Tubular | ++ | ++ | ++ | +++ | − | WT |
| 749 | +/+ | 428 | 1 | Solid+Tubular | + | − | SF | +++ | − | Δ985–1089 |
| 826 | +/+ | 430 | 1 | Solid→Sarcomatoid | + | − | + | +++ | − | N.D. |
| 824 | +/+ | 431 | 1 | Solid+Tubular | + | − | +++ | +++ | +++ | T391C/F131L |
| 808 | +/+ | 450 | 1 | Sarcomatoid+Tubular | − | − | − | +++ | − | N.D. |
| 882 | +/+ | 453 | 1 | Solid+Tubular | − | + | +++ | ++ | +++ | WT |
| 751 | +/+ | 456 | 1 | Tubular+Solid | − | − | SF | − | − | WT |
| 049 | +/+ | 498 | 1 | Solid+Tubular | − | − | +++ | +++ | − | Δ541–546, Δ664–771 |
| 912 | +/+ | 498 | 1 | Solid→Sarcomatoid | ++ | − | +++ | +++ | +++ | C808T/R270C |
| 915 | +/+ | 529 | 1 | Solid+Tubular | − | − | ++ | + | +++ | A632G/H211R |
| 742 | +/+ | 576 | 1 | Squamous+Tubular | + | ++ | − | +++ | − | N.D. |
| 827 | +/+ | 608 | T1 | Solid+Tubular | − | − | + | ++ | +++ | G829A/R277G |
| +/+ | 608 | T2 | Solid+Tubular | − | − | + | − | +++ | N.D. | |
| 825 | +/+ | 637 | 1 | Solid+Tubular | − | − | + | +++ | − | WT |
| 042 | +/+ | 665 | 1 | Squamous+Tubular | − | − | − | ++ | − | no PCR product |
| 747 | +/+ | 719 | 1 | Sarcomatoid+Solid | − | − | +++ | +++ | + | T618A/V206D |
| 907 | +/− | 520 | 1 | Solid+Tubular | − | − | SF | + | − | WT |
| 163 | +/− | 522 | 1 | Solid+Tubular | − | ++ | +++ | +++ | − | WT |
| 890 | +/− | 529 | T1 | Solid | − | − | ++ | ++ | +++ | C404T/A135V |
| +/− | 529 | T2 | Squamous+Tubular | − | − | SF | ++ | − | N.D. | |
| 047 | +/− | 567 | 1 | Squamous+Solid+Tubular | − | − | − | + | − | N.D. |
| 723 | +/− | 588 | 1 | Solid+Tubular | ± | − | +++ | +++ | − | WT |
| 119 | +/− | 626 | 1 | Solid+Tubular | ± | + | + | +++ | − | WT |
| 153 | +/− | 665 | 1 | Tubular+Solid | − | − | + | + | ++ | WT |
The status of the estrogen receptor (ER) and progesterone receptor (PR) in the 18 tumors was analyzed by immunohistochemistry (IHC) (Fig. 2A). Eight (44%) tumors showed positive ER staining and 4 (22%) were PR-positive (Table 1). For ER, the positive tumors generally showed nuclear staining in greater than 30% of the cells but the overall signal strength appeared to be weaker than what is commonly seen in typical human ER+ cancers. Similar findings were made for PR except that higher background staining was observed in approximately half of the tumors, in which case a “-” status was assigned unless some of the cells showed strong nuclear signal clearly above the background. Taken together, these results demonstrates that somatic deletion of Palb2 driven by Wap-cre can give rise to both ER+ and ER− mammary tumors, a scenario similar to human PALB2-associated breast cancers (10).
Figure 2.

Characterization of Palb2-asscoiated mouse mammary tumors by IHC. A, Representative ER and PR staining patterns of the tumors. Since similar percentage of staining- positive cells were found in most of the positive tumors, assignment of “+” or “++” grades is purely based on the intensity of staining signals. B, Representative staining patterns of p53 in the tumors. Grade assignment is based on the relative staining intensity. C, Different staining patterns of γH2A.X. (upper panels) and 8-oxo-dG (lower panels) in the control and Palb2-null tumors.
Role of p53 in Palb2-associated Mouse Mammary Tumors
The prevalence of TP53 mutations in BRCA- and PALB2-associated human breast tumors led us to sequence the Trp53 gene (cDNA) in tumors that arose from Palb2f/f;Wap-cre mice. Among the 14 tumors analyzed, 9 (64%) contained missense mutations or internal deletions, 4 were wild type and the remaining one did not yield cDNA, presumably due to biallelic deletion or extremely low mRNA expression level (Table 1). This finding suggests that loss of p53 function is important for the development of Palb2-associated mammary tumors. In the 4 tumors with a wild type Trp53 transcript, it is still possible that the p53 pathway may be rendered nonfunctional by other mechanisms, such as hyperactivation of MDM2.
To further understand the status of p53 in the tumors, we analyzed its protein levels using IHC (Fig. 2B). Nine (50%) of the 18 tumors were positive, including all of the 7 tumors with missense mutations (Table 1). As expected, the 2 tumors with intragenic deletions/frameshift mutations both showed completely negative staining. Three of the 4 tumors with wt Trp53 were negative but one was, surprisingly, strongly positive (#882). Although it is unclear whether the p53 downstream pathway is active in this particular tumor, our findings overall indicate that loss of normal p53 function is critical for the development of Palb2-associated mammary tumors.
To study the genetic interaction between Palb2 and Trp53, we introduced a floxed Trp53 allele (26) into our model. As shown in Fig. 1B, combined deletion of Palb2 and Trp53 in MECs led to highly efficient tumor development that is much faster than that caused by Palb2 single deletion. The median tumor latency of the double CKO mice was also slightly shorter than that of the Trp53 single CKO mice (T50=246 vs 289 days), suggesting that two genes may synergistically suppress breast cancer development. However, the difference did not reach statistical significance (p=0.0647, log-rank analysis). Additionally, we also monitored a small number (n=9) of Palb2w/w;Trp53f/w;Wap-cre females, and 7 of them developed mammary tumors with latencies from 466 to 736 days, which were in the similar range as tumors arising in Palb2f/f;Trp53w/w;Wap-cre mice.
DNA Damage in Palb2-null Tumor Cells and Their Sensitivity to DNA Damaging Agents
Given the role of PALB2 in DNA repair, we assessed the extent of endogenous DNA damage in tumors by IHC analysis of γH2A.X, a marker of DSBs (Fig. 2C). Thirteen (72%) of them showed γH2A.X staining regardless of Trp53 status, indicative of the existence of unrepaired DSBs (Table 1). In contrast, little to no staining was detected in tumors arising from Palb2w/w;Trp53f/w;Wap-cre mice, which developed with similar latency (Fig. 2C). Consistent with our recent finding that PALB2 plays a role in the oxidative stress response (4), the Palb2-null tumors were found to have much higher levels of oxidative DNA damage as revealed by IHC staining of 8-oxo-dG, a marker of such damage, as compared with the above-noted Trp53-associated tumors (Fig. 2C).
We have shown earlier that PALB2-null FA fibroblasts, like BRCA1- and BRCA2-null cells, are sensitive to agents that target HR-mediated DSB repair, such as mitomycin C (MMC) and poly (ADP-ribose) polymerase (PARP) inhibitors (14, 44). However, to our knowledge no human PALB2-null breast cancer cells have been established. Thus, to better understand the function and “druggability” of PALB2 we attempted to generate cell lines from the mouse mammary tumors. Several attempts were made to generate cell lines from Palb2-single-null tumors, but only one useful line (PF741) was successfully established, which retained a wt Trp53 gene. In contrast, multiple lines were established from Trp53-single-null (Palb2-wt) and Palb2/Trp53-double-null tumors.
Above cells were tested for their ability to repair DNA damage elicited by olaparib, a PARP inhibitor, and MMC. Interestingly, the Palb2-null cells contained more DSBs as revealed by γH2A.X immunofluorescence (IF) even in the absence of drugs, which was particularly evident in the Palb2/Trp53-double-null cells (Figs. 3A and S2). By 3 hr following treatment, both agents resulted in increased γH2A.X staining signal in all 3 cell types, and distinct RAD51 foci colocalizing with those of γH2A.X was observed in Palb2-wt cells but not in either of the Palb2-null cells. By 8 hr post treatment, γH2A.X signals had returned to pre-treatment levels in the Palb2-wt cells, whereas the signals persisted in both types of Palb2-null cells. By this time, RAD51 foci had largely disappeared in the Palb2-wt cells, and Palb2-single-null cells showed a diffuse RAD51 staining pattern. Another p53-single-null (control) and 2 additional Palb2/Trp53-double-null cell lines were tested in parallel and the results were essentially the same.
Figure 3.

DNA repair defect of Palb2-null tumor cells. A, γH2A.X and RAD51 foci formation before and after DNA damage induced by olaparib. Tumors cells were treated with 25 μM olaparib for 1 hr and the drug was then removed. Cells were fixed at 3 and 8 hr after drug removal and analyzed by IF. B, Levels of DNA breaks before and after olaparib treatment. Cells were treated as above, collected at the same time points and analyzed by neutral comet assay. C–D, Sensitivity of the tumor cells to olaparib and MMC. Cells were seeded in 96-well plates, treated with the drugs for 4 days and analyzed by CellTiterGlo assay. E, Western blots showing PALB2 and p53 protein levels in the tumor cells analyzed in C–D.
Next, we performed neutral comet assays to further assess the levels of DNA breaks in the 6 cell lines. Compared with the 2 Palb2-wt cells, all 4 Palb2-null cells showed substantially higher levels of DNA breaks before drug treatment (Fig. 3B and S2), indicative of a significant defect in the repair of DNA breaks resulting from endogenous factors, such as collapse of replication forks, etc. At 3 hr after drug treatment, increased DNA fragmentation was seen in all cells. By 8 hr post treatment, the levels of DNA breaks were found to have decreased in the Palb2-wt cells but not the Palb2-null cells (Figs. 3B and S2), again indicating an inability of the mutant cells to execute HR-based repair. Consistently, both types of Palb2-deficient cells were hypersensitive to both agents (Fig. 3C–D). The deletion of the respective proteins in the cell lines were confirmed by Western blotting (Fig. 3E). Collectively, these results further underscore the critical role of PALB2 in HR repair and support the applicability of PARP inhibitors and DNA crosslinkers for PALB2-associated cancers.
Senescence and Apoptosis upon Palb2 Deletion and the Rescue by Co-deletion of Trp53
To test the immediate consequence of PALB2 loss in primary cells and the role of p53 in this process, we generated mouse embryonic fibroblasts (MEFs) from Palb2f/f and Palb2f/f;Trp53f/f mice. After the cells were infected with a Cre-encoding retrovirus to induce gene deletion and subjected to selection, virtually complete loss of the respective proteins was observed (Fig. 4A–B). As expected, Palb2-null MEFs showed much increased endogenous DSBs as evidenced by nuclear foci formation of 53BP1 (Fig. 4C). γH2A.X foci were not counted in all experiments. However, when we co-stained γH2A.X with 53BP1, all 53BP1-positive cells were found to be positive for γH2A.X whereas some cells showing multiple but weakly stained γH2A.X foci did not display distinct 53BP1 foci (Fig. S3). Thus, the actual extent of DNA breaks in the cells should be even greater.
Figure 4.

Senescence and apoptosis of mouse embryonic fibroblasts (MEFs) following Palb2 loss and the rescue by co-deletion of Trp53. A, Schematic timeline of the generation and passaging of the MEFs. Two different MEF lines of each genotype were generated and analyzed in parallel. B, Western blots showing loss of PALB2, p53 or both proteins in the MEFs at passage 2. C, 53BP1 nuclear foci formation in the control, Palb2 deletion and Palb2/Trp53 double deletion MEFs. The top panel shows representative immunofluorescence (IF) images of 53BP1 staining during passage 1, and the bottom panel shows quantification of foci-positive cells in all 3 passages. D, NRF2 localization in the MEFs during passage 1, as determined by IF. E, Cellular levels of reactive oxygen species (ROS) in the MEFs in all 3 passages. F–G, Cellular senescence induced by Palb2 inactivation and the rescue by co-deletion of Trp53. F shows representative images of beta-galactosidase (β-gal) staining of the wild type, Palb2-null and Palb2/Trp53-double-null MEFs, and the quantification is shown in G. H, Growth curves of the MEFs showing the growth arrest of the Palb2-null MEFs and the rescue by loss of p53. I, Cellular apoptosis following Palb2 inactivation and the rescue by co-deletion of Trp53. Apoptotic cells were measured by Annexin V assay. In all above analyses, values shown are the averages of the 2 independent MEFs lines for each genotype and error bars represent standard deviations. P values were determined by two-tailed t-test. *p≤0.05; **p≤0.01.
Consistent with our previous finding that PALB2 promotes the nuclear accumulation and function of the antioxidant transcription factor NRF2 (4), the protein was localized mostly in the nucleus in Palb2-wild type MEFs but showed a diffuse staining pattern in Palb2-null MEFs (Fig. 4D). Accordingly, Palb2-null MEFs had significantly higher ROS levels throughout the experimental period (Fig. 4E). Together with the fact that Palb2-null tumor cells contained higher levels of oxidative DNA damage (Fig. 2C), these results further underscore the importance of PALB2 in cellular defense against oxidative stress.
Notably, starting from passage 2, large numbers of Palb2-null MEFs appeared flat and enlarged, stained positive for beta-galactosidase (β-gal) and displayed poor growth (Fig. 4F–H), indicating that the cells were entering senescence. Moreover, an Annexin V assay revealed apoptosis occurring in a substantial fraction of the cells (Fig. 4I). Co-deletion of Trp53 completely rescued the slow growth, senescence and apoptosis phenotypes that resulted from Palb2 deletion. These observations indicate that loss of p53 is able to allow cells to overcome growth arrest or apoptosis induced by DNA damage and oxidative stress after PALB2 loss.
Effect of Becn1 Heterozygosity on Palb2-associated Mammary Tumorigenesis
Autophagy is particularly important for mitigating oxidative stress and suppressing DNA damage response activation during stresses. In addition, recent studies have demonstrated that autophagy can also facilitate cellular senescence (45). Therefore, we suspected that autophagy might play a role in PALB2-associated breast cancer development. To address this, we crossed the Palb2f/f;Wap-cre and Palb2f/f;Trp53f/f;Wap-cre mice to Becn1+/− mice (36). As shown in Fig. 5A, allelic loss of Becn1 significantly delayed mammary tumor formation in Palb2f/f;Wap-cre animals (p=0.0035, log-rank analysis). Moreover, only 7 of the 26 Becn1+/− animals developed mammary tumors. However, it did not affect tumorigenesis due to combined MEC-specific loss of Palb2 and Trp53, suggesting that the suppression of Palb2-mediated tumorigenesis upon allelic loss of Becn1 is mediated by p53.
Figure 5.

Role of autophagy in Palb2-associated mammary tumor development. A, Kaplan-Meier survival curves showing mammary tumor development in Palb2-single and Palb2;Trp53-double conditional knockout mice in Becn1+/+ and Becn1+/− backgrounds. B, Autophagosomes observed in tumors from mice with indicated genotypes. Note that tumor #827 contains a Trp53 point mutation (Table 1), although it developed in Trp53-wild type mice. C, IHC analysis of autophagy substrate p62 in tumors arising from the 4 different genetic backgrounds as indicated. Tumors #751 and #163 still retained wild type Trp53, whereas #915 and #890 had acquired somatic mutations in Trp53 (Table 1). D, IHC analysis of cleaved caspase 3, a marker of apoptosis, in the same tumors as in C.
The 8 tumors that formed in the 7 Palb2f/f;Wap-cre;Becn1+/− mice were similar to their Becn1+/+ counterparts in terms of histology and DNA damage levels (Table 1). Two (25%) of them were marginally positive for ER, showing weak staining signals that were only seen in some areas of the tumors. Moreover, only one of the 6 tumors (16.7%) sequenced was found to have a Trp53 mutation, as compared with the 64% mutation rate of the Becn1+/+ tumors. These findings imply that a defect in autophagy may force a different path of tumor evolution following PALB2 loss. Due to the small number of Becn1+/− tumors obtained in this work, a larger study may be needed to confirm the results and address the potential mechanisms.
Autophagy and Apoptosis in the Palb2-deficient Mammary Tumors
The finding that allelic loss of Becn1 suppressed Palb2-associated mammary tumorigenesis by a p53-depedent mechanism suggests that autophagy facilitates tumor development. To assess the levels of autophagy activity in the mammary tumors, we analyzed 12 tumor samples (6 Becn1+/+ and 6 Becn1+/−) using electron microscopy. A number of autophagosomes were identified (Fig. 5B), indicating that autophagy indeed occurs in Palb2-associated breast cancer even in the absence of external stress. Notably, autophagosomes were observed in 5 of the 6 Becn+/+ tumors but in only one of the 6 Becn+/− tumors, suggesting that allelic loss of Becn1 caused a partial, but appreciable, impairment of autophagy in the setting used. To further confirm the autophagy defect in the Becn1+/− tumors, we compared the levels of p62 (SQSTM1), which is an important substrate for autophagy and accumulates when autophagy is impaired (35). When necrotic areas were excluded, all Becn1+/+ tumors exhibited weak or virtually no staining signal, whereas distinct areas of strong staining were observed in tumors arising from Palb2f/f;Wap-cre;Becn1+/− mice (Fig. 5C). In mammary tumors from Palb2f/f;Trp53f/f;Wap-cre;Becn1+/− animals, positive p62 staining was observed, but was markedly weaker than in mammary tumors arising from Palb2f/f;Wap-cre;Becn1+/− mice.
Next, we analyzed the levels of cleaved (activated) caspase-3, which marks apoptotic cells, in the tumor tissues. As in the case of p62, tumors from Palb2f/f;Wap-cre;Becn1+/+ mice showed weak or no cleaved caspase-3 staining across non-necrotic areas, whereas pockets of positive staining were found in tumors that developed in Palb2f/f;Wap-cre;Becn1+/− animals (Fig. 5D). In the Palb2;Trp53-doubly-deleted tumors, the staining was all negative regardless of Becn1 status. Thus, combined deficits in DNA repair and autophagy appeared to elevate p53-dependent apoptosis in Palb2−/−;Becn1+/− mammary tumor cells. To further address the potential correlation between autophagy defect and cell death (apoptosis) in Palb2-associated mammary tumors, we analyzed 6 tumors with wild type Trp53 (3 Palb2−/−;Becn1+/− and 3 Palb2−/−; Becn1+/+) by IHC for p62, LC3B (another autophagy substrate) and cleaved caspase-3. As shown in Fig. S4, the 3 Becn+/− tumors stained positive for all 3 markers, whereas the 3 Becn1+/+ tumors showed virtually no staining.
Discussion
We demonstrated that ablation of Palb2 in MECs led to mammary tumor development with a median latency of 607 days. The tumors displayed diverse histology but were generally high grade and invasive. With respect to hormone receptors, 44% of tumors analyzed showed positive ER staining and 22% were PR-positive (Table 1). In comparison, human PALB2 cancers are also generally high grade, whereas ER/PR status of the tumors appears to vary significantly depending on mutations and/or populations (10). Overall, approximately 40% of human PALB2 tumors were triple negative for ER, PR and HER2, putting the clinical phenotypes of PALB2 between BRCA1 and BRCA2 (10, 46). Thus, although our model shows somewhat lower positivity for ER and PR, it still recapitulates some key features of the human PALB2 disease, namely the high grade and intermediate ER positivity.
Among the numerous Brca1- and Brca2- CKO breast cancer models that have been reported (19, 47), the most comparable to this model are the Wap-cre-driven ones reported by Ludwig and colleagues (24, 25). In these studies, somatic deletion of Brca1 resulted in tumors that were 91% (19/21) ER-negative and basal-like; while Brca2 ablation produced tumors that were 50% (15/30) ER+ and 7% (2/30) PR+. Thus, the 44% ER+ and 22% PR+ rates of the Palb2 mammary tumors in this study are more phenotypically similar to Brca2-associated ones, which is consistent with what appears to be a much stronger physical association of PALB2 with BRCA2 than with BRCA1 (9, 18).
Median latencies of 512 and 508 days were reported for the above Brca1f/f;Wap-cre and Brca2f/−;Wap-cre mice, respectively (24, 28). In a separate Brca1f/−;Wap-cre model, mammary tumors were found in 2 out of 13 animals sacrificed between ages of 10–13 months (27). Given the differences in backgrounds and experimental settings, it is impossible to strictly compare these latencies with that of the present Palb2 model. Still, the long latencies in all 4 models indicate that there is a strong barrier to tumor development in mammary epithelial cells (MECs) that have suffered the loss of any one of these tumor suppressor proteins. As noted before, accumulating evidence from both human and mouse studies suggest that the barrier may be mostly enforced by p53. Our finding that the majority of the Palb2-associated tumors analyzed here (9/14) were Trp53-mutated lends further support to the above notion.
The most prominent molecular function shared by BRCA1/2 and PALB2 proteins is their role in HR, which is the major mechanism to repair the type of DSBs that inevitably arise during normal DNA replication. Upon loss of any of these proteins, an inability of cells to prevent and repair collapsed replication forks leads to DSB accumulation and a DNA damage response that presumably activates p53. Depending on circumstances and the extent of p53 activation, cells may undergo G1/S arrest, senescence and/or apoptosis. It still remains to be seen which one(s) is the predominant consequence of BRCA or PALB2 loss in MECs in vivo. This knowledge is important for understanding the developmental path, as well as tissue specificity, of BRCA- and PALB2-associated cancers.
Based on existing knowledge and results obtained in this study, we propose a model of PALB2-associated hereditary breast cancer development as illustrated in Fig. 6. Under normal conditions, PALB2 functions together with BRCA1 and BRCA2 to maintain genome stability and cellular homeostasis to suppress cancer development. When PALB2 is lost, increased DNA damage and ROS cause activation of p53, which induces growth inhibition and perhaps senescence or apoptosis thereby suppressing tumor formation. Under such adverse conditions, autophagy facilitates cell survival and growth, which allows PALB2-null cells to accumulate further mutations and evolve into cancer cells. When autophagy is defective, increased cell death occurs and the potential for tumor development is reduced. If PALB2 is lost in a cell with already mutated p53, highly efficient tumor formation occurs under both normal and low autophagy conditions.
Figure 6.
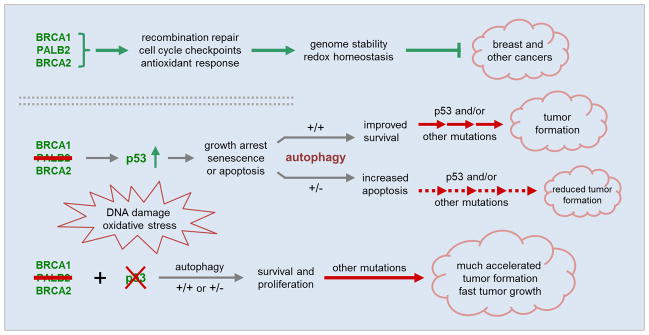
A model of the developmental paths of PALB2-associated breast cancer. Under normal conditions, PALB2 functions together with BRCA1 and BRCA2 to maintain genome stability and cellular homeostasis to suppress cancer development. Upon loss of PALB2, p53 is activated posing a strong barrier to tumor formation, whereas autophagy helps sustain cell viability and proliferation thereby facilitating tumor cell evolution. However, the impact of autophagy may only manifest when p53 is functional.
The role of p53 in regulating autophagy has been reported by multiple groups and appears to be complex. In particular, one study showed that nuclear p53 promotes autophagy by inducing relevant gene expression whereas cytoplasmic p53 inhibits autophagy (48). In the present study, allelic loss of Becn1 did not produce any difference in tumor formation in mice with Palb2;Trp53 double deletion in MECs. This finding may have at least two different implications. First, the strong growth advantage conferred by a complete p53 loss may override the reduced fitness elicited by impaired autophagy in Palb2−/− MECs or tumor cells. Second, p53 may negatively regulate autophagy in these cells so that loss of p53 leads to a compensation of autophagy function. However, it is important to note that real world cancers mostly harbor TP53 point mutations combined with loss of heterozygosity (LOH) instead of biallelic deletions, and it is known that point mutants may possess both loss and gain of functions. Therefore, the actual effect of TP53 mutations on the impact of autophagy on cancer may be variable and again context-dependent.
Our finding is consistent with a recent study which found that allelic loss of Becn1 delayed tumor development in ATM-deficient mice (3). However, while the above study suggests that Becn1 heterozygosity leads to a restoration of mitochondria health damaged by ATM deficiency, no gross difference was noted in mitochondria of the tumor samples analyzed by EM in the present study. Still, the Becn+/− tumors appeared to contain fewer vesiculated mitochondria and perhaps more fused ones than the Becn1+/+ tumors (Fig. S5). Yet, due to the small number of samples analyzed and the high degree of heterogeneity within each sample, it is unfeasible to draw any conclusions from the current study. Further investigation is needed to understand the potential involvement of mitochondria physiology in hereditary breast cancer.
Inhibiting autophagy as a potential cancer therapy has gained increasing attention. In this study, Palb2−/−;Becn1+/− tumors had reduced incidence and also seemed to grow slower compared with the corresponding Becn1+/+ tumors. Consistently, such (Becn1+/−) tumors were found to contain areas undergoing apoptosis. These results suggest that rational autophagy inhibition may selectively kill PALB2-deficient tumor cells. Given the close relationship and functional similarity between PALB2 and BRCA1/2, the same notion may apply to BRCA-deficient tumor cells as well.
Materials and Methods
To create a Palb2 conditional knockout mouse model, we targeted the Palb2 locus and generated a strain in which exons 2 and 3 of the gene are flanked by loxP sites (42). The Palb2flox/flox mice were crossed to strains carrying Trp53flox2-10 (26), Becn1-KO (36) and Wap-cre (43) alleles to generate all the genotypes in this study. Females of desired genotypes were mated to go through two rounds of pregnancy and lactation to induce Wap-cre expression and then monitored for tumor development. Tumors were collected when they reached ~1.0 cm in diameter. Primary mouse embryo fibroblasts (MEFs) were generated from E13.5 embryos. All experimental procedures involving animals were conducted in accordance with policies set forth by the Institutional Animal Care and Use Committee (IACUC) of the Robert Wood Johnson Medical School and under the protocol numbers I08-073-9 and I11-029-5. To delete Palb2 and Trp53 genes in MEFs, freshly generated cells with floxed alleles were infected with a Cre-encoding retrovirus and selected with puromycin. Mammary tumor cells were generated from tumor specimens dissociated with collagenase. Olaparib and mitomycin C (MMC) sensitivities were determined by the CellTiter Glo® cell proliferation assay (Promega). Levels of reactive oxygen species (ROS) were measured using the DCF (2′,7′-Dichlorofluorescin diacetate) assay. Cellular senescence and apoptosis were determined using the senescence-associated β-galactosidase (SA-β-gal) assay and Annexin V assay, respectively. Western blotting and immunofluorescence (IF) staining were performed using standard protocols. Neutral comet assay was performed using the CometAssay® kit from Trevigen following manufacturer’s protocol. For details see online methods in the Supporting Information (SI).
Supplementary Material
Significance.
Our findings directly demonstrate a tumor-promoting role of autophagy in a new model of hereditary breast cancer. Given the close functional relationship and the genetic similarity between PALB2 and BRCA1/2, our results further suggest that inhibition of autophagy may represent a new avenue to the prevention or treatment of a significant portion of hereditary breast cancers, namely ones associated with DNA damage and oxidative stress.
Acknowledgments
We thank Drs. David Livingston and Chrysi Kanellopoulou for supporting the initial stage of the work and for their critical comments on the manuscript. We also thank Dr. Shoreh Miller for valuable assistance in mouse breeding. This work was supported by the National Cancer Institute (R01CA138804 to B.X.), the American Cancer Society (RSG #TBG-119822 to B.X.) and The Cancer Institute of New Jersey (to B.X.).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the authors.
References
- 1.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Saha T, Rih JK, Rosen EM. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS letters. 2009;583:1535–43. doi: 10.1016/j.febslet.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–500. doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Molecular and cellular biology. 2012;32:1506–17. doi: 10.1128/MCB.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free radical biology & medicine. 2008;44:1529–35. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ, et al. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64:7893–909. doi: 10.1158/0008-5472.CAN-04-1119. [DOI] [PubMed] [Google Scholar]
- 7.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature reviews. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 12.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 14.Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–61. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 15.Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–9. doi: 10.1158/0008-5472.CAN-10-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science (New York, NY. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–9. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106:7155–60. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–97. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- 20.Rantakari P, Nikkila J, Jokela H, Ola R, Pylkas K, Lagerbohm H, et al. Inactivation of Palb2 gene leads to mesoderm differentiation defect and early embryonic lethality in mice. Hum Mol Genet. 2010;19:3021–9. doi: 10.1093/hmg/ddq207. [DOI] [PubMed] [Google Scholar]
- 21.Bouwman P, Drost R, Klijn C, Pieterse M, van der Gulden H, Song JY, et al. Loss of p53 partially rescues embryonic development of Palb2 knockout mice but does not foster haploinsufficiency of Palb2 in tumour suppression. J Pathol. 2011;224:10–21. doi: 10.1002/path.2861. [DOI] [PubMed] [Google Scholar]
- 22.Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High Incidence of Protein-Truncating TP53 Mutations in BRCA1-Related Breast Cancer. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-3426. [DOI] [PubMed] [Google Scholar]
- 23.Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res. 2001;61:4092–7. [PubMed] [Google Scholar]
- 24.Ludwig T, Fisher P, Murty V, Efstratiadis A. Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene. 2001;20:3937–48. doi: 10.1038/sj.onc.1204512. [DOI] [PubMed] [Google Scholar]
- 25.Shakya R, Szabolcs M, McCarthy E, Ospina E, Basso K, Nandula S, et al. The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc Natl Acad Sci U S A. 2008;105:7040–5. doi: 10.1073/pnas.0711032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 27.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104:12111–6. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung AM, Elia A, Tsao MS, Done S, Wagner KU, Hennighausen L, et al. Brca2 deficiency does not impair mammary epithelium development but promotes mammary adenocarcinoma formation in p53(+/−) mutant mice. Cancer Res. 2004;64:1959–65. doi: 10.1158/0008-5472.can-03-2270. [DOI] [PubMed] [Google Scholar]
- 30.Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115–24. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes & development. 1997;11:1226–41. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 33.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes & development. 2007;21:1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes & development. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of clinical investigation. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & development. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes & development. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes & development. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowman-Colin C, Xia B, Bunting S, Klijn C, Drost R, Bouwman P, et al. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1305362110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic acids research. 1997;25:4323–30. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–54. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes & development. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15:3214–22. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Cruz ES, Cabrera MC, Nakles R, Rutstein BH, Furth PA. BRCA1 deficient mouse models to study pathogenesis and therapy of triple negative breast cancer. Breast Dis. 2010;32:85–97. doi: 10.3233/BD-2010-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–4. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.