

Abstract
Background
The microtubule associated protein tau accumulates in neurodegenerative diseases known as tauopathies, the most common being Alzheimer’s disease (AD). One way to treat these disorders may be to reduce abnormal tau levels through chaperone manipulation, thus subverting synaptic plasticity defects caused by tau’s toxic accretion.
Methods
Tauopathy models were used to study the impact of YM-01 on tau. YM-01 is an allosteric promoter of triage functions of the most abundant variant of the Hsp70 family in the brain, Hsc70. The mechanisms by which YM-01 modified Hsc70 activity and tau stability were evaluated with biochemical methods, cell cultures and primary neuronal cultures from tau transgenic mice. YM-01 was also administered to acute brain slices of tau mice; changes in tau stability and electrophysiological correlates of learning and memory were measured.
Results
Tau levels were rapidly and potently reduced in vitro and ex vivo upon treatment with nanomolar concentrations of YM-01. Consistent with Hsc70 having a key role in this process, over-expression of Hsp40 (DNAJB2), an Hsp70 co-chaperone, suppressed YM-01 activity. In contrast to its effects in pathogenic tauopathy models, YM-01 had little activity in ex vivo brain slices from normal, wildtype mice unless microtubules were disrupted, suggesting that Hsc70 acts preferentially on abnormal pools of free tau. Finally, treatment with YM-01 increased long-term potentiation in from tau transgenic brain slices.
Conclusions
Therapeutics that exploit the ability of chaperones to selectively target abnormal tau can rapidly and potently rescue the synaptic dysfunction that occurs in AD and other tauopathies.
Keywords: tau, Alzheimer’s disease, chaperones, Hsc70, rhodocyanine, YM-01
Introduction
Abnormal tau accumulation in the brain is a critical contributing factor to pathogenesis in more than fifteen neurodegenerative diseases, including traumatic brain injury (TBI) and Alzheimer’s disease (AD) (1–9). Early pathological assessments link the cognitive decline that occurs in these diseases to synaptic plasticity defects, such as dystrophic neurites and neuropil threads (10). This hypothesis of altered synaptic activity in AD has more recently been confirmed by studies showing that the amyloid β (Aβ) peptide impairs axonal transport in conjunction with tau (11). Moreover, post-synaptic defects linked to disease progression are also evident in AD and tauopathy models (12–15). The main source thought to contribute to this synaptic dysfunction is not pathological tau tangles, but rather soluble tau intermediates. Aβ can generate this abnormal tau in AD, splice errors in tauopathies, or over-expression in transgenic tau models. In fact, a number of studies have shown that neuronal and cognitive defects of AD and mouse models of tauopathy can be reversed by genetically reducing soluble tau intermediates instead of stopping or reversing tau aggregation (8, 11, 16–20). Finding ways to chemically reduce these soluble tau intermediates would be beneficial as potential therapeutic strategies for AD and other tauopathies.
One way of controlling tau reduction may lie in affecting triage for tau degradation. Normal tau homeostasis is in part maintained through the actions of heat shock protein 70 variants. The inducible for of Hsp70 is termed Hsp72 and the constitutively expressed form of Hsp70 is termed heat shock cognate 70 protein (Hsc70) (21–23). These proteins are very similar structurally, but they are regulated in different ways. Hsp72 and Hsc70 cooperate with distinct subsets of co-chaperones to differentially affect whether aggregation-prone or misfolded tau should be degraded or retained. Although Hsp72 can facilitate tau degradation, the vast majority of tau in the brain is controlled by the more abundant Hsc70 (23). Unlike Hsp72, Hsc70 actually slows tau clearance, particularly after microtubule disruption (21–23). Altering the association of tau with Hsc70 could facilitate tau degradation; consistent with this idea, we have recently found that first generation general Hsp70 protein inhibitors, such as methylene blue (MB), rapidly reduce tau levels (22, 24–26) and partially suppress learning defects in the rTg4510 tauopathy mouse model (17, 26). Although these initial results were encouraging, the first generation compounds lacked sufficient selectivity.
The search for more potent and selective inhibitors yielded MKT-077, which has also been extensively studied as an anti-cancer agent (27, 28). The compound was shown to bind to an allosteric site on Hsc70 and stabilize the ADP-bound form of the chaperone (29). The binding site of MKT-077 is highly conserved amongst members of the Hsp70 family of chaperones; this molecule appears to bind Hsc70, mitochondrial Hsp70 (mtHsp70, mortalin), as well as other family members (27, 29, 30). An analog of MKT-077, YM-01, was recently found to have much more potent anti-cancer activity in tamoxifen-resistant cancer cell lines, based on cytoplasmic localization (31). Together, these observations suggest that MKT-077 and YM-01 might be valuable chemical probes to explore the relationship between Hsc70 and tau homeostasis and to test the potential of this chaperone as a drug target in tauopathy.
Based on these data, we predicted that YM-01 inhibition could promote tau clearance in tau transgenic models. Not only did YM-01 reduce tau potently in cell models at sub-micromolar concentrations, but also the reductions were extremely rapid, occurring within the first 60 minutes of treatment in brain tissue. Further, YM-01 significantly improved long-term potentiation (LTP) in the rTg4510 transgenic model of tauopathy within this 60-minute window. This compound had little effect on tau levels in brain slices from wildtype mice unless microtubules were damaged, suggesting that YM-01 takes advantage of the normal triage activity of Hsc70 to selectively promote the degradation of pathogenic tau. This finding has important implications for the expected safety of this class of small molecules and provides a new mechanism-of-action (MoA) for exploration. These results also show that abnormal tau is poised for clearance, but Hsc70 subverts this fate, further implicating Hsc70 as therapeutic target for AD and tauopathies.
Materials and Methods
Antibodies and Reagents
PHF1 tau antibody (pS396/S404) was provided by Dr. Peter Davies. Tau5 was provided by Dr. Lester Binder. Hsc70 antibody was purchased from Stressgen. Total tau antibody was purchased from Santa Cruz Biotechnology. Flag and actin antibodies, phenylmethylsulfonyl fluoride (PMSF), phosphatase inhibitor cocktails (I & III), mammalian protease inhibitor and nocodazole were purchased from Sigma. GAPDH antibody was from Biodesign International. M-PER was used as cell lysis buffer and purchased from Fisher Scientific. Secondary antibodies were purchased from Southern Biotech Inc. Lipofectamine 2000 was purchased from Invitrogen. Plasmids for human Hsc70 and DnaJ proteins were generated using pCMV6 vector with a FLAG tag where indicated (Origene). All tau clones were in pCDNA3.1 plasmid. Epoxomicin was purchased from ELAN pharmaceuticals.
Single-Turnover ATPase Activity
Single turnover ATPase assays were performed using a purified Hsp70 chaperone, Ssa1, which was pre-bound to α32P-ATP, as described (32). The compound or the vehicle control (DMSO) were incubated with the chaperone and reaction aliquots were removed at the indicated times. Select reactions also contained a fusion protein containing the J-domain of Hlj1, which exhibits promiscuous and robust activation of every Hsp/c70 examined (33, 34).
Compound synthesis
Compounds MKT-077 and YM-01 were synthesized and characterized as previously described (28, 30). The inactive control compound, YM-18, was synthesized by a similar synthetic method (Figure S1) and its characterization is shown in the Supplemental Information (Figures S2 and S3).
Cell Culture and Immunoblotting
Human neuroblastoma BE(2)M17, HeLa, and HeLa cells stably-transfected with tau (HeLaC3) (35) were maintained in OptiMEM media supplemented with 10% heat inactivated FBS and 1% penicillin/streptomycin antibiotic solution. Primary cortical neuronal cultures were developed from P0 mouse pups and maintained in neurobasal media with B27 supplement. All transfections were performed using Lipofectamine 2000 reagent (Invitrogen) as previously described (36). Transfected cells were harvested using M-PER buffer (Fisher Scientific) supplemented with mammalian protease inhibitor, phosphatase Inhibitor cocktails I & III, and PMSF. Samples were processed for immunoblotting as described earlier (36). Briefly, protein concentration was estimated using BCA assays (Pierce) and 20μg of protein for each sample were loaded in each well of NuPAGE gels (Invitrogen). Gels were transferred onto PVDF membranes, which were blocked for 60 min in 7% milk solution. Primary antibodies were incubated with the membranes overnight at 4°C. Protein signals were evidenced using chemiluminescence reaction (Pierce ECL).
Mice
All animal studies were approved by the University of South Florida’s Institutional Animal Care and Use Committee and abided by that Committee’s Policies on Animal Care and Use in accordance with the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Regulations Title 9 Code of Federal Regulations Subchapter A, “Animal Welfare”, Parts 1–3, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. This USF program and the facilities for animal care and use are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The animals are in standard housing on a 12-hour light dark cycle and have food and water ad lib. The rTg4510 and parental mice were maintained and genotyped as described previously (18).
Slice cultures and electrophysiology
Mice were decapitated and brains were rapidly removed and briefly submerged in ice-cold cutting solution (110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.5 mM CaCl2, 5 mM D-glucose, and 0.6 mM ascorbate). All solutions were saturated with 95% O2 and 5% CO2. Whole brains were dissected on cutting solution-soaked filter paper and mounted on a glass platform resting on ice. Hippocampal slices (400 μm) for electrophysiology and cortical slices for biochemistry were prepared on a vibratome and allowed to equilibrate in a 50% cutting saline and 50% artificial cerebrospinal fluid solution (aCSF; 125 mM NaCl, 2.5 mM KCl, 1.24 mM NaH2PO4, 25 mM NaHCO3, 10 mM D-glucose, 2 mM CaCl2, and 1 mM MgCl2) at room temperature for a minimum of 10 min. For biochemical studies, cortical slices were treated as indicated for ~4 hours unless otherwise noted and harvested for Western blot. Electrophysiology experiments were performed as previously described (20). Slices were transferred to an interface chamber supported by a nylon mesh and allowed to recover for a minimum of 1.5 h prior to recording. Slices were perfused in aCSF at 1 ml/min. Field excitatory post-synaptic potentials (fEPSPs) were obtained from area CA1 stratum radiatum. Stimulation was supplied with a bipolar Teflon-coated platinum electrode and recordings were obtained using a glass microelectrode filled with aCSF (resistance 1–4 mΩ). fEPSPs were generated using a 0.1 msec biphasic pulse delivered every 20 s. After a consistent response to a voltage stimulus was established for 5–10 min, the threshold voltage for evoking a fEPSP was determined and the voltage was then increased incrementally by 0.5 mV until the maximum amplitude of the fEPSP was reached. The data were used to create the I/O curve. A fEPSP baseline response, defined as 50% of the stimulus voltage used to produce the maximum fEPSP amplitude as determined by the I/O curve, was then recorded for 20 min. aCSF containing YM-01 or vehicle was then infused in the system at the same rate of 1 ml/min. Another fEPSP baseline was recorded for 40 min. The brain slices were exposed to the treatment 20 min post-baseline until completion of the experiment (~80 minutes total). The tetanus used to evoke CA1 LTP was a theta-burst stimulation (tbs) protocol, consisting of five trains of four pulse bursts at 200 Hz separated by 200 ms, repeated six times with an inter-train interval of 10 s. Following tbs, fEPSPs evoked by baseline stimulus were recorded for 60 min. Potentiation was measured as the normalized increase of the mean fEPSP descending slope following tbs normalized to the mean fEPSP descending slope for the duration of the baseline recording. Experimental results were obtained from slices that exhibited stable baseline synaptic transmission for a minimum of 20 min before the delivery of the LTP-inducing stimulus. Individual signals that were one standard deviation from the mean were considered outliers and removed from the data set. 2-way ANOVA analysis with Bonferroni post-test was used to determine significance between the slopes of each condition.
Results
To explore whether YM-01 or MKT-077 (Fig. 1A) might be valuable probes for further exploration, we first compared their abilities to inhibit ATP turnover by an Hsp70-Hsp40 system in vitro. In these studies, both MKT-077 and YM-01 inhibited the ATPase activity of Hsp70-Hsp40, with no significant activity against the Hsp70 alone (Fig. 1B). This activity is consistent with the known allosteric mechanism of MKT-077 (29). Thus, in vitro these compounds were largely equivalent. However, MKT-077 localizes to the mitochondria (27, 31), whereas most Hsc70 and tau is cytosolic. Recently, we found that YM-01 has improved cytosolic localization (31). These observations suggested that YM-01 might be better positioned to act through Hsc70 to reduce tau levels. Indeed, in side-by-side comparisons in HeLaC3 cells that stably over-express tau, YM-01 had improved potency compared to MKT-077, reducing tau at sub-micromolar concentrations (EC50 ~ 0.9 μM; ~8-fold better than MKT-077) (Fig. 1C). Based on these results, we selected YM-01 for further testing in cellular and animal tauopathy models.
Figure 1. Synthesis and evaluation of MKT-077 and YM-01.

(A) Chemical structures of MKT-077 and YM-01. Both compounds were synthesized and characterized as previously described (30). (B) MKT-077 and YM-01 (50 μM) reduce the ATPase activity of yeast Hsc70-Hsp40 system (see Materials and Methods for details). Results are the average of triplicates and error bars represent standard deviations. (C) YM-01 reduces total tau levels in HeLaC3 cells at sub-micromolar concentrations. Half maximal values (EC50) were approximately 0.9 μM for YM-01 and 8 μM for MKT-077. Results are representative of experiments performed in at least independent triplicates.
In the Be(2)-M17 neuronal cells and HeLaC3 cells, YM-01 dramatically reduced tau levels by ~75% with EC50 values between 1.5 and 0.9 μM (Fig. 2A and B). In primary hippocampal neurons derived from rTg4510 tau transgenic mice, YM-01 also reduced tau levels by ~65% with an EC50 value of ~6 μM (Fig. 2C). The loss of tau in these cells was rapid, with significant reduction observed after only 15 minutes post-YM-01 (10 μM) addition (Fig. 2D). The rapid reduction of tau prompted us to test compound efficacy in brain slice cultures from rTg4510 tau transgenic mice (20, 22). In this model, YM-01 reduced phospho-tau and total tau levels by more than 70% after 4 hours (Fig. 3A). Moreover, tau reductions were evident in brain slices from rTg4510 mice between 20 and 60 minutes of YM-01 treatment (Fig. 3B).
Figure 2. YM-01 rapidly and potently reduces tau levels in cell models and primary neurons from tau transgenic mice.

(A) Representative Western blot of lysates from M17 cells. Quantifications of endogenous tau levels, normalized to the actin loading control and shown as a percentage of the mock-treated control, are shown. Cells were treated with YM-01 for 24 hours. (B) Representative Western blot of lysates from HeLaC3 cells over-expressing tau. Quantifications of tau levels, normalized to the actin loading control and shown as a percentage of the mock-treated control, are shown. Cells were treated for 24 hours. (C) Representative Western blot of lysates from primary neurons derived from tau transgenic mice treated with the indicated concentrations of YM-01 for 24 hours. (D) Representative Western blot of M17 neuronal cells treated with YM-01 (10μM) for 0, 15 or 60 minutes. Tau levels were reduced within 15 minutes.
Figure 3. YM-01 rapidly and potently reduces tau levels in ex vivo slice cultures from tau transgenic mice and nocodazole-treated wildtype mice.

(A and B) Representative Western blots and quantification graph of ex vivo slice cultures from rTg4510 tau transgenic brains treated with indicated concentrations of YM-01 for 4 hours (A) or with 30μM YM-01 for the indicated times (B). Bands correspond to pS396/S404 (A) and total tau bands (A and B). (C) Tau levels as measured by Western blot in brain slices from non-transgenic mice treated with nocodazole (50 μM) for 30 minutes followed by YM-01 for 4 hours. Quantification of tau levels is shown as a percentage of vehicle-treated slices ± standard deviation (SD) after actin normalization.
Any therapy for tauopathies should ideally demonstrate selective reduction of pathogenic tau, with less impact on functional, cycling tau. To understand whether YM-01 affected normal tau, we tested its activity in slice cultures from wildtype mice. In this model, YM-01 had a more mild effect, reducing tau levels by 40% at very high concentrations (100 μM; Fig. 3C). One way to mimic tau pathology is to treat with microtubule destabilizers, such as nocodazole, which artificially increase the pool of free tau and enhance Hsc70 recognition (37). Combining YM-01 with nocodazole caused the compound to trigger tau loss in the wildtype slice cultures (Fig. 3C). These results suggest that a major function of YM-01 is to promote the normal triage functions of Hsc70, focusing the chaperone on potentially proteotoxic, free tau.
We then explored the mechanism of tau clearance by YM-01. The proteasome inhibitor epoximicin (Epox), blocked YM-01-mediated loss of tau in HeLaC3 cells (Fig. 4A) and hippocampal neurons from transgenic mice (Fig. 4B), suggesting that YM-01 directs Hsc70 to shuttle tau to the proteasome for degradation. Next, we explored the effects of Hsp40 (DnaJB2) on the activity of YM-01. Hsp40 co-chaperones stimulate the ATPase activity of Hsp70s (38) and, in vitro, we had found that YM-01 inhibited ATP turnover by the Hsp70-Hsp40 complex, but not free Hsp70 (see Fig. 1B). These findings suggest that the compound acts on the Hsc70-Hsp40 complex and that over-expression of Hsp40 might partially suppress the activity of YM-01. Indeed, over-expression of Flag-DnaJB2 in cells over-expressing tau for 48 hours suppressed YM-01 activity at 3 μM (Fig. 5). However, over-expression of another Hsp40 variant, DnaJB1, significantly reduced tau levels by itself and did suppress YM-01 activity (Fig. 5). Together, these results suggest that YM-01 acts through the Hsp70-Hsp40 system to promote proteasomal degradation of tau, but the specific Hsp40 component might tune this activity.
Figure 4. YM-01 reduces tau levels by a mechanism dependent on Hsc70 and the proteasome.
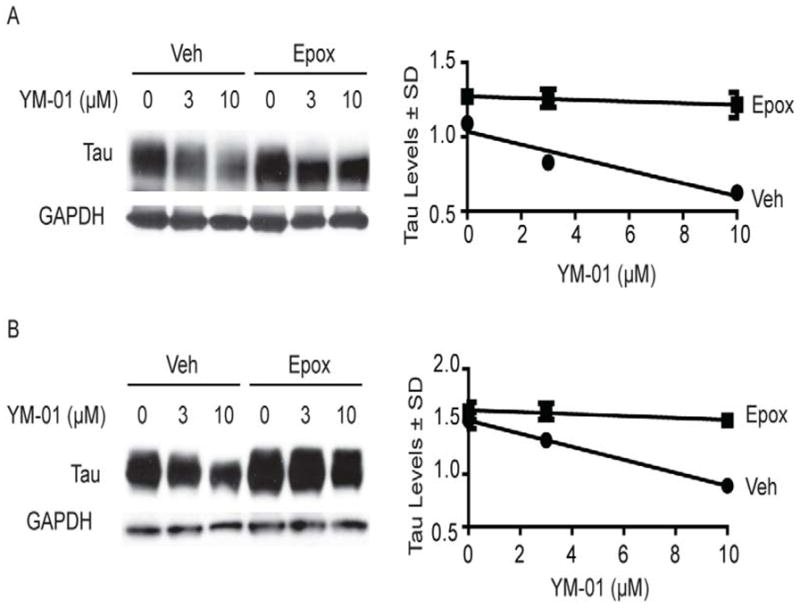
(A) Representative Western blots from HeLa cells over-expressing tau, treated with YM-01 and the proteasome inhibitor epoxomycin (Epox) (400 nM) for 4 hrs. Results are the average of triplicates and the error bars represent standard deviations. (B) Representative Western blots of neuronal M17 cells treated with YM-01. Quantification of tau levels is shown as a percentage of vehicle (0) treated cells ± standard deviation after GAPDH normalization.
Figure 5. Hsp40 (DnaJB2) suppresses YM-01 efficacy in a cell model of tau accumulation.
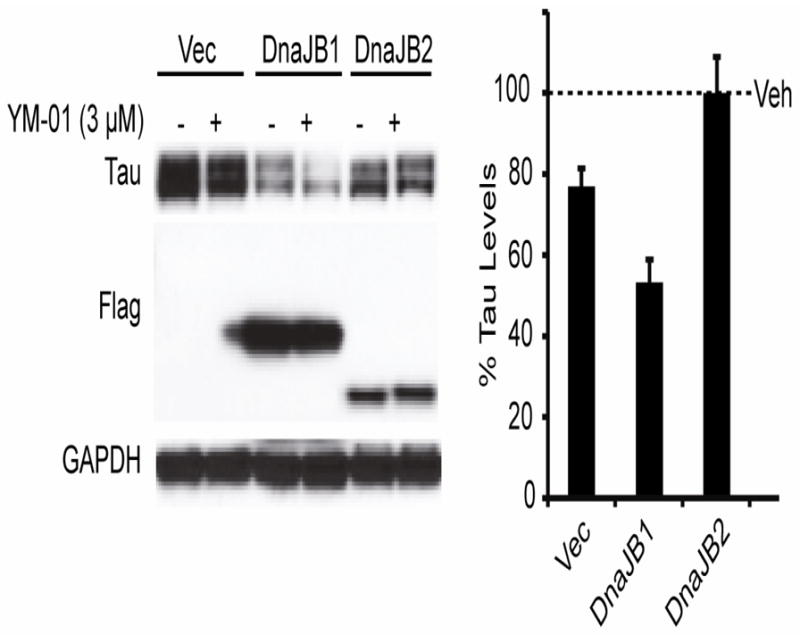
Representative M17 neuronal cells were transfected with DnaJB1 or DnaJB2 for 48 hours and then treated with 3 μM YM1 for 1 hour. Vec indicates empty vector transfection. Quantification shows the values for tau levels as a percent of each vehicle (Veh) control after GAPDH normalization ± standard deviation. DnaJB1 significantly reduced tau by 55% (p<0.0005).
An important goal of this study is to better understand the suitability of Hsc70 as a potential drug target for tauopathies. Therefore, we wished to evaluate whether YM-01 could improve neuronal function in a tau mouse model (20). Toward that goal, we evaluated the effects of YM-01 on long-term potentiation (LTP) in the rTg4510 mice. These mice develop robust LTP deficits by as early as three months of age (20). Acute hippocampal slices from 3.5- month-old rTg4510 and non-transgenic littermate control mice (Non) were treated with YM-01 (30 μM) for 20 minutes prior to theta burst stimulation and then maintained in YM-01 or vehicle solution for an additional hour. YM-01-treated slices from rTg4510 mice showed an improvement in LTP deficits compared to vehicle-treated rTg4510 mice (Fig. 6A). No change in potentiation was observed in wildtype slices following YM-01 treatment (Fig. 6B). Conversely, slices from rTg4510 mice treated with an inactive control compound, YM-18, had no effect (see Fig. S3B in the Supplement). These data strongly suggest that LTP rescue by YM-01 was caused by acute tau reduction. These results suggest that reducing pathogenic tau intermediates by promoting Hsc70-mediated triage may be a viable strategy to improve neuronal function in tauopathies.
Figure 6. LTP deficits in rTg4510 mice are rescued with YM-01 treatment.
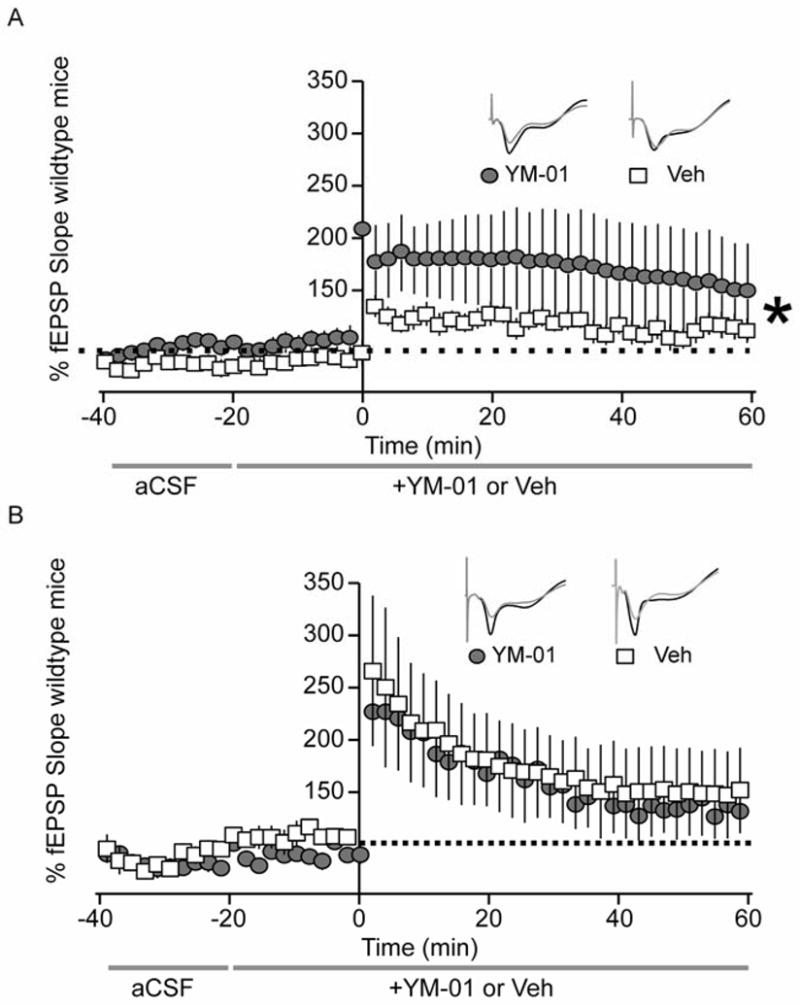
Graphical representation of electrophysiology experiments in hippocampal slices of 3.5 month old (A) rTg4510 and (B) non-transgenic wildtype mice. After recording baseline signal for 20 min, hippocampal slices were perfused with 30μm YM-01 or vehicle control in aCSF continuously for the remainder of the experiment. Baseline signal was recorded for 20 min, LTP was induced with TBS (5 bursts of 200 Hz separated by 200 ms, repeated 6 times with 10 s between the 6 trains), and LTP was recoded for 60 min. Changes in fEPSP slope are expressed as a percentage of baselines. (A) Gray circles and white boxes represent fEPSP traces of YM-01- (n=9) or vehicle-treated (n=16) hippocampal slices from 3.5 month old tau transgenic mice. (B) Gray circles and white boxes correspond to fEPSP traces of YM-01- (n=4) or vehicle-treated (n=7) hippocampal slices from 3.5 month old non-transgenic mice. Statistical analysis was performed using a 2-way ANOVA with Bonferroni post-test. Tau transgenic hippocampi treated with YM-01 displayed a significantly different LTP curve compared to vehicle-treated transgenic controls (***p=0.00097). LTP slopes for non-transgenic hippocampi were not significantly different (p=0.98). Gray traces represent baseline recordings, while black traces correspond to representative LTP recordings of each condition.
Discussion
Reducing tau levels as a therapeutic strategy is not without controversy. While several studies suggest that reducing tau could be advantageous, others show that a complete loss of tau can have toxic consequences. Tau knockout mice in certain genetic backgrounds seem quite normal (39), and restored memory function was evident when they were crossed with amyloid-based AD mouse models (8). However, humans with chromosomal deletions encompassing the MAPT gene exhibit severe mental retardation (40). Loss of tau has also been associated with iron retention, potentially contributing to toxic neuronal iron deposits in tauopathies (41) and axonal degeneration (42). These dichotic results have led most drug discovery efforts to focus on the abnormal tau species, such as phosphorylated or aggregated forms of the protein (43–48).
The Hsp70 chaperone system seems intrinsically capable of discriminating between functional, microtubule-bound tau and abnormal, free tau (37). All Hsp70 variants are able to recognize free tau because the binding site is located within the microtubule binding domain (23, 49). Therefore when tau is engaged with microtubules, it is inaccessible to Hsp70 proteins. Moreover, phosphorylation sites that alter the association of tau with microtubules have also been shown to influence its interaction with chaperones (21, 37). While this suggests a common mechanism for binding to tau, distinct Hsp70 variants control whether tau is triaged for degradation or preserved in the cell despite their structural similarities. For example, the inducible variant of the Hsp70 family, termed Hsp72, has high binding affinity for tau and promotes its association with pro-degradation co-factors, yet it is expressed at very low levels in the brain (23). Conversely, the main constitutively expressed variant of the Hsp70 family, termed Hsc70, is highly expressed in the brain and preserves tau despite its reduced affinity for tau. This suggests that Hsc70 is a major contributor to tau accumulation and possibly its pathogenesis. Whether tau is degraded or preserved by this Hsc70 complex depends on its association with specific co-chaperones, such as TPR-containing proteins (i.e. CHIP, FKBP51, etc) and distinct members of the DnaJ family (36, 50) (51). These co-chaperones may be the key for successful drug design targeting the Hsc70 complex, as suggested by our results with YM-01.
YM-01, despite increasing the affinity of Hsc70 for tau by stabilizing the ADP-bound conformation (52), enables Hsc70 to promote tau degradation. Thus, Hsc70 is able to locate and recognize abnormal tau, but is for some reason unable to process tau for degradation on its own. YM-01 seems to simply provide guidance to Hsc70, in the form of changing its co-chaperone repertoire or its structure in some way. This would not be the first time that a chaperone was incapable of properly triaging a disease-associated client protein. Aha1 mistakenly triages the mutant cystic fibrosis transmembrane receptor (CFTR) towards degradation despite its ability to still sufficiently function (53). Grp94 mistakenly interacts with mutant myocilin, a protein that causes glaucoma, allowing it to accumulate and form amyloids (51). In this way, chaperones like Hsc70 may be incapable of properly triaging a subset of abnormal clients, contributing to the manifestation of disease.
Our data also indicate that YM-01 will be a useful research tool to study the role of Hsc70 in tau homeostasis and quality control. In addition, this molecule serves as a lead for further preclinical development. Deployment of this idea will require optimization of its potency and pharmacokinetic properties, as well as overcoming the challenge of blood-brain barrier (BBB) permeability and the renal toxicity that have hindered development of MKT-077 (27, 54, 55). Another difficulty is that rhodacyanines represent a significant obstacle for medicinal chemistry efforts, given their functional group density and light sensitivity (56–59). In addition, due to the promiscuous nature of Hsp70 family members, there are concerns about the potential toxicity of an approach that targets these proteins. Despite these challenges, YM-01 rapidly and potently depleted Akt levels in breast cancer lines, inducing cytotoxicity; however Akt levels were unaffected by YM-01 in immortalized control lines and no toxicity was evident (31). YM-01 can also selectively target poly–glutamine expanded androgen receptor, preserving neuronal function (30). Therefore, it appears that targeting Hsp70 proteins may be similar to Hsp90 inhibitors, in that they can selectively target only aberrant proteins in diseased tissues (21, 60).
The current findings define an intracellular MoA that is intriguing. The allosteric binding site of MKT-077 is known (29) and it seems likely that additional scaffolds targeting this region may share the ability of YM-01 to promote triage functions associated with Hsc70. Cell- or tissue-based assays of tau levels may be a particularly appropriate platform to screen and identify these molecules. Together, these studies clearly identify Hsc70 as a promising drug target in tauopathies. Moreover, they provide an interesting, new MoA that may exploit natural tendencies in the chaperone-mediated quality control system to selectively reduce pathogenic tau levels. Allosteric molecules, such as YM-01, might promote the right “decision” and restore normal tau homeostasis.
Supplementary Material
Acknowledgments
We would like to thank Dr. Peter Davies for providing the PHF1 antibody. CAD was supported by grants from NIH/NIA (R00AG031291), NIH/NINDS (R01NS073899), the Alzheimer’s Association and CurePSP. JEG was supported by NIH/NINDS (NS059690) and the American Health Assistance Foundation (AHAF). JLB acknowledges NIH grants GM75061 and DK79307 (University of Pittsburgh Kidney Research Center). ZTY was supported by pre-doctoral fellowships from GM007767 and the Rackham Merit Program. JFA was supported by CurePSP and the Alzheimer’s Association.
Footnotes
Financial disclosures
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- 2.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 3.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 4.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nature genetics. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet. 1998;7:1825–1829. doi: 10.1093/hmg/7.11.1825. [DOI] [PubMed] [Google Scholar]
- 7.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 9.Hoshino S, Tamaoka A, Takahashi M, Kobayashi S, Furukawa T, Oaki Y, et al. Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport. 1998;9:1879–1883. doi: 10.1097/00001756-199806010-00039. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 11.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 13.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ardiles AO, Tapia-Rojas CC, Mandal M, Alexandre F, Kirkwood A, Inestrosa NC, et al. Postsynaptic dysfunction is associated with spatial and object recognition memory loss in a natural model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13835–13840. doi: 10.1073/pnas.1201209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickey CA, Loring JF, Eastman PS, Montgomery JR, Gordon M, Morgan DG. Selectively reduced expression of synaptic plasticity-related genes in APP+PS1 transgenic mice. JNeurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 17.O’Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abisambra JF, Blair LJ, Hill SE, Jones J, Kraft C, Rogers J, et al. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. Journal of Neuroscience. 2010 doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinwal UK, Akoury E, Abisambra JF, O’Leary JC, Thompson AD, Blair LJ, et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB Journal. 2013 doi: 10.1096/fj.12-220889. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AD, Scaglione KM, Prensner J, Gillies AT, Chinnaiyan A, Paulson HL, et al. Analysis of the Tau-Associated Proteome Reveals That Exchange of Hsp70 for Hsp90 Is Involved in Tau Degradation. ACS Chem Biol. 2012 doi: 10.1021/cb3002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, et al. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chemistry & biology. 2012;19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8:609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–6821. [PubMed] [Google Scholar]
- 28.Deocaris CC, Widodo N, Shrestha BG, Kaur K, Ohtaka M, Yamasaki K, et al. Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer letters. 2007;252:259–269. doi: 10.1016/j.canlet.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 29.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric Drugs: The Interaction of Antitumor Compound MKT-077 with Human Hsp70 Chaperones. Journal of molecular biology. 2011;411:614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang AM, Miyata Y, Klinedinst S, Peng H-M, Chua JC, Li X, Pratt WB, Osawa Y, Collins CA, Gestwicki JE. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nature Chemical Biology. 2013 doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koren IIIJ, Miyata Y, Kiray J, O’Leary JC, Nguyen L, Guo J, et al. Rhodacyanine Derivative Selectively Targets Cancer Cells and Overcomes Tamoxifen Resistance. PloS one. 2012 doi: 10.1371/journal.pone.0035566. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, et al. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–51140. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- 33.Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Molecular biology of the cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang AN, Valderramos JC, Balachandran R, Chovatiya RJ, Mead BP, Schneider C, et al. Select pyrimidinones inhibit the propagation of the malarial parasite, Plasmodium falciparum. Bioorg Med Chem. 2009;17:1527–1533. doi: 10.1016/j.bmc.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jinwal UK, Trotter JH, Abisambra JF, Koren J, 3rd, Lawson LY, Vestal GD, et al. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J Biol Chem. 2011;286:16976–16983. doi: 10.1074/jbc.M110.182493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jinwal UK, Koren J, 3rd, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, et al. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:591–599. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jinwal UK, O’Leary JC, 3rd, Borysov SI, Jones JR, Li Q, Koren J, 3rd, et al. Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem. 2010;285:16798–16805. doi: 10.1074/jbc.M110.113753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nature neuroscience. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 40.Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 41.Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 42.Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, et al. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballatore C, Brunden KR, Piscitelli F, James MJ, Crowe A, Yao Y, et al. Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of tau aggregation. Journal of medicinal chemistry. 2010;53:3739–3747. doi: 10.1021/jm100138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wischik C, Staff R. Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J Nutr Health Aging. 2009;13:367–369. doi: 10.1007/s12603-009-0046-5. [DOI] [PubMed] [Google Scholar]
- 45.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 46.Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 47.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 49.Sarkar M, Kuret J, Lee G. Two motifs within the tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J Neurosci Res. 2008 doi: 10.1002/jnr.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abisambra JF, Jinwal UK, Suntharalingam A, Arulselvam K, Brady S, Cockman M, et al. DnaJA1 Antagonizes Constitutive Hsp70-Mediated Stabilization of Tau. J Mol Biol. 2012 doi: 10.1016/j.jmb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szabo A, Langer T, Schroder H, Flanagan J, Bukau B, Hartl FU. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10345–10349. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 54.Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, et al. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–543. [PubMed] [Google Scholar]
- 55.Propper DJ, Braybrooke JP, Taylor DJ, Lodi R, Styles P, Cramer JA, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol. 1999;10:923–927. doi: 10.1023/a:1008336904585. [DOI] [PubMed] [Google Scholar]
- 56.Kawakami M, Koya K, Ukai T, Tatsuta N, Ikegawa A, Ogawa K, et al. Synthesis and evaluation of novel rhodacyanine dyes that exhibit antitumor activity. Journal of medicinal chemistry. 1997;40:3151–3160. doi: 10.1021/jm9702692. [DOI] [PubMed] [Google Scholar]
- 57.Delaey E, van Laar F, De Vos D, Kamuhabwa A, Jacobs P, de Witte P. A comparative study of the photosensitizing characteristics of some cyanine dyes. J Photochem Photobiol B. 2000;55:27–36. doi: 10.1016/s1011-1344(00)00021-x. [DOI] [PubMed] [Google Scholar]
- 58.Brooker LG. Chemistry of the cyanine dyes. Annals of the New York Academy of Sciences. 1948;50:108. doi: 10.1111/j.1749-6632.1948.tb39843.x. [DOI] [PubMed] [Google Scholar]
- 59.Carney RW, Wojtkunski J, Konopka EA, de Stevens G. The chemical, spectral, and biological properties of monomethine cyanine dyes containing 1,3-benzoxazine and quinazoline nuclei. Journal of medicinal chemistry. 1966;9:758–762. doi: 10.1021/jm00323a027. [DOI] [PubMed] [Google Scholar]
- 60.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.