

Abstract
Background
Smooth muscle has the distinctive ability to maintain force for long periods of time and at low energy costs. While it is generally agreed that this property, called the latch-state, is due to the dephosphorylation of myosin while attached to actin, dephosphorylated-detached myosin can also attach to actin and may contribute to force maintenance. Thus, we investigated the role of calponin in regulating and enhancing the binding force of unphosphorylated tonic muscle myosin to actin.
Methods
To measure the effect of calponin on the binding of unphosphorylated myosin to actin, we used the laser trap assay to quantify the average force of unbinding (Funb) in the absence and presence of calponin or phosphorylated calponin.
Results
Funb from F-actin alone (0.12±0.01pN; mean±SE) was significantly increased in the presence of calponin (0.20±0.02pN). This enhancement was lost when calponin was phosphorylated (0.12±0.01pN). To further verify that this enhancement of Funb was due to cross-linking of actin to myosin by calponin, we repeated the measurements at high ionic strength. Indeed, the Funb obtained at a [KCl] of 25mM (0.21±0.02pN; mean±SE) was significantly decreased at a [KCl] of 150mM, (0.13±0.01pN).
Conclusions
This study provides direct molecular level-evidence that calponin enhances the binding force of unphosphorylated myosin to actin by cross-linking them and that this is reversed upon calponin phosphorylation. Thus, calponin might play an important role in the latch-state.
General Significance
This study suggests a new mechanism that likely contributes to the latch-state, a fundamental and important property of smooth muscle that remains unresolved.
Keywords: latch-state, actin regulatory proteins, in vitro motility assay, laser trap
1. INTRODUCTION
Tonic smooth muscle is well known to maintain force for long periods of time at low energy levels. It is generally agreed that this property of smooth muscle, called the latch-state, is due to the dephosphorylation of myosin molecules while attached to actin filaments [1]. However, evidence is accumulating to suggest that calponin, a 35 kDa [2] actin binding protein [3, 4] may also play a role in smooth muscle force maintenance, presumably through its effects on unphosphorylated myosin [5–7].
Calponin has an inhibitory effect on the ATPase activity of smooth muscle myosin [8] and on the velocity (νmax) of actin propulsion in the in vitro motility assay [3, 9]. However, contrary to caldesmon, the inhibitory effect of calponin is more of an all-or-none mechanism [10], i.e. the filaments are either moving or they are stopped. At the cellular level, it has also been shown that calponin is necessary to inhibit the slow cycling of unphosphorylated myosin, stopping the shortening and force production of resting smooth muscle [5]. The inhibitory action of calponin on the actomyosin ATPase rate is due to its binding to actin which can be suppressed by calponin phosphorylation [8]. However, calponin also binds to unphosphorylated myosin [7] so it presumably cross-links unphosphorylated myosin to actin. This interaction between calponin and unphosphorylated myosin is dependent on ionic strength and it gets weaker at high [NaCl] [7].
We previously demonstrated, at the single molecule level, that unphosphorylated myosin purified from both tonic and phasic smooth muscle can bind to unregulated actin filaments with a binding force of approximately 1/10th of the force generated by phosphorylated myosin [11]. Unphosphorylated, and presumably dephosphorylated detached myosin, could therefore participate in force maintenance during the latch state. The actin regulatory proteins could possibly potentiate this binding force. Thus, in the current study, we investigated the role of calponin in the enhancement of this binding force as well as its regulation by phosphorylation.
2. MATERIALS AND METHODS
2.1 Proteins
Myosin was purified from pig stomach fundus following a previously published protocol [12]. For the protocols requiring myosin activation, myosin was thiophosphorylated [13]. Actin was purified from chicken pectoralis acetone powder [14] and fluorescently labeled by incubation with tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (P1951, Sigma-Aldrich Canada) [15]. Turkey gizzard calponin was purified as a by-product of caldesmon purification [16]. For the protocols requiring calponin activation, calponin was phosphorylated using Ca2+/calmodulin-dependent protein kinase II and MgATP (1 mol of Pi/mol of calponin) [8]. After 10 min, Ca2+ chelation was performed by adding 10mM EGTA to avoid contamination of the next assays with Ca2+.
2.2 Buffers
Myosin buffer (300 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 30 mM DTT; pH adjusted to 7.4); Actin buffer (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 30 mM DTT, with an oxygen scavenger system consisting of 0.25 mg/ml glucose oxidase, 0.045 mg/ml catalase, and 5.75 mg/ml glucose; pH adjusted to 7.4); Assay buffers: The in vitro motility assay buffer consisted of actin buffer to which methylcellulose (0.5%) was added, to favor binding of myosin to actin, and MgATP (2 mM). The laser trap assay buffer consisted of actin buffer to which methylcelullose (0.3%) and MgATP (200 μM) were added.
2.3 In vitro motility assay
The velocity (νmax) of actin filament propulsion by myosin was measured in the in vitro motility assay as previously described [11] with minor changes. Briefly, a flow-through chamber (20 μl) was constructed from a nitrocellulose-coated coverslip and a glass microscope slide [15]. Non-functional myosin molecules were removed by ultracentrifugation (Optima ultracentrifuge L-90K and 42.2 Ti rotor, Beckman Coulter, Fullerton, CA) of myosin (500 μg/ml) with equimolar filamentous actin and 1 mM MgATP in myosin buffer. Myosin was then perfused in the flow through chamber at a concentration of 125 μg/ml and allowed to randomly attach to the nitrocellulose for 2 min. The following solutions were then perfused sequentially in the flow through chamber (all in actin buffer): BSA (0.5 mg/ml), unlabeled G-actin (1.33 μM) to bind to any remaining non-functional myosin followed by MgATP (1 mM) to remove the unlabeled actin from the functional heads. Then two washes of actin buffer were followed by TRITC labeled actin (0.03 μM), with or without calponin (0.3μM), incubated for 1 min, and finally, motility buffer. All molecular mechanics measurements were performed at 30°C. Motility was then assessed using an inverted microscope (IX70, Olympus, Melville, NY) equipped with a high numerical aperture objective (X100 magnification Ach 1.25 numerical aperture, Olympus, Melville, NY) and rhodamine epifluorescence. An image intensified video camera (KP-E500 CCD Camera, Hitachi Kokusai Electric, Woodbury, NY, 720 × 480 resolution, 68.6 μm × 45.7 μm real frame size, 29.94 frame/s, 8 bit grayscale) was used to visualize and record the actin filament movement on computer (Custom Built by Norbec Communication, Montreal, QC) using a frame grabber (Pinnacle Studio AV/DV V.9 PCI Card) and image capturing software (AMCap software V9.20) at 29.94 Hz. νmax was determined from the total path described by the filaments divided by the elapsed time using our automated version of the National Institutes of Health tracking software (NIH macro in Scion Image 4.02, Scion) coded in Matlab (R2009b). Only filaments present for at least 20% of the recorded video time (50 s) and describing a path of at least 3μm were considered. To calculate the percentage of stopped filaments, a threshold of 0.1 μm/s was set below which, the filaments were considered immobile and moving only due to Brownian motion. This threshold was estimated by analyzing the frame-to-frame velocity of actin filaments in the absence of ATP [17]. Finally, filaments that were not moving continuously were eliminated from this analysis [10].
2.4 Laser trap assay
Our single beam laser trap assay was built around the Laser Tweezers Workstation (Cell Robotics, Albuquerque, NM) and the motility assay described above and as previously reported [11]. Briefly, pedestals were created by spraying 4.5 μm polystyrene microspheres (Polybead, Polysciences, Warrington, PA) on the coverslips before coating with nitrocellulose. 3 μm polystyrene microspheres (Polybead, Poly-sciences, Warrington, PA) coated by 30 min incubation at room temperature with N-ethylmaleimide-modified (NEM) skeletal myosin [15] were used for trapping. The perfusion of proteins and solutions in the flow-through chamber followed the same sequence as for the motility assay except that the myosin was unphosphorylated and at a concentration of 16.7μg/ml, TRITC-labeled actin was mixed with microspheres (13×103 microspheres/μl) in laser trap assay buffer, and there were no unlabeled G-actin and MgATP steps. A diode pumped Nd:YAG solid-state laser (TEM00, 1.5 W, 1064nm) was used to create the trap. To perform the assay, a microsphere visualized in bright field by a charge coupled device (CCD) camera (XC-75, Sony Corporation of America, New York, NY) was captured in the laser trap, and its position was recorded on computer as described above. An actin filament, visualized by fluorescence imaging (described above for the motility assay) was attached to the microsphere and brought in contact with unphosphorylated myosin molecules randomly adhered to a pedestal (Fig. 1A). Contact between myosin and actin was allowed for approximately 10s. During that time, the microsphere baseline position in the trap was recorded. The pedestal was then moved away from the trap at a slow and constant velocity of 0.5 μm/s. The microsphere initially followed the pedestal (Fig.1B) until the force exerted on it by the trap became greater than that exerted by the myosin molecules on the actin filament. At this point, the microsphere sprang back to its unloaded baseline position in the center of the trap (Fig. 1C). The total unbinding force (Total Funb) of the myosin molecules was calculated as follows:
| (1) |
where k is the trap stiffness and Δd is the maximal displacement of the trapped microsphere from its baseline position. k was calibrated using the Stokes force (Ff) approach, as previously reported [11]. Briefly, a viscous drag was applied to a trapped microsphere by moving it at a constant velocity (v) in 0.3% methylcellulose while measurements of Δd were performed. According to Stokes’ law, the frictional force exerted on spherical objects with very small Reynolds numbers is calculated as follows:
| (2) |
where η is the dynamic viscosity and r is the microsphere radius. The viscosity of 0.3% methylcellulose was measured with a viscometer (DV-I at 60 rpm, Brookfield, Middleboro, MA), using a UL (ultra low viscosity) adapter and was equal to 10.4 cP, at 30°C. Thus,
Figure 1.

A single beam laser trap assay is used to capture a polystyrene bead to which is attached a TRITC-labeled actin filament regulated by calponin. A: The regulated actin is brought in contact with unphosphorylated myosin that randomly coats a pedestal on a coverslip. ℓ: length of actin in contact with myosin. B: The pedestal/coverslip is then moved away from the laser trap at a constant and slow velocity, thereby dragging the trapped bead away from the trap center as a result of the attachment of unphosphorylated myosin to actin. C: When the force exerted by the trap on the bead is greater than the force of binding of unphosphorylated myosin to actin, the bead snaps back into the trap, its unloaded position. The unbinding force (Funb) is then calculated as the maximal distance between the bead and the trap center (max Δd) multiplied by the trap stiffness calculated by the Stokes force method. (See section 2.4 for details).
| (3) |
The value of k (0.013 pN/nm, R2=0.95) was averaged from several measurements performed at different velocities and then used to perform the force measurements (see example of trap calibration in figure 3 from [11]).
Figure 3.

Velocity of actin filament (νmax) when propelled by phosphorylated myosin as measured in the in vitro motility assay. The measurements were performed with naked actin (N=4) and in the presence of calponin (N=6) and phosphorylated calponin (N=5). Cp: calponin, phos Cp: phosphorylated calponin, *: p<0.05. Inset: Western blot analysis of calponin and phosphorylated calponin as assessed by probing with an antibody that recognizes top panel: calponin, and bottom panel: phosphorylated calponin.
The average binding force per myosin head (Funb) was obtained as follows:, First, we measured the length of actin filament in contact with the pedestal (ℓ, Fig.1A) by fluorescence imaging. That is, the portion of the pedestal where the actin filament was bound, was brought in focus and the bound actin filament length (ℓ) was measured using the National Institutes of Health analysis software (NIH macro in Scion Image 4.02, Scion). Unbound actin was readily detected because it moved in and out of focus due to Brownian motion, so it was discarded from the length measurements. We then used the estimates of the number of active myosin heads on the motility surface previously obtained by Warshaw and co-workers [18, 19] by performing NH4-EDTA ATPase assays directly on the cover slip. (Note that similar results have also been obtained by two other groups [20, 21]). The density of active myosin heads was obtained by dividing this number by the surface area. The number of active myosin head per actin filament length was calculated by assuming that all myosins could interact with actin within a 26 nm wide band [18, 20, 21]. For example, at a concentration of myosin of 20 μg/ml, a value of 26 heads/μm of actin filament was estimated [18].
2.5 Microsphere displacement analysis software
Bead motion tracking was performed in Matlab using optimal fitting of a reference image. In short, in the first frame of the acquired video (720×480 resolution, 68.6×40.3 μm real frame size, 29.94 frame/s, 8 bit grayscale), the largest area of connected area pixels with a gray value above 90% of the highest pixel value in the image was identified (Figure 2A). A 60×60 pixel section of the frame, centered on the center of mass of the area of connected pixels, was defined as the reference image (Figure 2B). While this centre of mass was not necessarily the centre of mass of the bead, it was visually confirmed that the entire bead was always fully contained in the reference image. In each subsequent frame the reference image was matched to a section of the frame of the same size by finding the location at which the summed absolute difference in pixel grey values (δz) between the current frame and the reference image was minimized (sample of difference image in Figure 2C). To achieve sub pixel resolution of the bead position, the δz values at the found location and the surrounding 8 pixels were interpolated with a cubic interpolation algorithm (Matlab, Figure 2D) and the coordinates of the global minimum point on this interpolation surface was stored as the position of the bead. After analysis of the entire video a frame range where no movement occurs was manually chosen to calculate a baseline location of the bead and distances were plotted relative to this baseline location (Figure 2E). The relevant local maxima, which correspond to the detachment of myosin from actin, were found by searching for maxima in user defined regions. From these maxima the associated total Funb were calculated.
Figure 2.
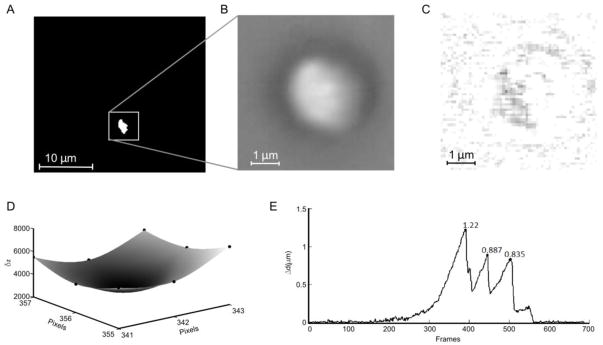
Video analysis for the calculation of microsphere displacement (Δd) (see section 2.5 for detailed discription). A) A treshold map of the first video frame. The white box indicates the reference region. B) The reference image as found from A. C) Example of the absolute difference image in optimal location (differences amplified 10×, white equals no difference). D) The summed pixel value differences (δz) in 9 points centered around the found optimal location were mapped (black dots) and interpolated with a cubic interpolation algorithm to find subpixel resolution of the coordinates of the bead (x and y are coordinates, δz equals the summed pixel value difference). E) The resulting displacement plot with marked local maxima (which correspond to the detachment of myosin from actin). In this sample the baseline position was calculated as the mean position over the first 100 frames.
2.6 Western Blot analysis of purified calponin
The phosphorylation of calponin was assessed by Western blot analysis. Proteins were separated by SDS-PAGE using a 4–15% ready gradient gel (Bio-Rad, Hercules, CA). Protein concentration was estimated by a standard Bradford assay, and 1.7 μg of calponin and phosphorylated calponin were loaded in each well. Proteins were transferred electrophoretically onto PVDF membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat milk and probed with the monoclonal Ab65827 antibody that recognizes calponin (Abcam Inc, Cambridge, MA) or with the rabbit polyclonal SC16717R antibody that recognizes phosphorylated calponin (Santa Cruz Inc., Santa Cruz, CA). Antibody detection was done by Super Signal West Dura substrate (ThermoFisher, Waltham, MA).
2.7 Co-sedimentation assay
To confirm the effect of unphosphorylated calponin on the cross-linking of unphosphorylated myosin to actin, a co-sedimentation assay was performed. The proteins and calponin phosphorylation protocol were described in section 2.1. Combinations of actin (6 μM), myosin (1 μM) and calponin (2 μM) (phos- or unphosphorylated) were incubated in actin buffer (see section 2.2) for 1 h at 26ºC with continuous mixing at 400 rpm. The samples were than centrifuged at low speed (13,300 rpm) for 20 min, at 4ºC in order to sediment the myosin and cross-linked actin and calponin. The supernatants and re-suspended pellets were then separated by SDS-PAGE using a 4–20% ready gradient gel (Bio-Rad, Hercules, CA) and visualized by Coomassie blue.
2.8 Statistical analysis
Differences in νmax and Funb between multiple conditions were tested using one way ANOVAs, with a Bonferroni correction. Differences in νmax and Funb with only two conditions were tested using the Student’s t-test. In cases when the test for equal variance failed, a Mann-Whitney U test was performed. A value of P<0.05 was considered significant. The Systat Software Inc., (San Jose, CA) was used. For the motility assay, N represents the number of flow-through chambers studied. A minimum of three locations in each flow through chambers were analyzed; each location contained at least 10 filaments. Thus, a minimum of 30 filaments were analyzed per chamber to make up an N of 1. For the laser trap assay, N represents the number of actin filaments analyzed.
3. RESULTS
3.1 Maximal velocity of actin propulsion
To ascertain that our actin filaments are regulated by physiological levels of calponin, we measured νmax when propelled by pig stomach fundus myosin in the in vitro motility assay. As expected from the literature [9, 22], νmax for actin (0.54±0.01 μm/s; mean±SE) was slightly but statistically significantly decreased in the presence of calponin (0.43±0.01 μm/s; p<0.001,) and this inhibitory effect was suppressed when calponin was phosphorylated (0.50±0.01 μm/s; p=0.04), as shown in Fig. 3. The percentage of stopped filaments was also increased from 12.7 % for regular actin to 28.9 % in the presence of calponin and back to 12.5% in the presence of phosphorylated calponin. The phosphorylation of calponin was assessed by Western blot analysis as shown in the inset of Fig. 3.
3. 2 Cross-linking of unphosphorylated myosin to actin
To verify if calponin alters the average binding force of unphosphorylated myosin to actin filaments, we measured Funb using the laser trap assay. Funb in presence of actin only (0.12± 0.01 pN; mean±SE) was increased in the presence of calponin (0.20± 0.02 pN; p=0.009). To investigate if this enhancement was due to the binding of calponin to actin, we repeated the Funb measurements in the presence of phosphorylated calponin, i.e. detaching calponin from actin [8]. Indeed, this enhancement was lost when calponin was phosphorylated (0.12± 0.01 pN; p=0.006), as shown in Fig.4A.
Figure 4.




Figure 4A. Unbinding force (Funb) of unphosphorylated myosin to actin as measured with the single beam laser trap assay. The measurements were performed with naked actin (N=10) and in the presence of calponin (N=9) and phosphorylated calponin (N=10). Funb is reported as the average force per myosin molecule (see section 2.4 for details). *: p<0.05.
Figure 4B. Unbinding force (Funb) at regular (25mM) and at high KCl (150mM) of unphosphorylated myosin to calponin regulated actin. Funb is reported as the average force per myosin molecule (see section 2.4 for details). N=11 and 7, respectively. *: p<0.05.
Figure 4C. Unbinding force (Funb) of unphosphorylated myosin to actin, in the absence of calponin, at regular (25mM) and at high KCl (150mM). Funb is reported as the average force per myosin molecule (see section 2.4 for details). N= 17 and 5, respectively.
Figure 4D. Total unbinding force normalized per actin length (Total Funb/ℓ) in the absence (N=12) or presence (N=12) of unphosphorylated myosin but in the presence of calponin regulated actin.*: p<0.05.
To confirm that calponin enhances Funb by cross-linking unphosphorylated myosin to actin, we repeated the unbinding experiments at high ionic strength because the calponin-myosin interactions are known to be weak in such conditions in vitro. Funb obtained at the regular [KCl] of 25mM (0.21± 0.02 pN; mean±SE) was significantly decreased at [KCl] of 150mM, (0.13± 0.01 pN; p=0.007), as shown in Fig.4B. To control for the effect of high [KCl] on the Funb measurements, we also measured Funb at a [KCl] of 150mM but in the absence of calponin. The results were not significantly different from those obtained without calponin at a [KCl] of 25mM (Fig.4C).
3.3 Control measurements in the absence of myosin
To eliminate the possibility that calponin regulated actin was binding in an unspecific manner to the pedestal, the Total Funb was also measured in the absence of myosin but in the presence of actin and calponin. The resulting force was normalized per length of actin in contact with the pedestal (Total Funb/ℓ). In the absence of unphosphorylated myosin, a significantly smaller Total Funb/ℓ (0.28±0.04 pN/μm; mean±SE) was observed than in its presence (4.19±0.42 pN/μm; p<0.001), as shown in Fig.4D.
3.4 Co-sedimentation assay
As previously reported in the literature [8], we confirmed that the phosphorylation of calponin by Ca2+/calmodulin protein kinase II decreases its binding to actin, by performing a co-sedimentation assay in the presence of phosphorylated or unphosphorylated calponin. A low-speed centrifugation in actin buffer was used because under such conditions, actin and calponin are soluble but will pellet more when cross-linked together. Indeed, we found more calponin and actin in the supernatant (S) when the calponin was phosphorylated (Fig. 5 lanes 1 & 2) and more in the pellet (P) when calponin was unphosphorylated (Fig. 5 lanes 3 & 4). To then confirm the cross-linking of unphosphorylated myosin to actin by unphosphorylated calponin, a co-sedimentation assay was performed in the presence of phosphorylated or unphosphorylated calponin. The above centrifugation and buffer conditions were used again because they allow for myosin filaments to sediment whereas actin and calponin remain soluble. In the presence of phosphorylated calponin, less myosin-actin-calponin complex was found in the pellet (Fig. 5, lanes 5 & 6). (Note that the ATP used to phosphorylate the calponin made the myosin filaments more soluble so more myosin was also found in the supernatant than would have been otherwise expected (Fig. 5 lane 5), as also observed in [8]). To the contrary, in the presence of unphosphorylated calponin, more myosin-actin-calponin complex was found in the pellet (Fig. 5, lanes 7 & 8). These results demonstrate that the unphosphorylated calponin enhances the binding of unphosphorylated myosin to actin compared to phosphorylated calponin.
Figure 5.

Co-sedimentation assay results shown after separation by SDS-PAGE. L: ladder, S: supernatant, P: pellet, MHC: myosin heavy chain, A: actin, Cp: calponin, phos: phosphorylated calponin, unphos: unphosphorylated calponin.
4. DISCUSSION
In this study, we provided direct molecular level-evidence that calponin enhances the average force of binding of unphosphorylated myosin to actin and that this is reversed upon calponin phosphorylation. Furthermore, we showed that this enhancement in binding force is accomplished by the action of calponin cross-linking unphosphorylated myosin with actin. These findings suggest that calponin could be a key player in the force maintenance capacity of smooth muscle, known as the latch state.
Several studies have suggested a role for calponin in the binding of unphosphorylated myosin to actin [5–7]. In a technically challenging study performed at the whole cell level, Malmqvist and co-workers [5] showed that when calponin is extracted from the smooth muscle cell and the myosin is exchanged for a non-phosphorylatable mutant, approximately 65% of the maximal cell force can still develop at approximately 30% of the maximal shortening velocity, following photolysis of caged ATP. Upon re-addition of calponin, the velocity and force development are prevented [5]. Our results showed that this calponin-induced inhibition in movement and force production is not due to the lack of binding of unphosphorylated myosin to actin because, to the contrary, calponin enhances the binding. Thus, it is likely that calponin decreases the active cycling of the myosin heads [9, 22] promoting force maintenance. Indeed, Horiuchi and Chacko [23] reported that calponin inhibits the catalytic step in the actin-activated ATP hydrolysis. Furthermore, because calponin also binds to unphosphorylated myosin [7], it most likely cross-links myosin to actin, thus explaining the enhancement of the binding force and the lack of cycling. We confirmed this possibility by measuring the Funb at different ionic strengths because the binding of calponin to unphosphorylated myosin is known to be weak at high ionic strength [7]. Indeed, we found that the calponin-induced enhancement of Funb was lost at high [KCl]. These data support the concept that calponin cross-links unphosphorylated myosin to actin, thus enhancing their binding force and preventing active force generation.
One point worth noting is that the high [KCl] conditions in the in vitro motility and laser trap assays correspond to normal ionic strength conditions inside the cell. Indeed, these assays have always been performed at relatively low ionic strength. Several papers have reported best motility below 60 mM KCl and it is the standard procedure to perform molecular mechanics measurements at 25 mM KCl [15, 24–28]. Whereas these conditions are not physiological, good quality molecular mechanics measurements are not possible otherwise. It is generally believed that the crowded cell environment provides conditions that are quite different from what can be obtained in the motility or laser trap assays, which leads to better protein binding conditions in vivo than in vitro. This has to be compensated for in vitro by working at lower ionic strengths. Nonetheless, the goal of working at high ionic strength was to change the conditions in such a way that calponin would detach from myosin in order to prove that this binding was indeed responsible for the increase in Funb. Szymanski and Tao [7] demonstrated by a sedimentation assay that the calponin/myosin interaction was ionic strength dependent; they showed a strong interaction at 50 mM NaCl which they said started to weaken at 100 mM NaCl and was abolished at 150 mM NaCl. Thus, using their approach in a molecular mechanics assay was the obvious procedure to follow. Because of such technical limitations, in vitro molecular mechanics studies cannot prove theories unequivocally but can suggest mechanisms that remain to be confirmed in vivo.
Calponin can be phosphorylated by protein kinase C and Ca2+/calmodulin-dependant protein kinase II [29]. Furthermore, it has been shown from ATPase measurements that phosphorylation of calponin inhibits its binding to actin [8]. Thus, in the presence of phosphorylated calponin both phosphorylated or dephosphorylated myosin heads can cycle. Our data showed that upon phosphorylation of calponin, the binding force of unphosphorylated myosin to actin is reduced. Because calponin is likely to be phosphorylated during smooth muscle cell activation, it must detach from actin [8] and not interfere with muscle shortening and force generation. Following calponin dephosphorylation, the binding of myosin to actin must be enhanced, due to their cross-linking by calponin, leading to force maintenance. The binding of unphosphorylated calponin to actin and the cross-linking of myosin to actin by unphosphorylated calponin were also demonstrated by our co-sedimentation results (Fig. 5).
It is interesting to note that in intact tissue measurements, the phosphorylation of calponin has not been observed in the carotid artery [30], a clearly tonic muscle, while it has been reported in the stomach and in airways [31, 32]. It is not clear why calponin would not be phosphorylated in the carotid artery but if as a general rule, calponin is less phosphorylated in the tonic than in the phasic muscles, it could certainly be a mechanism that contributes to the tonic muscle force maintenance. Furthermore, while the content of calponin is approximately the same in phasic and tonic smooth muscle, the effect of calponin is believed to be more prominent in tonic muscle because of the higher concentrations of caldesmon in phasic smooth muscle, and the fact that caldesmon and calponin compete for binding to actin [6].
Knocking-out calponin is an approach that has also been used to address its role in vivo [33–35]. Although the results are not entirely clear due to concomitant alterations in α-actin and h-caldesmon expression [33, 34], Takahashi and co-workers [35] demonstrated that calponin inhibits shortening velocity during the tonic phase of contraction. They suggested that this was due to direct regulation of the cross-bridge cycling rate, as can be seen in the in vitro motility assay (as we also reported in figure 3). Our results suggest that in addition to the slowing effect of calponin on the phosphorylated and cycling cross-bridges, the decrease in shortening velocity may also be due to its enhancement of the binding force between dephophorylated myosin and actin. Thus, instead of cycling, the dephosphorylated myosin molecules maintain the force that is actively generated by the few remaining phoshorylated myosin molecules.
We previously reported that the average binding force of unphosphorylated myosin to actin is approximately 10% of that generated by phosphorylated myosin [36]. This may seem low but our current data show that it is increased by approximately 40% in the presence of unphosphorylated calponin. In addition, the contribution of other actin regulatory proteins such as caldesmon and tropomyosin may also play a role in increasing this binding force further. However, it is also important to note that our measurements do not allow us to distinguish between stronger individuals cross-bridges in the presence of calponin versus more cross-bridges of similar strength. This is why our data are reported in terms of average binding force per myosin molecule. The only way to distinguish between these two possibilities would be to perform molecular mechanics measurements on single myosin molecules in the presence of calponin, which is beyond the scope of the current study.
Another limitation of our study is that all experiments have been performed with unphosphorylated and not dephosphorylated myosin. Nonetheless, there is no data in the literature to suggest that dephosphorylated myosin is different from unphosphorylated myosin. However, if they were different structurally and functionally, we would have missed that effect. Because of the high sensitivity of our laser trap assay, if we were to work with dephosphorylated myosin, any remaining phosphorylated myosin heads would lead to overestimated force values.
Finally, although we believe that calponin plays an important role in force maintenance in the latch-state, it is not sufficient to explain the whole phenomenon. Instead we suggest that a combination of factors must be present to lead to force maintenance. For one, tonic muscle is known to express a greater amount of the slower myosin heavy chain [37, 38]. This myosin lacks the 7 amino acid insert found in the surface loop above the ATP binding pocket [38, 39] and thus is referred to as the (-)insert or SMA myosin isoform. Lacking the insert increases its time of attachment to actin [36] by increasing its affinity for ADP [40, 41]. The fact that the (-)insert isoform remains attached longer to actin presumably increases its chances of getting dephosphorylated while attached. This is likely to contribute to the greater propensity of tonic muscle to get into the latch state. Then, along with the few remaining phosphorylated cross-bridges that still generate active force, the attached and potentially reattaching dephosphorylated myosin molecules, cross-linked by calponin and most-likely by other actin regulatory proteins, cooperate to lead to force maintenance. The fact that calponin does not appear to get phosphorylated in tonic muscle [30] may also promote its cross-linking action.
5. CONCLUSION
In conclusion, the dephosphorylation of calponin provides a mechanism to enhance the binding of unphosphorylated myosin, and presumably dephosphorylated myosin, to actin. This enhanced binding is likely to contribute to the force maintenance observed in tonic smooth muscle.
Highlights.
Molecular level myosin force measurements.
Molecular mechanical mechanism to explain the latch-state.
The latch-state, a smooth muscle property that may prove to be important in disease.
Acknowledgments
We thank Professor John M. Dealy, from the department of Chemical Engineering, McGill University, for performing the methylcellulose viscosity measurements, Genevieve Bates for the art work of figure 1 and Michel Alveis from Marvid Poultry for the procurement of the chicken breasts for the purification of skeletal muscle actin and myosin for NEM preparation.
7. GRANTS
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) grant, National Heart, Lung, and Blood Institute grant RO1-HL 103405-02, and the Costello Fund. The Meakins-Christie Laboratories (McGill University Health Centre Research Institute) are supported in part by a center grant from Le Fonds de la Recherche en Santé du Québec (FRSQ). GI is a recipient of the P.T. Macklem Memorial Fellowship. LH is a recipient of the Canadian Institutes of Health Research Systems Biology Training Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495–497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- 2.Mezgueldi M, Fattoum A, Derancourt J, Kassab R. Mapping of the functional domains in the amino-terminal region of calponin. J Biol Chem. 1992;267:15943–15951. [PubMed] [Google Scholar]
- 3.Kolakowski J, Makuch R, Stepkowski D, Dabrowska R. Interaction of calponin with actin and its functional implications. Biochem J. 1995;306(Pt 1):199–204. doi: 10.1042/bj3060199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K, Hiwada K, Kokubu T. Isolation and characterization of a 34,000-dalton calmodulin- and F-actin-binding protein from chicken gizzard smooth muscle. Biochem Biophys Res Commun. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- 5.Malmqvist U, Trybus KM, Yagi S, Carmichael J, Fay FS. Slow cycling of unphosphorylated myosin is inhibited by calponin, thus keeping smooth muscle relaxed. Proc Natl Acad Sci U S A. 1997;94:7655–7660. doi: 10.1073/pnas.94.14.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szymanski PT. Calponin (CaP) as a latch-bridge protein--a new concept in regulation of contractility in smooth muscles. J Muscle Res Cell Motil. 2004;25:7–19. doi: 10.1023/b:jure.0000021349.47697.bf. [DOI] [PubMed] [Google Scholar]
- 7.Szymanski PT, Tao T. Interaction between calponin and smooth muscle myosin. FEBS letters. 1993;334:379–382. [PubMed] [Google Scholar]
- 8.Winder SJ, Walsh MP. Smooth muscle calponin. Inhibition of actomyosin MgATPase and regulation by phosphorylation. J Biol Chem. 1990;265:10148–10155. [PubMed] [Google Scholar]
- 9.Haeberle JR. Calponin decreases the rate of cross-bridge cycling and increases maximum force production by smooth muscle myosin in an in vitro motility assay. J Biol Chem. 1994;269:12424–12431. [PubMed] [Google Scholar]
- 10.Shirinsky VP, Biryukov KG, Hettasch JM, Sellers JR. Inhibition of the relative movement of actin and myosin by caldesmon and calponin. J Biol Chem. 1992;267:15886–15892. [PubMed] [Google Scholar]
- 11.Leguillette R, Zitouni NB, Govindaraju K, Fong LM, Lauzon AM. Affinity for MgADP and force of unbinding from actin of myosin purified from tonic and phasic smooth muscle. Am J Physiol Cell Physiol. 2008;295:C653–660. doi: 10.1152/ajpcell.00100.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobieszek A. Smooth muscle myosin. Molecule conformation, filament assembly and association of regulatory enzymes. In: Giembycz DRaMA., editor. Airways smooth muscle: Biochemical Control of Contraction and Relaxation. Birkhauser; Bassel: 1994. pp. 1–29. [Google Scholar]
- 13.Trybus KM, Lowey S. Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J Biol Chem. 1984;259:8564–8571. [PubMed] [Google Scholar]
- 14.Pardee JD, Spudich JA. Purification of muscle actin. 1982. [DOI] [PubMed] [Google Scholar]
- 15.Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J Cell Biol. 1990;111:453–463. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobieszek A, Sarg B, Lindner H, Seow CY. Phosphorylation of caldesmon by myosin light chain kinase increases its binding affinity for phosphorylated myosin filaments. Biological chemistry. 391:1091–1104. doi: 10.1515/BC.2010.105. [DOI] [PubMed] [Google Scholar]
- 17.Homsher E, Wang F, Sellers JR. Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am J Physiol. 1992;262:C714–723. doi: 10.1152/ajpcell.1992.262.3.C714. [DOI] [PubMed] [Google Scholar]
- 18.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem. 1993;268:14764–14768. [PubMed] [Google Scholar]
- 19.VanBuren P, Work SS, Warshaw DM. Enhanced force generation by smooth muscle myosin in vitro. Proc Natl Acad Sci U S A. 1994;91:202–205. doi: 10.1073/pnas.91.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kishino A, Yanagida T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 1988;334:74–76. doi: 10.1038/334074a0. [DOI] [PubMed] [Google Scholar]
- 21.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J Mol Biol. 1990;214:699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 22.Marston SB, Fraser ID, Bing W, Roper G. A simple method for automatic tracking of actin filaments in the motility assay. J Muscle Res Cell Motil. 1996;17:497–506. doi: 10.1007/BF00123365. [DOI] [PubMed] [Google Scholar]
- 23.Horiuchi KY, Chacko S. The mechanism for the inhibition of actin-activated ATPase of smooth muscle heavy meromyosin by calponin. Biochem Biophys Res Commun. 1991;176:1487–1493. doi: 10.1016/0006-291x(91)90455-g. [DOI] [PubMed] [Google Scholar]
- 24.Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci U S A. 1986;83:6272–6276. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kron SJ, Toyoshima YY, Uyeda TQ, Spudich JA. Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 1991;196:399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 26.Nagy A, Takagi Y, Billington N, Sun SA, Hong DK, Homsher E, Wang A, Sellers JR. Kinetic characterization of nonmuscle myosin IIb at the single molecule level. J Biol Chem. 2013;288:709–722. doi: 10.1074/jbc.M112.424671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umemoto S, Sellers JR. Characterization of in vitro motility assays using smooth muscle and cytoplasmic myosins. J Biol Chem. 1990;265:14864–14869. [PubMed] [Google Scholar]
- 28.Wang Y, Ajtai K, Burghardt TP. Qdot Labeled Actin Super-Resolution Motility Assay Measures Low Duty Cycle Muscle Myosin Step-Size. Biochemistry. 2013 doi: 10.1021/bi301702p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winder SJ, Walsh MP, Vasulka C, Johnson JD. Calponin-calmodulin interaction: properties and effects on smooth and skeletal muscle actin binding and actomyosin ATPases. Biochemistry. 1993;32:13327–13333. doi: 10.1021/bi00211a046. [DOI] [PubMed] [Google Scholar]
- 30.Adam LP, Haeberle JR, Hathaway DR. Calponin is not phosphorylated during contractions of porcine carotid arteries. Am J Physiol. 1995;268:C903–909. doi: 10.1152/ajpcell.1995.268.4.C903. [DOI] [PubMed] [Google Scholar]
- 31.Pohl J, Winder SJ, Allen BG, Walsh MP, Sellers JR, Gerthoffer WT. Phosphorylation of calponin in airway smooth muscle. Am J Physiol. 1997;272:L115–123. doi: 10.1152/ajplung.1997.272.1.L115. [DOI] [PubMed] [Google Scholar]
- 32.Winder SJ, Allen BG, Fraser ED, Kang HM, Kargacin GJ, Walsh MP. Calponin phosphorylation in vitro and in intact muscle. Biochem J. 1993;296(Pt 3):827–836. doi: 10.1042/bj2960827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babu GJ, Celia G, Rhee AY, Yamamura H, Takahashi K, Brozovich FV, Osol G, Periasamy M. Effects of h1-calponin ablation on the contractile properties of bladder versus vascular smooth muscle in mice lacking SM-B myosin. J Physiol. 2006;577:1033–1042. doi: 10.1113/jphysiol.2006.118828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matthew JD, Khromov AS, McDuffie MJ, Somlyo AV, Somlyo AP, Taniguchi S, Takahashi K. Contractile properties and proteins of smooth muscles of a calponin knockout mouse. J Physiol. 2000;529(Pt 3):811–824. doi: 10.1111/j.1469-7793.2000.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi K, Yoshimoto R, Fuchibe K, Fujishige A, Mitsui-Saito M, Hori M, Ozaki H, Yamamura H, Awata N, Taniguchi S, Katsuki M, Tsuchiya T, Karaki H. Regulation of shortening velocity by calponin in intact contracting smooth muscles. Biochem Biophys Res Commun. 2000;279:150–157. doi: 10.1006/bbrc.2000.3909. [DOI] [PubMed] [Google Scholar]
- 36.Lauzon AM, Tyska MJ, Rovner AS, Freyzon Y, Warshaw DM, Trybus KM. A 7-amino-acid insert in the heavy chain nucleotide binding loop alters the kinetics of smooth muscle myosin in the laser trap. J Muscle Res Cell Motil. 1998;19:825–837. doi: 10.1023/a:1005489501357. [DOI] [PubMed] [Google Scholar]
- 37.Babij P. Tissue-specific and developmentally regulated alternative splicing of a visceral isoform of smooth muscle myosin heavy chain. Nucleic Acids Res. 1993;21:1467–1471. doi: 10.1093/nar/21.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White S, Martin AF, Periasamy M. Identification of a novel smooth muscle myosin heavy chain cDNA: isoform diversity in the S1 head region. Am J Physiol. 1993;264:C1252–1258. doi: 10.1152/ajpcell.1993.264.5.C1252. [DOI] [PubMed] [Google Scholar]
- 39.Kelley CA, Takahashi M, Yu JH, Adelstein RS. An insert of seven amino acids confers functional differences between smooth muscle myosins from the intestines and vasculature. J Biol Chem. 1993;268:12848–12854. [PubMed] [Google Scholar]
- 40.Fuglsang A, Khromov A, Torok K, Somlyo AV, Somlyo AP. Flash photolysis studies of relaxation and cross-bridge detachment: higher sensitivity of tonic than phasic smooth muscle to MgADP. J Muscle Res Cell Motil. 1993;14:666–677. doi: 10.1007/BF00141563. [DOI] [PubMed] [Google Scholar]
- 41.Khromov A, Somlyo AV, Trentham DR, Zimmermann B, Somlyo AP. The role of MgADP in force maintenance by dephosphorylated cross-bridges in smooth muscle: a flash photolysis study. Biophys J. 1995;69:2611–2622. doi: 10.1016/S0006-3495(95)80132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]