

Abstract
In addition to amyloid beta (Aβ) and tau, α-synuclein, best known for its role in Parkinson’s disease (PD), has been suggested to be involved in cognition and pathogenesis of Alzheimer’s disease (AD). We investigate the potential of α-synuclein in cerebrospinal fluid (CSF) as a biomarker of cognitive decline in AD, and its prodromal phase, mild cognitive impairment (MCI). Using an established, sensitive Luminex assay, we measured α-synuclein levels in the CSF of a cohort of close to 400 healthy control, MCI and AD subjects obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and factored in APOE genotype in data analysis. CSF α-synuclein levels were significantly higher in the MCI (P = 0.005) and AD (P < 0.001) groups, compared to controls. However, receiver operating characteristic (ROC) curve analysis suggests that CSF α-synuclein level on its own only offered modest sensitivity (65 %) and specificity (74 %) as a diagnostic marker of AD, with an area under the curve (AUC) value of 0.719 for AD vs controls. The effect of APOE genotype, if any, was quite subtle. However, there was a significant correlation between α-synuclein and cognition (P = 0.001), with increased α-synuclein levels associated with decreased MMSE scores. Our results support a role for α-synuclein even in MCI, the early phase of AD, in addition to being a potential contributor in MCI and AD diagnosis or monitoring of disease progression.
Keywords: alpha-synuclein, Alzheimer’s disease, Mild cognitive impairment, biomarkers, cerebrospinal fluid
1. Introduction
α-Synuclein, though best characterized for Parkinson’s disease (PD), might also be involved in Alzheimer’s disease (AD), given that its fragment, non-Aβ component (NAC) was identified in extracellular plaques in the brains of AD patients [1]. Indeed, it was found subsequently that 40–50% of AD patients also present with α-synuclein positive Lewy bodies [2–4] and these patients demonstrate an accelerated cognitive decline [5, 6].
The interest on α-synuclein in AD has been further developed by evidence suggesting α-synuclein might interact with Aβ and tau, two critical proteins in AD pathogenesis, promoting their mutual aggregation [7–10] amplifying neuronal damage and accelerating cognitive decline [11, 12]. Importantly, the accumulation of α-synuclein alone has also been shown to significantly disrupt cognition in mice [12, 13], as well as be associated with cognitive impairment or dementia in synucleinopathies [14, 15].
Several groups have attempted to investigate α-synuclein levels in the brain and cerebrospinal fluid (CSF) of patients with AD, PD, and dementia with Lewy bodies (DLB). While many studies demonstrated lower CSF α-synuclein in PD and DLB patients [24, 28, 29], results related to AD patients are quite variable, with no change, lower or higher CSF α-synuclein in AD reported [16–23]. These inconsistent findings may be due to methodological differences, limited number of patients, heterogeneous inclusion criteria for patients, or a lack of appropriate control of blood contamination in CSF, which influences CSF α-synuclein levels substantially [24].
In the current study, we measured α-synuclein levels in CSF of a large cohort of well-characterized subjects, using an established and sensitive assay, and with rigorous controls for blood contamination, as well as consideration of APOE genotype. We asked whether: (1) there are differences in CSF α-synuclein levels between AD, mild cognitive impairment (MCI), a prodromal phase of AD, and control groups, and (2) individual α-synuclein CSF levels are correlated with cognitive impairment as measured by the Mini Mental Status Exam (MMSE).
2. Materials and methods
2.1 Subjects and samples
CSF samples from subjects with MCI or AD, as well as age matched, healthy controls were obtained from the original Alzheimer’s Disease Neuroimaging Initiative (ADNI-1). ADNI-1 was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations. The initial enrollment target for ADNI-1 was 400 patients with MCI, 200 patients with mild AD and 200 healthy controls. The ADNI protocol was approved by the human studies committees at 58 institutions in the United States and Canada. Written and verbal informed consents were obtained from participants at screening and enrollment. Subjects underwent clinical and cognitive assessments, including the MMSE, at screening. According to the ADNI1 protocol, lumbar punctures for CSF collection took place at the baseline visit, which was scheduled no more than 28 days after the screening visit. For details regarding inclusion/exclusion criteria, cognitive assessment and CSF collection methods, please see http://adni.loni.ucla.edu/wp-content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf. Briefly, AD subjects had MMSE scores of 20–26, a CDR of 0.5 or 1.0 and met the NINCDS/ADRDA criteria for probable AD. MCI subjects had MMSE scores of 24–30, a memory complaint, objective memory loss measured by education adjusted scores on Wechsler Memory Scale Logical Memory II, a CDR of 0.5, as well as absence of significant impairment in other cognitive domains and absence of dementia. Control subjects had MMSE scores of 24–30 and a Clinical Dementia Rating (CDR) of 0.
Only a subset of the ADNI-1 CSF sample set (available at the time of request) was included in the current study (see Figure 1 for details) and their demographic data, along with average levels of CSF α-synuclein, with versus without restriction of hemoglobin levels, are summarized in Table 1. It should be mentioned that, based on the ADNI database, a few cases in our cohort did convert to DLB after initial diagnosis as control (n = 1), MCI (n = 2) or AD (n = 2).
Figure 1.
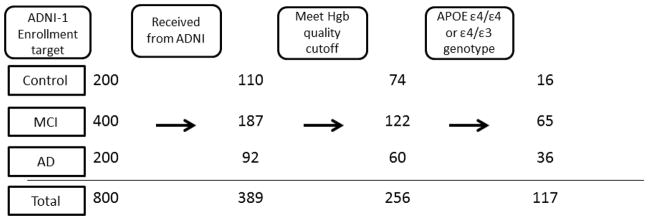
Flowchart depicting for control, MCI and AD subjects: the target enrollment number of ADNI-1, the number of CSF samples received for α-synuclein measurement in the current study, the number of CSF samples remaining after controlling for blood contamination and the number of CSF samples from patients with APOE genotype ε4/ε4 and ε4/ε3. Of note, among the subjects enrolled in ADNI, initial CSF tap rate is only about 50%, and we received all cases with >10 aliquots CSF samples (the cut-off set by ADNI Repository).
Table 1.
Summary of demographics and α-synuclein values of donors
| Control | Mild cognitive impairment | Alzheimer’s disease | |
|---|---|---|---|
| Number of cases | |||
| Total | 110 | 187 | 92 |
| After elimination of cases with hemoglobin >200 ng/ml | 74 | 122 | 60 |
| APOE ε4/ε4 & ε4/ε3 | 16 | 65 | 36 |
| Gender (F/M) | |||
| Total | 54/56 | 62/125 | 40/52 |
| After elimination of cases with hemoglobin >200 ng/ml | 36/38 | 41/81 | 27/33 |
| Age (years; all cases) | |||
| Mean±SD | 75.4±5.2 | 74.0±7.6 | 74.4±7.8 |
| Range | 62–89 | 54–89 | 56–89 |
| Age (years; after elimination of cases with hemoglobin >200 ng/ml) | |||
| Mean±SD | 75.2±0.6 | 73.6±0.7 | 74±1.1 |
| Range | 63–89 | 55–89 | 56–89 |
| MMSE score (all cases) | |||
| Mean±SD | 29.1±1.0 | 27.0±1.8 | 23.6±1.9 |
| Range | 25–30 | 23–30 | 20–27 |
| MMSE score (after elimination of cases with hemoglobin >200 ng/ml) | |||
| Mean±SD | 29.1±0.1 | 26.9±0.2 | 23.7±0.2 |
| Range | 25–30 | 23–30 | 20–26 |
| MMSE score (APOE ε4/ε4 & ε4/ε3) | |||
| Mean±SD | 29.1±0.9 | 26.8±1.7 | 23.7±1.9 |
| Range | 28–30 | 23–30 | 20–26 |
| CSF hemoglobin (ng/ml) | |||
| Mean±SD | 684.5±2462.0 | 585.2±1325.0 | 500.6±1055.1 |
| Range | 0–23870.6 | 0–10488.3 | 0–5208.9 |
| CSF α-synuclein (ng/ml; all cases) | |||
| Mean±SD | 0.83±1.40 | 0.76±0.81 | 0.83±0.85 |
| Range | 0.10–12.24 | 0.15–7.17 | 0.22–6.0 |
| CSF α-synuclein (ng/ml; after elimination of cases with hemoglobin >200 ng/ml) | |||
| Mean±SD (all) | 0.42±0.14 | 0.50±0.18 | 0.56±0.20 |
| Range (all) | 0.10–0.82 | 0.15–1.0 | 0.22–1.17 |
2.2 CSF Hemoglobin and α-synuclein assays
Hemoglobin levels were measured with a human hemoglobin ELISA quantitation kit from Bethyl Lab Inc (Montgomery, TX, USA) and used as an index of the degree of blood contamination, which has previously been shown to influence CSF α-synuclein levels significantly [24]. Total α-synuclein levels were determined by Luminex assay as previously described [24]. Briefly, CSF samples were thawed on ice, treated 1:1 with 2× radioimmunoprecipitation assay buffer for 0.5–1 hr and centrifuged at 15 000 g for 10 minutes at 4 °C. Supernatants were removed and 50 μl diluted in 50 μl of assay diluent (0.1% bovine serum albumin/phosphate buffered saline) loaded per well containing the capturing antibody-coupled beads. Plates were then incubated for 3 hrs, washed, and the diluted biotinylated detection antibody added to wells. After another 3 hr incubation, plates were washed, diluted streptavidin-R-PE added to wells and incubated for 30 minutes. The plates were then washed and read on a LiquiChip Luminex 200TM Workstation (Qiagen). The sensitivity of the assay ranged as low as 0.009 ng/ml and the accuracy, as determined by recovery of spiked α-synuclein protein was about 93 %. The assay also demonstrated low plate-to-plate, as well as day-to-day variability, with high signal reproducibility and linear performance in the low pg range.
2.3 Statistical analysis
Statistical analysis was performed using PASW 18.9 Statistics software (SPSS, Inc., Chicago, IL, USA). Correlations between CSF α-synuclein and hemoglobin, age and gender were evaluated using Spearman’s nonparametric correlation. For all subsequent analysis, CSF α-synuclein data was log transformed to obtain a normal distribution. To compare CSF α-synuclein levels between diagnostic and APOE genotype groups, a one-way analysis of variance (ANOVA) followed by the post hoc Tukey honestly significant difference (HSD) test was performed. A receiver operating characteristic (ROC) curve was used to calculate the relationship between sensitivity and specificity for the disease group versus healthy controls, and hence evaluate the diagnostic performance of the analyte. The optimum cut-off value from the ROC curve is the point at which the sum of sensitivity and specificity is maximal. Pearson’s correlation was used to evaluate the relationship between CSF α-synuclein levels and MMSE scores.
3. Results
3.1 Effect of blood contamination, age and gender on CSF α-synuclein levels
Because α-synuclein levels in human blood is much higher than in CSF, it is to be expected that even minimal blood contamination of CSF could have significant impact on CSF α-synuclein levels [24]. Consistent with our previous reports, there was a significant correlation (rho = 0.550; P<0.001) between CSF hemoglobin and α-synuclein levels, with α-synuclein levels increasing substantially when hemoglobin was >200 ng/ml (Figure 2 A). When a cut off value of 200 ng/ml was set for hemoglobin, there was no significant correlation between CSF hemoglobin and α-synuclein levels (Figure 2 B). As a result, all subsequent statistical analyses were performed in cases with hemoglobin levels ≤200 ng/ml. There was no significant relationship between CSF α-synuclein levels and age or gender in this cohort (Supplementary Figure 1).
Figure 2. Correlation of CSF α-synuclein levels with hemoglobin levels.

CSF α-synuclein and hemoglobin levels were measured in individual healthy controls and cases with mild cognitive impairment (MCI) and Alzheimer’s disease (AD). Data shown are before (A) or after (B) elimination of samples with blood contamination (200 ng/ml hemoglobin was used as a cut-off). Spearman’s correlation coefficients (rho) and P-values were rho = 0.550 (P<0.001) and rho = 0.066 (P = 0.292), for before and after elimination of contaminated cases, respectively.
3.2 CSF α-synuclein levels in controls and patients with MCI and AD
After exclusion of cases with hemoglobin levels >200 ng/ml, there was a significant difference in CSF α-synuclein levels between groups (P<0.001; Figure 3). CSF α-synuclein levels were significantly higher in the MCI group (P = 0.005) and AD group (P<0.001) compared to the control group. Although there was a trend towards an increase in CSF α-synuclein in AD compared to MCI groups, it did not reach statistical significance (P = 0.083). As mentioned earlier, a few cases in our cohort did convert to DLB after initial diagnosis as control, MCI or AD. However, exclusion of these cases from the analysis did not alter the results (data not shown).
Figure 3. Cross-sectional examination of CSF α-synuclein levels.
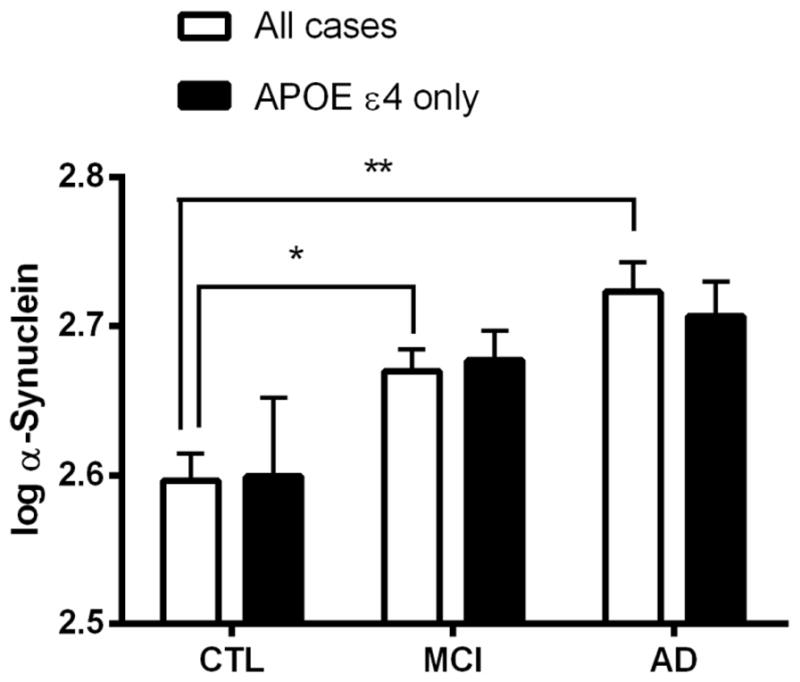
Quantitative Luminex analysis was performed to measure CSF α-synuclein in healthy controls (CTL) and patients with mild cognitive impairment (MCI) and Alzheimer’s disease (AD). Data shown are after elimination of samples with blood contamination (200 ng/ml hemoglobin was used as a cut-off). To obtain a normal distribution, α-synuclein levels were log transformed. Data are shown for all cases irrespective of APOE genotype (clear bars) and for cases with APOE genotype ε4/ε4 and ε4/ε3 (solid bars). Data shown are mean ± SEM. *P = 0.005 and **P<0.001 versus control group in all cases. Number of cases can be found in Table 1.
Given that APOE is the most important gene associated with sporadic AD risk, an additional analysis was performed in cases with APOE genotype ε4/ε4 and ε4/ε3. In this subset of cases, there was no significant difference in CSF α-synuclein levels between diagnostic groups (P = 0.087; Figure 3). In addition, because it has been suggested that gender might influence APOE ε4 associated risk for AD [25, 26], the analysis was also done separately in males and females with APOE genotype ε4/ε4 and ε4/ε3. In males with APOE genotype ε4/ε4 and ε4/ε3, there was no significant difference in CSF α-synuclein levels between diagnostic groups (data not shown; P = 0.786). However, in females with APOE genotype ε4/ε4 and ε4/ε3 there was a significant difference between groups (P = 0.012), with higher CSF α-synuclein levels in the MCI (P = 0.029) and AD (P = 0.009) groups compared to controls (data not shown). Finally, a one-way ANOVA showed no significant difference in CSF α-synuclein levels between APOE genotype ε4/ε4, ε4/ε3, ε4/ε2, ε3/ε3 and ε2/ε3 (data not shown).
ROC curve analysis was performed to further evaluate the usage of CSF α-synuclein in AD/MCI diagnosis. When a cut-off value of 0.49 ng/ml was chosen for α-synuclein, the sensitivity and specificity for distinguishing AD from control were 65 and 74 %, respectively (Figure 4 A; Supplementary Table 1). When a cut-off value of 0.41 ng/ml was selected, the sensitivity and specificity for distinguishing MCI from control were 63 and 61 %, respectively (Figure 4 B; Supplementary Table 1). Performing the ROC curve analysis on cases with APOE genotype ε4/ε4 and ε4/ε3 did not improve the sensitivity or specificity for distinguishing AD from control or MCI from control (data not shown).
Figure 4. ROC curves to evaluate CSF α-synuclein level as a biomarker of MCI and AD.

ROC curve for α-synuclein of (A) mild cognitive impairment (MCI) and (B) Alzheimer’s disease (AD) versus controls, after elimination of blood contaminated samples (200 ng/ml hemoglobin was used as a cut-off). To obtain a normal distribution, α-synuclein levels were log transformed. AUCs, P values, as well as sensitivity and specificity can be found in Supplementary Table 1.
3.3 Correlation of CSF α-synuclein with cognitive impairment and disease duration
When considering only cases with hemoglobin levels ≤200 ng/ml, there was a statistically significant correlation (R = −0.204; P = 0.001) between CSF α-synuclein levels and MMSE score in control, MCI and AD patients together (Figure 5 A). When the analysis was replicated n control, MCI, and AD separately, while the general trends remained same, statistical significance was lost, probably because of reduced power with reducing case numbers. Additional analysis was performed on control, MCI and AD cases with APOE genotype ε4/ε4 and ε4/ε3. In these select cases, there was no significant correlation between CSF α-synuclein levels and MMSE score (Figure 5 B). There was also no significant correlation between CSF α-synuclein levels and disease duration in years in all AD patients or in AD patients with APOE genotype ε4/ε4 and ε4/ε3 (data not shown).
Figure 5. Correlation of CSF α-synuclein levels with cognition as measured by MMSE.

CSF α-synuclein levels were measured in healthy controls (CTL) and patients with mild cognitive impairment (MCI) and Alzheimer’s disease (AD). Data shown are after elimination of samples with blood contamination (200 ng/ml hemoglobin was used as a cut-off). To obtain a normal distribution, α-synuclein levels were log transformed. Data are shown for all cases irrespective of APOE genotype (A) and for cases with APOE genotype ε4/ε4 and ε4/ε3 (B). The correlation coefficients (R) and P-values were R = −0.204 (P<0.01) and R = −0.71 (P = 0.449), for all cases and cases with APOE genotype ε4/ε4 and ε4/ε3, respectively.
4. Discussion
We find a significant difference in CSF α-synuclein levels between cognitive normal and impaired groups. Importantly, not only was CSF α-synuclein levels different in the AD versus control group, but also the MCI versus control group, with a trend toward an increase in AD compared to MCI groups. However, ROC curve analysis shows that CSF α-synuclein on its own only offers modest sensitivity and specificity as a diagnostic marker. Additionally, factoring in APOE genotype did not affect outcome substantially. On the other hand, there was a significant, negative correlation between CSF α-synuclein levels and cognitive function as measured by the MMSE. Together, these data suggest that although increased CSF α-synuclein levels alone might not be robust enough for AD or MCI diagnosis, its increase may be an early feature of cognitive decline, and it may increase further with the onset of AD dementia, indicating its critical role in AD pathogenesis. A few important issues are further discussed below.
Age and gender dependence
Like our previous study, there was no correlation between CSF α-synuclein and gender [27]. However, whereas we previously reported a significant correlation between CSF α-synuclein and age in normal controls, in the current study, we find no such correlation. This contradictory finding might be explained by the difference in age range for the two studies. In the current study, the age range for controls were 62–89 (Table 1), compared to an age range of 21–90 for our previous study [27].
Caveats associated with CSF α-synuclein
α-Synuclein has been measured in several previous studies with contradicting results. Reesink and colleagues [19] measured CSF α-synuclein levels in 35 DLB, 63 AD, 18 PD and 34 subjective complaint controls using an ELISA and found no significant difference between groups. In the DLB but not the AD group, they found a significant positive correlation between CSF α-synuclein and MMSE. Öhrfelt et al [23] measured CSF α-synuclein in 66 AD, 15 PD, 15 DLB and 55 control subjects using an ELISA, demonstrating no differences between controls and PD or DLB groups, but significantly decreased α-synuclein levels in the AD group versus controls. Moreover, they report that AD patients with MMSE scores below 20 had significantly lower CSF α-synuclein levels than AD patients with MMSE scores of 20 or higher. Finally, Hall and colleagues [22] measured CSF α-synuclein levels in 90 PD, 48 AD, 33 PDD, 70 DLB and 107 control subjects using a Luminex assay. In contrast to the previous 2 studies, they controlled for blood contamination of CSF samples by measuring hemoglobin levels. Similar to the results of our current and previously published studies [24], they found an increase in CSF α-synuclein levels in patients with AD and decreased levels in patients with PD, PDD and DLB, respectively. However, the authors did not report any correlation between CSF α-synuclein and MMSE in any of the groups in this study.
These conflicting results may be the result of methodological differences in α-synuclein quantification (antibody and assay platform used), lack of appropriate and rigorous control for blood contamination of CSF samples (varying from no control or a cut-off of <500 RBC/μl or 1000 ng Hgb/ml [19, 23]), limited number of patients (in some of the studies) or heterogeneous inclusion criteria for patients. In the current study, we used an established and sensitive Luminex assay to investigate a relatively large cohort of participants subjected to rigorous clinical and neuropsychological assessments. In addition, samples were collected, stored and handled according to standardized procedures, tested for hemoglobin levels and excluded from the analysis if found to be significantly contaminated with blood (>200 ng/ml), as this has been shown to influence α-synuclein levels [24]. In fact, if we did not control for hemoglobin, the conclusion would be totally different, i.e. there were no statistical difference in CSF α-synuclein levels when comparing AD or MCI vs. controls (data not shown).
α-Synuclein as a cognition modulator
Only a few groups investigated the correlation between CSF α-synuclein and cognition, and several caveats of these reports are discussed above. The results of our CSF study are in agreement with a recent study by Larson and colleagues [16] indicating a role for α-synuclein in modulation of cognitive impairment. In the study, soluble α-synuclein was increased approximately 1.7-fold and 2.2-fold in tissue from the inferior temporal gyrus of AD patients compared to MCI patients and controls, respectively. Strikingly, increased α-synuclein was a stronger correlate of cognitive impairment than soluble Aβ and tau levels. In contrast, an earlier study reported decreased soluble α-synuclein in the frontal cortex of AD, but not MCI patients compared to controls and this decrease correlated with MMSE score even in controls, suggesting that α-synuclein levels may be a sensitive index of synaptic integrity [17].
The mechanism of CSF α-synuclein elevation and its pathological role in AD is unknown. This is especially intriguing, because diseases with other synucleinopathy, e.g. PD, DLB and MSA are typically associated with decreased CSF α-synuclein [18, 23, 24, 28, 29]. Although alterations in CSF α-synuclein cannot be explained readily by a simplistic model, one may speculate that in synucleinopathies with profound Lewy body pathology, the lower CSF α-synuclein concentrations might be mainly due to trapping α-synuclein in Lewy bodies, but in AD, the higher CSF α-synuclein may be a result of the release of the protein from damaged neuron cell bodies or their processes after neurodegeneration. To this end, exclusion of few cases that converted to DLB (four with <200 ng Hgb/ml) after initial diagnosis did not change the results or conclusions. However, given presence of Lewy body pathology at autopsy of a significant number of AD cases [2–4], it is expected that, in a subset of ADNI subjects, there will be opposite forces to drive CSF α-synuclein levels. This issue needs to be investigated further by factoring in typical AD biomarkers or when autopsy results of the cohort become available.
Assuming increased CSF α-synuclein levels in AD reflects increased brain tissue α-synuclein levels shown by others [16], whether released from damaged cells or not, this could mean that CSF α-synuclein levels might correlate with AD severity or progression. This hypothesis is supported by the fact that higher CSF α-synuclein is associated with lower MMSE scores (Figure 5). Physiologically, the high presynaptic concentration of α-synuclein and its association with synaptic vesicles suggest a role of the protein in the modulation of neurotransmitter release [30]. The correlation of higher CSF α-synuclein levels with lower MMSE scores is consistent with the observation that abnormal elevation of soluble α-synuclein results in decreases in selected synaptic vesicle proteins, alteration of the protein composition of synaptic vesicles [16] and subsequently, impaired neurotransmitter release by neuronal cells [31, 32]. On the other hand, the relative weak association between CSF α-synuclein and MMSE in the current investigation suggests significant heterogeneity of the population, and it is imperative to assess the relationship again in the samples collected longitudinally.
APOE genotype and limitations
A largely negative yet important observation is that APOE genotype did not affect CSF α-synuclein levels. To this end, studies have shown that subjects with the APOE ε4/ε4 and ε4/ε3 genotype are at increased risk for developing late onset AD, whereas the APOE ε2 allele is considered protective [33, 34]. Both the APOE ε2 and ε4 alleles have been associated with increased risk for PD [35, 36] and the ε4 allele has been associated with an increased risk for developing dementia in PD [37], although contradictory findings have also been published [38, 39]. In transgenic mice, α-synuclein induced neurodegeneration involves a massive increase in ApoE levels and accumulation of insoluble Aβ. Furthermore, ApoE deletion delays α-synuclein induced neurodegeneration and suppresses accumulation of Aβ [40]. In the current study, when selecting subjects with the APOE ε4/ε4 and ε4/ε3 genotype, we do not find a significant difference in CSF α-synuclein levels between control, MCI and AD groups, although a similar trend of increasing levels in MCI and AD versus control groups remain. When selecting only these cases, the significant correlation between CSF α-synuclein levels and MMSE scores also disappears. However, the results need to be interpreted with caution because selecting for APOE ε4/ε4 and ε4/ε3 genotype substantially reduces the number of subjects (cases are reduced to 16 control, 65 MCI and 36 AD; see Figure 1 & Table 1). When analyzing the 2 genders separately, we find that although there is no significant difference in CSF α-synuclein levels for male groups, there is a significant difference between female groups, with both MCI and AD groups having higher levels compared to controls (data not shown). This is interesting, given studies suggesting that women, but not men that are APOE ε4 heterozygous have a higher risk of developing AD compared to those without a ε4 allele [25, 26]. Then again, because selecting for APOE ε4/ε4 and ε4/ε3 genotype and restricting gender substantially reduces the number of subjects (4 control, 26 MCI and 15 AD females; 13 control, 42 MCI and 22 AD males), this observation on gene/gender interaction is suggestive at the best.
A limitation of our investigation is the potential confounding effect of pharmacotherapy as subjects were not drug naïve at the time of CSF collection, though the use of many CNS-active drugs such as antidepressants with anti-cholinergic properties, narcotic analgesics, neuroleptics with anti-cholinergic properties or anti- Parkinsonian medications were excluded. Data from our previous study in PD suggest that α-synuclein CSF levels are not influenced by treatment with dopamine-specific drugs [24]. However, the effect of other drugs, including anti-AD drugs such as galantamine, donepezil, memantine and rivastigmine, on α-synuclein CSF levels remains an unknown, potentially confounding factor.
In conclusion, our results support the argument that α-synuclein plays a significant role in AD pathogenesis even during its early phase, i.e. MCI. Additionally, when in combination with other markers, α-synuclein likely contributes to AD diagnosis or monitoring of its progression. The exact role of the protein in this regard is not known, and definitive interpretations of changes in CSF analyte levels require precise knowledge of the site of its formation, as well as all processes that control its concentration in CSF [41]. The effect of APOE genotype on CSF α-synuclein is not readily observed. Additional, longitudinal studies on the pathological role and biomarker potential of CSF α-synuclein levels in MCI and AD are needed.
Supplementary Material
Acknowledgments
Data collection and sharing was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health (NIH) Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. Finally, the authors’ efforts were supported by the NIH: P42 ES004696-5897 and P30 ES007033-6364; NIA: R01 AG033398; NIEHS: R01 ES016873 and R01 ES019277; NINDS: R01 NS057567, P50 NS062684-6221 and U01 NS082137.
Footnotes
The authors declare no conflict of interest.
References
- 1.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol. 1999;45:353–357. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–296. doi: 10.1016/s0006-8993(00)03082-1. [DOI] [PubMed] [Google Scholar]
- 5.Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, Thal LJ. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology. 1998;51:351–357. doi: 10.1212/wnl.51.2.351. [DOI] [PubMed] [Google Scholar]
- 6.Parkkinen L, Soininen H, Alafuzoff I. Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:363–367. doi: 10.1093/jnen/62.4.363. [DOI] [PubMed] [Google Scholar]
- 7.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 8.Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Goncalves SA, Pera M, Suarez-Calvet M, Clarimon J, Outeiro TF, Lleo A. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One. 2011;6:e26609. doi: 10.1371/journal.pone.0026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jellinger KA. Interaction between alpha-synuclein and other proteins in neurodegenerative disorders. Scientific World Journal. 2011;11:1893–1907. doi: 10.1100/2011/371893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Yuan JX, Masliah E. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freichel C, Neumann M, Ballard T, Muller V, Woolley M, Ozmen L, Borroni E, Kretzschmar HA, Haass C, Spooren W, Kahle PJ. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol Aging. 2007;28:1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100:285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- 15.van den Berge SA, Kevenaar JT, Sluijs JA, Hol EM. Dementia in Parkinson’s Disease Correlates with alpha-Synuclein Pathology but Not with Cortical Astrogliosis. Parkinsons Dis. 2012;2012:420957. doi: 10.1155/2012/420957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesne SE. Soluble alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang DS, Bennett DA, Mufson E, Cochran E, Dickson DW. Decreases in soluble alpha-synuclein in frontal cortex correlate with cognitive decline in the elderly. Neurosci Lett. 2004;359:104–108. doi: 10.1016/j.neulet.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 18.Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, Ng J, Schulz-Schaeffer W, Kretzschmar HA, McLean PJ, Trenkwalder C, Sarracino DA, Vonsattel JP, Locascio JJ, El-Agnaf OM, Schlossmacher MG. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213:315–325. doi: 10.1016/j.expneurol.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Reesink FE, Lemstra AW, van Dijk KD, Berendse HW, van de Berg WD, Klein M, Blankenstein MA, Scheltens P, Verbeek MM, van der Flier WM. CSF alpha-synuclein does not discriminate dementia with Lewy bodies from Alzheimer’s disease. J Alzheimers Dis. 2010;22:87–95. doi: 10.3233/JAD-2010-100186. [DOI] [PubMed] [Google Scholar]
- 20.Tateno F, Sakakibara R, Kawai T, Kishi M, Murano T. Alpha-synuclein in the cerebrospinal fluid differentiates synucleinopathies (Parkinson Disease, dementia with Lewy bodies, multiple system atrophy) from Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26:213–216. doi: 10.1097/WAD.0b013e31823899cc. [DOI] [PubMed] [Google Scholar]
- 21.Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, Hansson O, Nielsen HM. Low CSF Levels of Both alpha-Synuclein and the alpha-Synuclein Cleaving Enzyme Neurosin in Patients with Synucleinopathy. PLoS One. 2013;8:e53250. doi: 10.1371/journal.pone.0053250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall S, Ohrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, Nilsson C, Widner H, Decraemer H, Nagga K, Minthon L, Londos E, Vanmechelen E, Holmberg B, Zetterberg H, Blennow K, Hansson O. Accuracy of a Panel of 5 Cerebrospinal Fluid Biomarkers in the Differential Diagnosis of Patients With Dementia and/or Parkinsonian Disorders. Arch Neurol. 2012;69:1445–1452. doi: 10.1001/archneurol.2012.1654. [DOI] [PubMed] [Google Scholar]
- 23.Ohrfelt A, Grognet P, Andreasen N, Wallin A, Vanmechelen E, Blennow K, Zetterberg H. Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett. 2009;450:332–335. doi: 10.1016/j.neulet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Leverenz JB, Baird G, Montine TJ, Hancock AM, Hwang H, Pan C, Bradner J, Kang UJ, Jensen PH, Zhang J. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133:713–726. doi: 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bretsky PM, Buckwalter JG, Seeman TE, Miller CA, Poirier J, Schellenberg GD, Finch CE, Henderson VW. Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis Assoc Disord. 1999;13:216–221. doi: 10.1097/00002093-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Payami H, Zareparsi S, Montee KR, Sexton GJ, Kaye JA, Bird TD, Yu CE, Wijsman EM, Heston LL, Litt M, Schellenberg GD. Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: a possible clue to the higher incidence of Alzheimer disease in women. Am J Hum Genet. 1996;58:803–811. [PMC free article] [PubMed] [Google Scholar]
- 27.Shan S, Hong-Min T, Yi F, Jun-Peng G, Yue F, Yan-Hong T, Yun-Ke Y, Wen-Wei L, Xiang-Yu W, Jun M, Guo-Hua W, Ya-Ling H, Hua-Wei L, Ding-Fang C. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiol Aging. 2011;32:443–458. doi: 10.1016/j.neurobiolaging.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69:570–580. doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, Locascio JJ, Schlossmacher MG, El-Agnaf OM. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun. 2006;349:162–166. doi: 10.1016/j.bbrc.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Bellani S, Sousa VL, Ronzitti G, Valtorta F, Meldolesi J, Chieregatti E. The regulation of synaptic function by alpha-synuclein. Commun Integr Biol. 2010;3:106–109. doi: 10.4161/cib.3.2.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 34.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 35.Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, Walter JW, Nance MA, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Small GW, Mastaglia F, Haines JL, Pericak-Vance MA, Vance JM. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62:2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Chen PC, Poole C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology. 2004;62:2198–2202. doi: 10.1212/01.wnl.0000130159.28215.6a. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Chen P, Kaufer DI, Troster AI, Poole C. Apolipoprotein E and dementia in Parkinson disease: a meta-analysis. Arch Neurol. 2006;63:189–193. doi: 10.1001/archneur.63.2.189. [DOI] [PubMed] [Google Scholar]
- 38.Williams-Gray CH, Goris A, Saiki M, Foltynie T, Compston DA, Sawcer SJ, Barker RA. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J Neurol. 2009;256:493–498. doi: 10.1007/s00415-009-0119-8. [DOI] [PubMed] [Google Scholar]
- 39.Ezquerra M, Campdelacreu J, Gaig C, Compta Y, Munoz E, Marti MJ, Valldeoriola F, Tolosa E. Lack of association of APOE and tau polymorphisms with dementia in Parkinson’s disease. Neurosci Lett. 2008;448:20–23. doi: 10.1016/j.neulet.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 40.Gallardo G, Schluter OM, Sudhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- 41.Potter WZ. Mining the secrets of the CSF: developing biomarkers of neurodegeneration. J Clin Invest. 2012;122:3051–3053. doi: 10.1172/JCI65309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.