

Abstract
The pathogenic protozoa responsible for malaria lack enzymes for the de novo synthesis of purines and rely on purine salvage from the host. In Plasmodium falciparum (Pf), hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT) converts hypoxanthine to inosine monophosphate and is essential for purine salvage making the enzyme an anti-malarial drug target. We have synthesized a number of simple acyclic aza-C- nucleosides and shown that some are potent inhibitors of Pf HGXPRT while showing excellent selectivity for the Pf versus the human enzyme.
Keywords: Phosphoribosyltransferase, malaria, HGXPRTase, purine salvage, protozoa
Introduction
Malaria is endemic in many parts of the world and there were over 200 million cases resulting in >600,000 deaths in 2010. 1 The disease is caused by Plasmodium protozoa of which Pf is the most deadly. Drug resistance 2 and the limitations of current vaccines 3 makes new drug development important for effective malaria control. Several pathogenic protozoa lack the enzymes for de novo synthesis of purines and are dependent on the purine salvage pathway. 4-6 In Pf purine salvage requires purine nucleoside phosphorylase (PfPNP) to generate hypoxanthine and then hypoxanthine-guanine-xanthine phosphoribosyltransferase (Pf HGXPRT) to convert the hypoxanthine to inosine monophosphate. 7 Inhibition of PfPNP has been shown to kill Pf in cell culture8 and in an Aotus monkey model. 9 We have been interested in extending this work to the discovery of inhibitors of Pf HGXPRT as they are also expected to be lethal to the parasite. A number of purine analogues were reported some time ago as potential inhibitors of Plasmodium berghei, but none showed significant inhibition of the enzyme.10-11
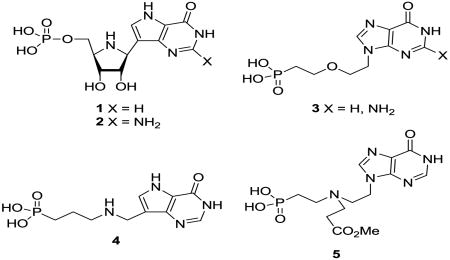
Pf HGXPRT is an N-ribosyltransferase and the transition states of such enzymes have been characterized with strong ribooxacarbenium ion properties and low bond order to both the purine and the nucleophile. 12-13 Kinetic commitment factors have prevented the transition state analysis of Pf HGXPRT to date but it would be reasonable to predict a dissociative transition state with ribooxacarbenium ion character similar to that shown (Scheme 1). Enzyme inhibitor design based on incorporating features of the transition state will often lead to exceptionally potent compounds. 14-18 Immucillin-H (and G) 5′-phosphate 1 and 2 are transition state analogue inhibitors of Pf HGXPRT and also of the human hypoxanthine-guanine phosphoribosyltransferase (HGPRT). 19-21 They mimic the ribooxacarbenium ion positive charge as well as other substrate properties. However, they are not selective for the parasite enzyme, would be expensive to synthesize and are unlikely to penetrate the erythrocyte and parasite cell membranes – so ruling them out as potential malaria therapeutics.
Scheme 1.

Possible transition state of the HGXPRTase-catalysed reaction.
We have been interested in extending inhibitors 1 and 2 to compounds that are easier to synthesize and with selectivity for the parasite over the human enzyme and were intrigued by a report of some acyclic nucleoside phosphonates (such as 3) which were moderate inhibitors of Pf HGXPRT with some selectivity over the human enzyme. 22 We envisaged that acyclic nucleoside phosphonates such as 4 with a nitrogen to mimic the ribooxacarbenium ion with a methylene bridge to a 9-deazapurine might capture some transition state features. After this work was complete, some nitrogen-containing acyclic nucleoside phosphonates were described (e.g 5)23 which offered some improvements but failed to capture the full benefits of the nitrogen. Here we present the synthesis and enzyme inhibition properties of a number of acyclic aza-C-nucleoside phosphonates and phosphates; some of which are potent and selective inhibitors of Pf HGXPRT. A preliminary account of the biology of some of these compounds has been reported.24
Results and Discussion
Target Selection and Synthesis
We decided to prepare a series of linear aminophosphonates and to this end the ccommercially available bromoalkyl phthalamides 6 were treated with triethyl phosphite and then hydrazine hydrate25 to give the known aminophosphonates 7. 26 Coupling of aldehyde 827 with these aminophosphonates and reduction o the resulting imines afforded 9 – 13 (Scheme 2). Acidic hydrolysis of the O-benzyl group in the deazapurines provided 14 – 18 and then dealkylation of the phosphonate ester groups gave the target aza-C-nucleoside mimics 4, 19 – 22. In a similar manner the 8-azapurine analogue 24 was also prepared from 23. While it was likely that the positioning of the nitrogen in compounds 4, 19 – 22 was optimal for mimicking the positive charge of a ribooxacarbenium ion transition state, we decided to explore the effect of moving the nitrogen along the chain. To this end, treatment of 2528 with dimethylamine and formaldehyde in a Mannich reaction afforded 26 (Scheme 3), which after reaction with methyl iodide and then basic nitromethane gave 27. Reduction of the nitro group generated amine 28 which underwent reductive alkylation with phosphonate aldehydes 29 to give 30. Deprotection as before gave the targets 31 and 32. It was possible that substitution of the aminophosphonate alkyl chain with hydroxy or hydroxymethyl groups would offer improved properties as ribose mimics and so the known azide (±)-3329 was reduced to the aminophosphonate (±)-34 (Scheme 4). In addition the monobenzyl ether 3530 was converted into bromide (±)-36 and then the phosphonate (±)-37, from which the aminophosphonate (±)-41 was derived. Reductive alkylation of aminophosphonates (±)-34 and (±)-41 with 8 gave, after deprotection, the targets (±)-44 and (±)-45.
Scheme 2.

(a) triethyl phosphite, 120°C; (b) H2NNH2, EtOH; (c) NaBH4, EtOH; (d) 35% aq HCl, 60 °C; (e) 48% HBr 90 °C; (f) 2-picoline borane, MeOH.
Scheme 3.

(a) HCHO, Me2N.HCl, NaOAc; (b) MeI; (c) CH3NO2, NaOMe; (d) CoCl2, NaBH4; (e) (EtO)3P, 160 °C; (f) 1M HCl, reflux; (g) NaBH4, EtOH; (h) conc HCl, 60 °C; (i) 48% HBr, 90 °C.
Scheme 4.

(a) H2, Pd/C; (b) i, NaH, TBDMSCl, ii, Ph3P, CBr4; (c) (EtO)3P, 175 °C; (d) MeOH/35% aq HCl; (e) MsCl, Et3N; (f) NaN3, DMF, 80 °C; (g) 2-picoline borane, 8, EtOH; (h) i, 35% aq HCl, 70 °C, ii, 48% HBr, 90 °C.
We have observed when designing inhibitors of nucleoside phosphorylases and hydrolases that serinol can be a good mimic of a ribooxacarbenium ion. 14,31-32 Consequently we wanted to make serinol phosphonate analogues. For these compounds we chose to derivatise 9-deaza-2,6-dichloropurine (46) via 47 and 48 into aldehyde 49 (Scheme 5). This was done with the intention that both 9-deazahypoxanthine and 9- deazaguanine derivatives would be synthesized from 49 or its derivatives. Unfortunately attempted displacements of the Cl by NH3 were unsuccessful for those compounds (data not given). The D-serine-derived vinyl phosphonate 5033 was reduced and deprotected to aminophosphonate 51 which was coupled with aldehyde 49 to give 52. This was hydrogenolyzed and further deprotected to the target 53. The L-serine-derived phosphonate 54 and the fluorophosphonate 58 were processed in the same way to the enantiomer 57 and the monofluoro compound 61, while the difluorophosphonate 6234-35 similarly afforded 65. We wanted to be sure that partial racemization had not occurred during reduction of the double bond in 50 and to this end we prepared 53 by an alternative method. Treatment of di-tert-butylphosphite with sodium hydride and methyl iodide gave 66, and after deprotonation of 66 the anion was condensed with epoxide 67 to give 68. This was converted to the amine 69 under conditions for inversion of configuration and the amine was converted into 53 as before. Both preparations of 53 had the same [α]D value. Additionally the 9-deazaguanine derivative 7036 was coupled to difluorophosphonate 63 (Scheme 6) to afford, after deprotection, the alternative deazaguanine-containing inhibitor 72.
Scheme 5.

(a) KO tBu, THF; (b) K2CO3, HCHO, aq dioxane; (c) Dess-Martin periodinane, Et3N, THF; (d) H2, Pd/C; (e) aq HCl; (f) 49, 2-picoline borane, MeOH; (g) 8, 2-picoline borane, MeOH; (h) 48% HBr, 80 °C; (i) NaH, MeI; (j) BuLi, BF3.OEt2; (k) i, iPrOCON2COOiPr, (PhO)2P(O)N3, Ph3P; ii, LiAlH4.
Scheme 6.

(a) 2-picoline borane, Et3N, MeOH; (b) 48% HBr 80 °C.
We decided to make the corresponding phosphate esters of some of the phosphonates described above to compare the relative inhibitory properties. Reductive amination of aldehyde 8 with ethanolamine afforded 73 (Scheme 7), which was phosphorylated to 74 and deprotected to phosphate ester 75. Treatment of the Fmoc-protected serinol 7637 with mixture of iodine and trimethylphosphite38 afforded the monophosphate (±)-77 in moderate yield which was deprotected to the serinol phosphate (±)-78. Reductive amination of aldehyde 8 with amine (±)-78 gave (±)-79 which was hydrogenolyzed to the serinol target (±)-80. In addition, use of aldehyde 49 in the reductive coupling step followed by acid hydrolysis gave the 2-chloropurine derivative (±)-81. The Tris derivative 8239 was also phosphorylated and deprotected to the aminoalkyl phosphate 83 which was treated as described for 78 to give 84. The chiral serinol phosphate esters were also prepared; phosphorylation of serinol derivative 8540-41 and deprotection gave aminoalcohol phosphate 86 which was coupled with the appropriate 9-carbaldehyde-9-deazapurine derivatives (8 and 70) under reductive conditions to afford, after deprotection, targets 87 and 88. The enantiomers 89 and 90 were also prepared from the corresponding enantiomer of 8641 in the same way.
Scheme 7.

(a) ethanolamine, 2-picoline borane; (b) i, Boc2O, ii, (BnO)2PN(iPr)2, tetrazole, iii, tBuOOH; (c) H2, Pd/C; (d) 80% aq TFA; (e) P(OMe)3, I2, py; (f) i, TmsBr, ii, piperidine; (g) NaBH3CN, 8, Et3N, MeOH 50 °C; (h) 49, 2-picoline borane, Et3N, MeOH; (i) i, (BnO)2PNiPr2, tetrazole, ii, MCPBA.
The aminoalcohol 9142 (Scheme 9) can be viewed as a chain extended serinol derivative. It was converted using standard procedures into carbamate 92 and then phosphate 93, the diol 94 and finally the chain-extended target 95.
Scheme 9.

(a) TBDPSCl, imidazole; (b) BnOCOCl, NaHCO3; (c) HOAc/H2O/THF; (d) BzCl, py; (e) Me2CO, Me2C(OMe)2, TsOH; (f) NaOMe, MeOH; (g) (BnO)2PNiPr2, tetrazole, then MCPBA; (h) Bu4NF, THF; (i) TFA/THF/H2O; (j) H2, Pd black; (k) NaBH3CN, 8, Et3N, MeOH.
Biological Results
The compounds were assayed as inhibitors of PfHGXPRT using spectrophotometric methods to observe the conversion of either xanthine or guanine to xanthosine-5′-monophosphate or guanosine-5′-monophosphate in the presence of 5-phospho-α-D-ribose-1-pyrophosphate. Analysis of the results from the simple phosphonates with different chain lengths (4, 19 – 22) showed that 4 is a potent inhibitor and with excellent selectivity for the parasite over the human enzyme. That 4 is the most potent is consistent with the fact that there are the same number of bonds between the phosphonate and the deazapurine in 4 as in immucillin-H phosphate 1. Moving the nitrogen along the chain (in 31) was not beneficial and neither was incorporating the 8-azapurine (in 24). Adding the hydroxyl group to the phosphonate alkyl chain (in 44) may have conferred a small benefit in that 44 was made as the racemate and so one of the enantiomers may be better. However, it is a small benefit compared to the extra costs of introducing the chirality. Interestingly the selectivity margin of 44 was even better than for 4. Adding a hydroxymethyl group (in 45) was not beneficial for inhibitor potency, however the serinol phosphonate 53 proved to be an exceptionally potent inhibitor, showing again that serinol can act as an effective mimic for the ribooxacarbenium ion while the selectivity for Pf over Hs enzymes was a factor of >500 for this compound. The enantiomer 57 was >30 times less active. Use of α-fluorophosphonates and α-α-difluorophosphonates has been proposed to change the pKa of the phosphonates to make them better mimics of phosphates, but in this situation the corresponding compounds 61 and 65 were less potent. Use of a deazaguanine moiety did not confer an advantage either, as in 72. Making the phosphate analogue 75 of phosphonate 4 resulted in a surprising loss in activity with the phosphonate clearly being superior in activity. The phosphate analogues of the serinol phosphonates showed a similar but less dramatic trend with all being less potent than the corresponding phosphonates.
Conclusions
We have synthesized and tested a range of acyclic aza-C-nucleoside phosph(on)ates and we discovered some relatively simple compounds that are potent and selective inhibitors of the Pf HGXPRT enzyme. These compounds offer promise as potential therapeutics for malaria by blocking purine salvage. The next step will be to devise a membrane permeable prodrug approach that will deliver drug through the erythrocyte and into the parasite. We have described34 some preliminary experiments to this end and further work will be published in due course.
Experimental
General Methods
Chromatography solvents are distilled prior to use. Anhydrous solvents are those commercially available. Organic solutions are dried over anhydrous MgSO4 and evaporated under reduced pressure. Air sensitive reactions are performed under Ar. Analytical TLC is performed on Merck pre-coated silica gel 60 F254, detection by UV absorption and/or by heating after dipping in a solution of (NH4)6Mo7O24·4H2O (5 wt%) and Ce(SO4)2 ·4 H2O (0.1 wt%) in 5% aq. H2SO4. Flash column chromatography is performed on silica gel (40-63 μm) or on an automated system with continuous gradient facity. 13C-, 31P- and 19F-NMR spectra are 1H decoupled, chemical shifts are in ppm and coupling constants in Hz if not already stated. 1H NMR in CDCl3 (internal TMS, δ 0), CD3OD (internal TMS, δ 0), DMSOd6 (internal TMS, δ 0) or D2O (internal HOD)13C NMR in CDCl3 (centre line of CDCl3), DMSOd6 (centre line of DMSOd6) or D2O, 31P NMR in CDCl3 or D2O (external H3PO4, δ 0), 19F NMR in CDCl3 or D2O (external CHF3, δ 0). Assignments of 1H and 13C resonances are based on 2D (1H -1H DQF-COSY, 1H -13C HSQC, HMBC) and DEPT experiments. High resolution positive and negative electrospray mass spectra (ESI-HRMS) are recorded on a Q-TOF Premier tandem mass spectrometer. Melting points are uncorrected. Microanalyses are performed by the Campbell Microanalytical Laboratory, University of Otago, New Zealand.
Diethyl {[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}(methyl)amino]methyl}phosphonate (14)
Diethyl aminomethylphosphonate (7 n=1, Chromatography solvents are distilled prior to use. Anhydrous solvents are those 98.5 mg, 1.5 eq.) and the aldehyde 827 (100 mg, 1 eq.) are heated to 60 °C in EtOH (6 mL) for 30 min until dissolved. The solution is then cooled to room temperature, NaBH4 (29.5 mg, 2eq) is added and the mixture is stirred for 30 min. The mixture is evaporated onto silica gel and flash chromatography (9/1/0.1 v/v/v DCM/MeOH/conc. NH3), gives diethyl [({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7- yl]methyl}amino)methyl]phosphonate (9), (76 mg). This product is heated with conc. HCl (0.5 mL) at 60 °C for 1 h and the mixture is evaporated in vacuo twice from water and purified by flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc. NH3) to give 14, (45 mg, 36%). 1H NMR (500 MHz, MeOD) δ 7.93 (s, 1H), 7.51 (s, 1H), 4.20-4.14 (m, 4H), 3.26-3.35 (m, 4H), 3.34 (m, 6H). 13C NMR (125MHz, MeOD) δ155.8, 145.0,143.3, 129., 119.5, 111., 64.4, 44.5, 44.4, 43.5, 42.3, 16.7. 31P NMR (202 MHz, MeOD) δ 22.5. ESI-HRMS for C12H9N4O4NaP [M+Na] + calcd 337.1042; found 337.1039.
Diethyl {2-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7- yl}methyl)amino]ethyl}phosphonate (15)
Using the procedure above for the preparation of compound 14, compound 7, (n=2) is converted to title compound 15. 1H NMR (500 MHz, DMSO-d6) δ 7.77 (s,1H), 7.27 (s, 1H), 3.95 (m, 4H), 3.76 (s, 2H), 2.72 (m, 2H), 2.50 (s, 2H),1.92 (m, 2H), 1.19 (m, 6H). 13C NMR (125MHz, MeOD) δ153.6, 143.0, 141.3, 125., 117.6, 115.3, 60., 42.1, 26.0, 24.9, 16.2. 31P NMR (202 MHz, DMSO-d6) 630.3. ESI-HRMS or C13H21N4O4NaP [M+Na]+ calcd 351.119; found 351.1194.
Diethyl {2-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]propyl}phosphonate (16)
Using the procedure above for the preparation of compound 14, compound 7, (n=3) is converted to title compound 16. 1H NMR (500 MHz, D 2O) δ 8.79 (s,1H), 7.70 (s,1H), 4.29 (s, 2H), 3.98-3.92 (m, 4H), 3.07- 3.04 (m,2H), 1.86-183 (m, 2H), 1.16-1.10 (m,6H). 13C NMR (125MHz, D2O) δ 152.7, 144.8, 133.0, 132.6, 118.2, 103.3, 63.6, 46.7, 40.4, 21.8, 20.7, 18.9, 15.6. 31P NMR (202 MHz, D2O) δ 33.4. ESI-HRMS for C14H24N4O4P [M+H]+ calcd 343.1535; found 343.1528.
Diethyl {2-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]butyl}phosphonate (17)
Using the procedure above for the preparation of compound 14, compound 7, (n=4) is converted into first diethyl [4-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino) butyl]phosphonate (12). 1H NMR (500 MHz, CDCl3) δ 8.46 (s, 1H), 7.72 (s, 1H), 7.44-7.27 (bm, 5H), 5.53 (s, 2H), 4.19 (s, 2H), 4.03-3.98 (m, 4H), 2.80-2.77 (m, 2H), 1.72-1.51 (bm, 6H), 1.30-1.23 (m, 6H). 13C NMR (125MHz, CDCl3) δ 149.7, 139.4, 130.5, 129.3, 128.5, 128.4, 128.2, 128.1, 127.7, 115.0, 108.5, 67.7, 61.6, 47.1, 41.5, 27.8, 27.7, 25.6, 24.5, 20.9, 16.4. 31P NMR (202 MHz, CDCl3) δ 31.6. ESI-HRMS for C22H32N4O4P [M+H]+ calcd 447.2161; found 447.2153. his is then converted to title compound 17. 1H NMR (500 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.78 (s, 1H), 4.36 (s, 2H), 4.01-4.07 (m, 4H), 3.10-3.08 (m, 2H), 1.91-1.85 (m, 2H), 1.81-1.75 (m, 2H), 1.65-1.54 (m, 2H), 1.26-1.20 (m, 6H).
Diethyl {2-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]pentyl}phosphonate (18)
Using the procedure above for the preparation of compound 14, compound 7, (n=5) is converted first into diethyl [5-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino) pentyl]phosphonate (13). 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H), 7.33 (s, 1H), 7.47-7.28 (bm, 5H), 5.57(s, 2H), 4.09-4.00 (m, 4H), 4.01(s, 1H), 1/70-1.51 (bm, 6H), 1.39-1.35(m, 2H), 1.32-1.27 (m, 6H). 13C NMR (125MHz, CDCl3) δ 155.4, 149.5, 149.1, 136.3, 128.5, 128.4, 128.3, 128.0, 127.7, 115.3, 113.8, 67.8, 61.4, 48.8, 43.2, 29.0, 28.2, 26.0, 25.0, 22.2, 16.4. 31P NMR (202 MHz, CDCl3) δ 32.2. ESI-HRMS for C23H34N4O4P [M+H]+ calcd 461.2318; found 461.2311. This is then converted to title compound 18. 1H NMR (300 MHz, D2O) δ 8.53 (s, 1H), 7.68 (s,1H), 4.36 (s, 2H), 4.11-3.97(m, 4H), 3.09-3,01 (m, 2H), 1.90-1.40 (bm, 8H), 1.28-1.22 (m, 6H).
{[({4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]methyl}phosphonic acid (19)
The diethyl phosphonate (14, 43mg) is heated with 48% HBr for 5 h at 90 °C and the solution is evaporated in vacuo twice from water. Reverse phase (C18) chromatography eluting with water gives title compound (19, 40 mg) as a white water insoluble solid. 1H NMR (500 MHz, DMSO-d6) δ 8.75 (s, 1H), 7.73 (s, 1H), 3.39 (s, 2H), 3.36 (s, 2H). 13C (125 MHz, DMSO-d6) δ 152.3, 144.0, 137.2, 130.7, 117.7, 104.3, 42.2, 41.0. 31P (202 MHz, DMSO-d6) δ 11.7. ESI-HRMS f or C8H12N4O4P [M+H]+ calcd 259.0596; found 259.0599.
{[({4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]ethyl}phosphonic acid (20)
Using the procedure for the preparation of compound 19, compound 15 is converted to title compound 20. 1H NMR (500 MHz, D2O) δ 8.69 (s, 1H), 7.69 (s,1H), 4.34 (s,2H), 3.23-3.19 (m, 2H), 2.02-1.95 (m, 2H). 13C NMR (125MHz, D2O) δ 153.2, 144.5, 134., 132.3, 118.2, 103.6, 42.0, 40.2, 25.0, 23.9. 31P NMR (202 MHz, D2O) δ 20.6. ESI-HRMS for C9H12N4O4P [M-H]- calcd 271.0596; found 271.0594.
{[({4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]propyl}phosphonic acid (4)
Using the procedure for the preparation of compound 19, compound 16 is converted to title compound 4. 1H NMR (500 MHz, D2O) δ 8.68 (s, 1H), 7.69 (s, 1H), 4.30 (s, 2H), 3.07-3.01 (m, 2H), 1.88-1.80 (m, 2H), 1.74-1.68 (m 2H). 13C NMR (125MHz, D2O) δ 152.5, 144.9, 132.8, 131.8, 118.3, 103.0, 47.1, 40.5, 24.1, 23.0, 19.4. 31P NMR (202 MHz, D2O) δ 29.2. ESI-HRMS for C10H16N4O4P [M+H]+ calcd 287.0909; found 287.0904.
{[({4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]butyl}phosphonic acid (21)
Using the procedure for the preparation of compound 19, compound 17 is converted to title compound 21. 1H NMR (500 MHz, D2O) δ 8.93 (s, 1H), 7.75 (s, 1H), 4.31 (s, 2H), 3.02-3.06 (m, 2H), 1.75-1.67 (m, 4H), 1.55-1.51 (m, 2H). 13C NMR (125 MHz, D2O) δ 152.5, 144.9, 132.8, 131.7, 118.3, 103.0, 46.6, 40.4, 26. 3, 26.2, 25.0, 19.3. 31P NMR (202 MHz, D2O) δ 31.2. ESI-HRMS or C11H18N4O4P [M+H]+ calcd 301.1066; found 301.1064.
{[({4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]pentyl}phosphonic acid (22)
Using the procedure for the preparation of compound 19, compound 18 is converted to title compound 22. 1H NMR (500 MHz, D2O) δ 8.93 (s, 1H), 7.76 (s, 1H), 4.31 (s, 2H), 3.01-3.05 (m, 2H), 1.71-1.58 (bm, 4H), 1.49-1.42 (m, 2H), 1.38-1.32 (m, 2H). 13C NMR (125MHz, D2O) δ 152.4, 144.9, 132.8, 131.7, 118.3, 103.1, 46.9, 40.3, 26.6, 26.5, 26.3, 25.1, 21.4. 31P NMR (202 MHz, D2O) δ 32.3. ESI-HRM for C12H20N4O4P [M+H]+ calcd 315.1222; ound 315.1221.
3-((7-Hydroxy-1H-pyrazolo[4,3-d]pyrimidin-3-yl)methylamino)propylphosphonate ammonium salt (24)
Anhydrous HCl in MeOH (1.0 M, 0.11 mL, 0.11 mmol) is added dropwise to a solution of the diethyl phosphonate (7, n=3) (100 mg, 0.51 mmol) in anhydrous MeOH (2 mL) followed by the addition of the aldehyde 2324 (90 mg, 0.34 mmol) and 2-picoline borane complex (50 mg, 0.44 mmol). The resulting suspension is left to stir for 2 h and the then homogeneous solution absorbed onto silica gel and the resulting residue subjected to flash chromatography (EtOAc then 5 to 10% v/v MeOH in CHCl3) to afford diethyl 3-((7-methoxy-2-(tetrahydro-2H-pyran-2-yl)-2H-pyrazolo[4,3-d]pyrimidin-3-yl)methylamino)propylphosphonate (123 mg, 82 % yield) as a white solid, which is committed to the next step without further characterisation or purification. Concentrated HCl (1.5 mL, 18 mmol) is added to a solution of this material (90 mg, 0.204 mmol) in MeOH (3 mL, 0.204 mmol) and the resulting mixture heated to reflux for 6 h. The reaction mixture is concentrated in vacuo and partitioned between chloroform and water, the water layer is extracted with additional chloroform, the organic layers are combined, dried and concentrated in vacuo. The residue is dissolved in aqueous hydrobromic acid (2 mL, 17.68 mmol) and heated for a further 6 h at 80 °C. The reaction mixture is concentrated in vacuo and the resulting residue co-distilled with water (2 × 20 mL). Flash chromatography (1:1 v/v dioxane/conc. NH4OH) afforded the title compound 24 (35 mg, 60%) as a foam. 1H NMR (500 MHz, D2O): δ = 7.98 (s, 1H), 4.47 (s, 2H), 3.16 (t, J = 6.9 Hz, 2H), 1.91 (septet, J = 7.0 Hz, 2H), 1.56 (m, 2H). 13C NMR (125 MHz, D2O): δ = 155.2, 148.4, 137.5, 135.6, 128.6, 66.6, 48.7, 48.6 (d, J = 13.4 Hz), 40.8, 26.1 (d, J = 132 Hz), 25.6, 20.7(d, J = 3.8 Hz). P (202 MHz, D2O): δ = 21.9.
{[4-(Benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}dimethylamine (26)
The deazapurine 2528 (178 mg,1 eq.) in dioxane (1 mL) and water (1 mL) with NaOAc (64.6 mg, 1 eq.), dimethylamine hydrochloride (64.6 mg,1 eq.) and formaldehyde (65.5 μL, 1.1 eq., 37% aq) is heated at 95 °C for 4 h and then evaporated under reduced pressure. The residue is evaporated twice from ammonia in MeOH onto silica gel. Flash chromatography (DCM/MeOH-NH3 9:1 v/v) gives title compound 26 (163 mg). 1H NMR (500 MHz, MeOD) δ 8.42 (s, 1H), 7.54 (s, 1H), 7.53-7.31 (m, 5H), 5.68 (s, 2H), 3.75 (s, 2H), 2.28 (s, 6H). 13C NMR (125 MHz, MeOD) δ 157.2, 150.3, 137.8, 131.8, 129.4, 116.4, 112.1, 69.1, 52.4, 44.8. ESI-HRMS for C15H19N4O [M+H]+ calcd 283.1559; found 283.1560.
4-(Benzyloxy)-7-(2-nitroethyl)-5H-pyrrolo[3,2-d]pyrimidine (27)
The dimethylamine 26 (548 mg,1 eq.) in tetrahydrofuran (25 mL) is treated with methyl iodide (1.2 mL, 10 eq.) at room temperature for 18 h and then the solution is evaporated under high vacuum to give an orange foam of the quaternary ammonium salt of 26. Nitromethane (3.34 mL, 30 eq.) is added to methanol (32 mL) and methanolic sodium methoxide (7 mL, 30%, 20 eq.) and the mixture is stirred under argon for 10 min and then the crude quaternary ammonium salt is added. The mixture is stirred for 2 h and then the methanol is evaporated. The residue is diluted with water, the pH adjusted to 3 with 1 N HCl and extracted 3x with ethyl acetate. The organic extract is washed with brine, dried and evaporated. Flash chromatography (20-50% v/v EtOAc in hexanes) gives title compound 27 (310 mg, 53%). 1H NMR (500 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.54 (d, 1H), 7.42- 7.33 (m, 5H), 5.61 (s, 2H), 4.93 (m, 2H), 3.37 (m, 2H). 13C NMR (125MHz, DMSO-d6) δ 154.9, 148.7, 136.6, 128.8, 128.4, 114.4, 109.7, 75.0, 67.0, 22.1. ESI-HRMS for C15H4N4O3Na [M+Na]+ calcd 321.0964; found 321.0970.
2-[4-(Benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl] ethan-1-amine (28)
Sodium borohydride (393 mg, 10 eq.) is added in small portions to 27 (310 mg 1 eq.) in methanol (20 mL) with CoCl2.6H2O (495 mg, 2 eq.) at 0 °C and then the mixture is stirred at 0 °C for 30 min. The product is evaporated onto silica gel and flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc. NH3) gives title compound 28 (195 mg, 70%). 1H NMR (500 MHz, MeOD) δ 8.4 (s, 1H), 7.36 (s, 1H), 7.53-7.30 (m, 5H), 5.62 (s, 2H), 2.98-2.92 (m, 4H). 13C NMR (125MHz, MeOD) δ 157.1, 149.8, 137.9, 129,3,129.6,128.3, 116.8, 114.2, 69.1, 43.0, 28.1. ESI-HRMS for C15H17N4O [M+H]+ calcd 269.1402; found 269.1404.
Diethyl (2-oxoethyl)phosphonate (29, n=1) and diethyl (3-oxopropyl)phosphonate (29, n=2)
Triethylphosphite (8.4 mL) and 2-(2-bromoethyl)-1,3-dioxolane (10 g) are heated at 160 °C for 18 h and then evaporated under high vacuum at 100 °C . The residue is refluxed with 1 N HCl under argon for 15 min and cooled to room temperature. The product is salted out with solid NaCl and extracted with DCM, washed with saturated NaHCO3, brine, dried and concentrated. Flash chromatography (EtOAc-MeOH 95:5 v/v) gives title compound 29, n=2 (2.5 g). The NMR spectral data was identical with literature values.44 The same method applied to 2-(bromomethyl)-1,3-dioxolane afforded title compound 29, n=1 with the same NMR data as reported.45
Diethyl [2-({2-[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]ethyl}amino)ethyl]phosphonate (30, n=2)
The amine 28 (67 mg, 15 eq) and phosphonoaldehyde 29, n=1 (40 mg, 1 eq.) in ethanol (2 mL) are heated at 70 °C for 30 min and cooled to room temperature. NaBH4 (17 mg, 2 eq.) is added and the mixture stirred for 30 min. The mixture is evaporated onto silica gel and subjected to flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc.NH3) to give title compound 30, n=2 (30 mg). 1H NMR (500 MHz, CDCl3) δ 8.48 (s, 1H), 7.48-7.27 (m, 5H), 7.19 (s, 1H), 5.55 (s, 2H), 4.07-4.01 (m, 4H), 3.04-2.95 (m, 6H), 2.08-2.02 (m, 2H), 1.28-1.25 (m, 6H). 13C NMR (125 MHz, CDCl3) 155.3, 149.1, 148.8, 136.3, 128.4, 127.4, 115.4, 113.7, 67.9, 61.8, 49.1, 42.8, 26.1, 25.0, 24.0, 16.4. 31P NMR (202 MHz, CDCl3) 29.2. ESI-HRMS for C21H30N4O4P [M+H]+ calcd 433.2005; ound 433.1999.
Diethyl [3-({2-[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]ethyl}amino)propyl]phosphonate(30, n=3)
The amine 28 (60 mg, 1.5 eq.) and phosphonoaldehyde 29, n=2 (36 mg, 1 eq.) in ethanol (2 mL) are heated at 70 °C for 30 min and cooled to room temperature. NaBH4 (15 mg, 2 eq.) is added and the mixture stirred for 30 min. Evaporation onto silica gel and flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc. NH3) gives title compound 30, n=3 (50 mg). 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H), 7.26 (s, 1H), 7.49-7.19 (m, 5H), 5.57 (s, 2H), 4.09- 4.01 (m, 4H), 2.98 (s, 4H), 2.70 (m, 2H), 1.78-1.71 (m, 4H), 1.31-1.26 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 155.3, 149.3, 136.3, 128.5, 126.8, 115.4, 114.8, 67.9, 61.5, 49.7, 49.6, 24.5, 24.0, 22.8, 22.7, 16.4. 31P NMR (202 MHz, CDCl3) δ 32.1. ESI-HRMS for C22H32N4O4P [M+H]+ calcd 447.2161; found 447.2160.
{2-[(2-{4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}ethyl)amino]ethyl}phosphonic acid (31)
The diethyl phosphonate 30, n=2 (52 mg) is heated at 60 °C with conc. HCl (0.5 mL) for 1 h, evaporated under reduced pressure twice from water and the heated with 48% HBr (1 mL) at 90 °C for 5 h and evaporated again twice from water. The product is chromatographed on reverse phase C18 silica gel eluting with water to give title compound 31 (39 mg). 1H NMR (500M Hz, D2O) δ 8.78 (s, 1H), 7.50 (s, 1H), 3.29-3.24 (m, 2H), 3.22-3.18 (m, 2H), 3.05-3.02 (m, 2H), 2.04-1.97 (m, 2H). 13C NMR (125MHz, D2O) δ 152.8, 144.0, 132.2, 130.3, 117.9, 108.0, 47.0, 42.9, 24.9, 23.8, 20.3. 31P NMR (202 MHz, D2O) δ 21.3. ESI-HRMS for C10H16N4O4P [M+H]+ calcd 287.0909; found 287.0912.
{3-[(2-{4-Hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}ethyl)amino]propyl}phosphonic acid (32)
The diethyl phosphonate 30, n=3 (50 mg) is heated at 60 °C with conc. HCl (0.5 mL) for 1 h and then evaporated under reduced pressure twice from water. The residue is then heated with 48% aq. HBr (1 mL) at 90 °C for 5 h and evaporated again twice from water. The product is chromatographed on reverse phase C18 silica gel eluting with water to give title compound 32 (30mg). 1H NMR (500 MHz, D2O) δ 8.76 (s, 1H), 7.49 (s, 1H), 3.26-3.17 (m, 2H), 3.07-2.98 (m, 4H), 1.88-1.79 (m, 2H), 1.74-1.67 (m, 2H). 13C NMR (125MHz, D2O) δ 152.9, 144.0, 132.4, 130.2, 117.9, 108.2, 47.9, 47.2, 24.3, 23.2, 20.3, 19.5. 31P NMR (202 MHz, D2O) δ 28.3. ESI-HRMS or C11H18N4O4P [M+H]+ calcd 301.1066; found 301.1062.
Diethyl (3-amino-2-hydroxypropyl)phosphonate (34)
Diethyl (3-azido-2-hydroxypropyl)phosphonate (33)29 (620 mg) is reduced with 10% palladium on charcoal (250 mg) in ethanol (10 mL) under hydrogen at 1 atm. for 1 h, filtered and concentrated. Flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc. NH3) gives title compound 34 (388 mg). 1H NMR (500 MHz, CDCl3) δ 4.18-4.09 (m, 4H), 3,98-3.92 (m, 1H), 2.88-2.68 (m, 2H), 2.15-1.95 (bm, 3H),1.98-1.89 (m, 2H), 1.35-1.33 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 67.4, 61.9, 48.1, 31.6, 31.0, 16.4. 31P NMR (202 MHZ,CDCl3)δ 30.0. ESI-HRMS or C7H19NO4P [M+H]+ calcd 212.1052; found 212.1049.
[3-(Benzyloxy)-2-(bromomethyl)propoxy](tert-butyl)dimethylsilane (36)
A solution of 3530 (1.04 g,1 eq.) in dry THF (3 mL) is added to NaH (60%, 127 mg, 5.3 eq.) in dry THF (12 mL) with ice cooling. The mixture is stirred at room temperature for 30 min and then tBuMe2SiCl (800 mg, 5.3 eq.) in dry THF (2 mL) is added. After 2 h, water (6 mL) is added and the product extracted with EtOAc, washed with brine, dried and evaporated. Flash chromatography (hexanes-EtOAc 9:1 v/v) gives 2-[(benzyloxy)methyl]-3-[(tert-butyldimethylsilyl)oxy]propan-1-ol (1.25 g). 1H NMR (500 MHz, CDCl3) δ 7.37-7.30 (m, 5H), 4.51 (s, 2H), 3.80-3.71 (m, 4H), 3.64-3.53 (m, 2H), 2.07-2.01 (m, 1H), 0.86 (s, 9H), 0.06 (s, 6H). 13C NMR (125MHz,CDCl3) δ 138.0, 128.4, 127.7, 73.2, 70.0, 64.5, 63.4, 43.0, 25.9, 18.2, 0.0. ES-HRMS or C17H30O3NaSi [M+Na]+ calcd 333.1826; found 333.1858. To this material (1.25 g, 1 eq.) in dry DCM (2 mL) and dry pyridine (0.35 mL) under argon is added CBr4 (2 g, 1.5 eq.) followed by the dropwise addition of triphenylphosphine (1.07 g, 1.02 eq.) in dry DCM (2 mL). After 2.5 h, ice-cold hexanes (25 mL) are added and the precipitate filtered off. Flash chromatography (hexanes and then hexane with 5% v/v EtOAc) of the concentrated filtrate gives title compound 36 (788 mg). 1H NMR (500 MHz, CDCl3) δ 7.32-7.21 (m, 5H), 4.45 (s, 2H), 3.67-3.59 (m, 2H), 3.55-3.41 (m, 2H), 2.16-2.09 (m, 1H), 0.83 (s, 9H), 0.00 (s, 6H). 13C NMR (125 MHz,CDCl3) δ 138.3, 128.4, 127.6, 73.4, 69.1, 61.7, 43.8, 33.0, 25.9, 18.2, 0. ESI-HRMS or C17H29O2NaSi79Br [M+Na]+ calcd 395.1018; ound 395.1020.
Diethyl {2-[(benzyloxy)methyl]-3-[(tert-butyldimethylsilyl)oxy] propyl}phosphonate (37)
Compound 36 (2.46 g, 1 eq.) and triethylphosphite (3.5 mL, 3 eq.) are heated at 175 °C for 18 h. Concentration of the product under high vacuum at 100 °C and flash chromatography (hexanes-EtOAc 20%-100% v/v) of the residue gives title compound 37 (2.07g). 1H NMR (500 MHz, CDCl3) δ 7.29-7.22 (m, 5H), 4.45 (s, 2H), 4.04-4.03 (4H), 3.66-3.62 (m,2H), 3.52-3.50 (m,2H), 2.20-2.12 (m,1H), 1.87-1.72 (m, 2H), 1.28-1.24 (m, 6H), 0.84 (s, 9H), 0.0 (s, 6H). 13C NMR (125 MHz,CDCl3) δ 138.6, 128.3, 127.5, 73.0, 70.2, 63.0, 61.4, 36.6, 25.9, 24.3, 23.1, 18.3, 16.4, 0.0. 31P NMR (202 MHZ, CDCl3) δ.32.0. ESI-HRMS for C21H39O5NaSiP [M+Na]+ calcd 453.2202; found 453.2205.
Diethyl [2-(azidomethyl)-3-(benzyloxy)propyl]phosphonate (40)
Compound 37 (1.87 g) in MeOH (10 mL) and conc. HCl (10 mL) is stirred at room temperature for 1 h. Evaporation at reduced pressure gives crude diethyl {2-[(benzyloxy)methyl]-3-hydroxypropyl}phosphonate (38) (1.45 g) which is dissolved in DCM (30 mL) and triethylamine (1.9 mL, 3 eq.) and cooled to 0 °C. Methanesulfonyl chloride (0.72 mL, 2 eq.) is added and the mixture stirred at room temperature for 1 h, diluted with DCM and the organic extract washed with satd. NaHCO3, brine, dried and evaporated to give crude mesylate 39. Displacement of the mesylate is effected by heating a solution of crude 39 (1.93 g) in DMF (20 mL) with NaN3 (0.9 g) at 80 °C for 7 h. Addition of water and extraction with diethyl ether followed by flash chromatography (50%-100% v/v EtOAc in hexanes) gives title compound 40 (1.06 g). 1H NMR (500 MHz, CDCl3) δ 7.38-7.27 (m, 5H), 4.53 (s, 2H), 4.14-4.03 (m, 4H), 3.58-3.47 (m, 4H), 2.34-2.25 (m, 1H), 1.88-1.76 (m, 2H), 1.35-1.27 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 138.0, 129.6, 128.3, 73.2, 70.2, 61.6, 52.6, 34.4, 25.4, 24.3, 16.4. ESI-HRMS tor C15H24N3O4NaP [M+Na]+ calcd 364.1402; found 364 1393.
Diethyl [2-(aminomethyl)-3-hydroxypropyl]phosphonate (41)
Compound 40 (1.06 g) in MeOH (10 mL) with 20% Pd(OH)2/C (0.5 g) is hydrogenated at 1 atm for 18 h. Filtration and flash chromatography (9/1/0.1 and 8/2/0.2 v/v DCM/MeOH/conc. NH3) gives title compound 41 (300 mg). 1H NMR (500 MHz, CDCl3) δ 4.22-4.05 (m, 4H), 3.83-3.71 (m, 2H), 3.02-2.81 (m, 2H), 2.25-2.15 (m, 1H), 2.10-1.98 (m, 1H), 1.86-1.71 (m, 2H), 1.38-1.31 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 67.4, 61.7, 46.2, 37.1, 26.2, 25.1, 16.5. 31P NMR (202 MHz, CDCl3) δ.31.8. ESI-HRMS for C8H21NO4P [M+H]+ calcd 226.1208; found 226.1206.
Diethyl [3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)-2-hydroxypropyl]phosphonate (42)
The aminophosphonate 34 (95 mg,1.5 eq.) and aldehyde 8 (80 mg, 1 eq.) in ethanol (3 mL) are heated at 70 °C for 10 min and then picoline borane (64 mg, 2 eq.) is added and the mixture stirred for 3.5 h. The product is evaporated onto silica gel and subjected to flash chromatography (9/1/0.1 and 8/2/0.2 v/v/v DCM/MeOH/conc. NH3) to give title compound 42 (20 mg). 1H NMR (500 MHz,CDCl3) δ 8.53 (s, 1H), 7.49-7.35 (m, 5H), 7.26 (s, 1H), 5.58 (s, 2H), 4.16-3.96 (m, 6H), 2.83-2.64 (m, 2H), 2.02-1.87 (m, 2H), 1.34-1.30(m, 6H). 13C NMR (125 MHz, CDCl3)δ 155.4, 149.6, 149.0, 136.2, 128.5, 127.3, 115.4, 115.1, 67.9, 65.0, 61.8, 55.1, 43.0, 32.1, 31.0, 16.4. 31P NMR (202 MHz, CDCl3) δ.29.9. ESI-HRM for C21H30N4O5P [M+H]+ calcd 449.1954; found 449.1951.
Diethyl {2-[({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)methyl]-3-hydroxypropyl}phosphonate (43)
The aminophosphonat e 41 (100 mg, 1.5 eq.) and aldehyde 8 (80 mg, 1 eq.) are heated together in ethanol (3 mL) at 70 °C for 10 min and then picoline borane (64 mg, 2 eq.) is added and heated at 70 °C for 2 h. Evaporation and flash chromatography (9/1/0.1v/v/v DCM/MeOH/conc. NH3) gives title compound 43 (99 mg). 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 7.49-7.33 (m, 5H), 7.26 (s, 1H), 5.58 (s, 2H), 4.13-3.99 (m, 6H), 3.81-3.63 (m, 2H), 3.01-2.81 (m, 2H), 2.26-2.19 (m, 1H), 1.75-1.63 (m, 2H), 1.35-1.24 (m, 6H). 13C NMR (125MHz, CDCl3) δ 155.3, 149.8, 149.1, 136.2, 128.5, 127.3, 115.4, 114.3, 67.8, 61.7, 53.7, 43.3, 34.6, 26.6, 25.5, 16.4. 31P NMR (202 MHz, CDCl3) δ 31.3. ESI-HRMS for C22H32N4O5P [M+H]+ calcd 463.2110; ound 463.2103.
{2-Hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]propyl} phosphonic acid (44)
Compound 42 (20 mg) is heated with conc. HCl (0.5 mL) at 70 °C for 30 min and then evaporated under reduced pressure twice from water. The product is heated with 48% HBr (1 mL) at 90 °C for 5 h and then evaporated under reduced pressure twice from water. Reverse phase chromatography on C18 silica gel eluting with water gives title compound 44 (7 mg). 1H NMR (500 MHz, D2O) δ 8.89 (s, 1H), 7.44 (s, 1H), 4.38 (s, 2H), 4.24-4.10 (m, 2H), 3.27-2.99 (m, 2H), 2.04-1.97 (m, 2H). 13C NMR (125 MHz, D2O) δ 152.7, 144.9, 132.9, 132.4, 118.2, 103.3, 62.3, 52.0, 40.5, 33.2, 32.1. 31P NMR (202 MHz, D2O) δ 25.0. ESI-HRMS for C10H16N4O5P [M+H]+ calcd 303.0858; found 303.0862.
(3-Hydroxy-2-{[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7- yl}methyl)amino]methyl}propyl)phosphonic acid (45)
Compound 43 (45 mg) is heated at 70 °C in conc. HCl (0.5 mL) for 1 h and evaporated twice from water. The crude product is the heated with HBr (48%, 1 mL) at 90 °C for 3 h and then evaporated twice from water. Chromatography on reverse phase C18 silica gel eluting with water gives title compound 45 (23 mg). 1H NMR (500 MHz, D2O) δ 8.19 (s, 1H), 7.58 (s, 1H), 4.30-4.21 (m, 2H), 3.56-3.37 (m, 2H), 3.15-3.04 (m, 2H), 2.19-2.11 (m, 1H), 1.68-1.46 (m, 2H). 13C NMR (125 MHz, D2O) δ 152.6, 144.9, 132.8, 118.4, 103.1, 63.8, 49.8, 40.9, 33.0, 27.3, 26.2. 31P NMR (202 MHz, D2O) δ.27.5. ES-HRMS or C11H18N4O5P [M+H]+ calcd 317.1015; found 317.1007.
4-(tert-Butoxy)-2-chloro-5H-pyrrolo[3,2-d]pyrimidine (47)
2,4-Dichloro-5H- pyrrolo[3,2-d]pyrimidine (46)46 (0.5 g, 2.66 mmol) and sublimed potassium tert-butoxide (1.492 g, 13.30 mmol) are stirred in THF (25 mL) at 40 °C f or 16 h. he solvent is evaporated, H2O (10 mL) added and the mixture extracted with EtOAc (50 mL), washed with brine, then dried and the solvent evaporated to yield a cream coloured solid. Flash chromatography (EtOAc-Hexanes, 3:7 v/v) gives title compound 47 (0.33 g, 55%) as a colourless solid. 1H NMR (500 MHz, CDCl3) 8.58 (bs, exchanged to D2O, 1H), 7.37 (t, J = 3.1, after D2O became d, J = 3.1, 1H), 6.56 (dd, J = 3.1, 2.2, after D2O became d, J = 3.1, 1H), 1.70 (s, 9H). 13C NMR (125.7 MHz, CD3OD) 157.3 (C), 152.4 (C), 150.4 (C), 132.0 (CH), 116.3 (C), 102.0 (CH), 85.0 (C), 28.7 (CH3). Referenced to the centre line of CD3OD at δ 49.0. ESI-HRMS for C10H13 (35)ClN3O [M+H]+ calcd 226.0747; found 226.0752.
[4-(tert-Butoxy)-2-chloro-5H-pyrrolo[3,2-d ]pyrimidin-7-yl]methanol (48)
Compound 47 (0.330 g, 1.462 mmol) is dissolved in a mixture of 1,4-dioxane (6 mL) and H2O (3 mL) then potassium carbonate (0.525 g, 3.80 mmol) and formaldehyde solution (37% aq., 1.53 mL, 19.10 mmol) are added and the mixture heated at 80 °C for 19 h. The mixture is cooled, extracted with EtOAc (50 mL), washed with brine, dried then the solvent evaporated to a gum which is dissolved in 7M NH3-MeOH (10 mL) and left at room temperature for 30 min After evaporation of the solvent the residue is flash chromatographed (EtOAc-hexanes, 7:3 then EtOAc) to give title compound 48 (0.214 g, 57%) as a colourless solid. 1H NMR (500 MHz, CD3OD) 7.51 (s, 1H), 4.75 (s, 2H), 1.72 (s, 9H). 13C NMR (125.7 MHz, CD3OD) 157.3 (C), 150.5(C × 2), 131.0 (CH), 116.8 (C), 116.6 (C), 85.0 (C), 55.2 (CH2), 28.8 (CH3). Referenced to the centre line of CD3OD at 49.0 ppm. ESI-HRMS for C11H14 (35) ClN3NaO2 [M+Na]+ calcd 278.0672; found 278.0671.
4-(tert -Butoxy)-2-chloro-5H-pyrrolo[3,2-d ]pyrimidine-7-carbaldehyde (49)
Dess-Martin periodinane (0.390 g, 0.921 mmol) is added to a mixture of triethylamine (0.47 mL, 3.35 mmol) and alcohol 48 (0.214 g, 0.837 mmol) in THF (6 mL) and the mixture is stirred at room temperature for 40 min. EtOAc (50 mL), sat.aq. NaHCO3 (3 mL) and water (3 mL) are added successively and the mixture filtered through Celite. The organic layer is separated and washed with brine then dried and the solvent evaporated to leave a solid which is dissolved in a 1:1 v/v mixture of MeOH/CHCl3, silica gel added and the solvent evaporated. The residue is flash chromatographed (EtOAc-hexanes,4:6 then 1:1 v/v) to give title compound 49 as a colourless solid which is triturated with a little EtOAc and dried (0.179 g, 84%). A sample is recrystallized from EtOAc. 1H NMR (500 MHz, DMSO d6) 13.08 (bs, 1H), 10.06 (s, 1H), 8.41 (s, 1H), 1.70 (s, 9H). 13C NMR (125.7 MHz, DMSO d6) 183.8 (CH), 156.2 (C), 151.0 (C), 149.6 (C), 137.7 (CH), 116.8 (C), 115.9 (C), 84.5 (C), 28.2 (CH3). Referenced to the centre line of DMSOd6 at δ 39.7. ESI- HRMS for C11H12 (35) ClN3NaO2 [M+Na]+ calcd 276.0516; found 276.0511.
Diethyl [(3S)-3-amino-4-hydroxybutyl]phosphonate hydrochloride (51)
Compound 5033 (0.460 g, 1.27 mmol) and 10% Pd-C (50 mg) are stirred in EtOH (20 mL) under a hydrogen atmosphere at ambient pressure and temperature for 6 h. The mixture is filtered through Celite and the solvent evaporated. The residue is dissolved in a mixture of EtOH (3 mL) and 37% aq. HCl (2 mL) and left at room temperature for 1.5 h then the solvent is evaporated to give title compound 51 as a colourless gum (330 mg, 100%). (c 0.57 MeOH). 1H NMR (500 MHz, D2O) 4.28-4.17 (m, 4H), 3.91 (dd, J = 12.5, 3.7, 1H), 3.74 (dd, J = 12.5, 6.4, 1H), 3.48 (m, 1H), 2.14-1.93 (m, 4H), 1.40 (t, J = 7.1, 6H). Referenced to HOD at 4.79 ppm. 13C NMR (125.7 MHz, D2O), 64.3 (d, J = 6.6, CH2), 60.7 (CH2), 53.5 (d, J = 18.2, CH), 22.3 (d, J = 4.1, CH2), 21.0 (d, J = 141.2, CH2), 16.2 (d, J = 5.7, CH3). Referenced to internal CH3CN at δ 1.47. 31P NMR (202.4 MHz, D2O), 33.3 (s). ES-HRMS for C8H21NO4P [M-HCl+H]+ calcd 226.1208; found 226.1213.
Diethyl [(3R)-3-amino-4-hydroxybutyl]phosphonate hydrochloride (55)
Compound 5433 (0.35 g, 0.96 mmol) is treated as described above for the preparation of 51 to give title compound 55 (0.25 g, 99%). (c 0.63, MeOH). ESI-HRMS for C8H21NO4P [M-HCl+H]+ calcd 226.120; ound 226.1205. The 1H, 13C and31P NMR spectra are identical to those of the enantiomer 51.
Diethyl [(3S3-[({2-chloro-4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]-4-hydroxybutyl]phosphonate (52)
A mixture of 51 (0.055 g, 0.591 mmol), triethylamine (0.030 mL, 0.211 mmol), aldehyde 49 (0.1 g, 0.394 mmol) and 2-picoline borane complex (0.055 g, 0.512 mmol) are stirred together in MeOH (4 mL) at room temperature for 16 h. The solvent is evaporated and the residue chromatographed on silica gel (CHCl3-MeOH-7M NH3 in MeOH, 9.6:0.2:0.2 v/v/v) to give intermediate diethyl [(3S)-3-({[4-(tert-butoxy)-2-chloro-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)-4-hydroxybutyl]phosphonate as a colourless gum (108 mg) which is dissolved in 1:1 mixture of 37% aq. HCl-THF (3 mL) and left at room temperature for 10 min. The solvent is evaporated and the residue dissolved in EtOH and neutralized with Amberlyst A21 resin. After filtering off the resin, the filtrate is evaporated and the residue is chromatographed on silica gel (CHCl3-MeOH-7M NH3 in MeOH, 7.0:2.75:0.25 v/v/v) to give title compound 52 (0.070 g, 44%) as a colourless amorphous solid. (c 0.555, MeOH)1H NMR (500 MHz, CD3OD) 7.37 (s, 1H), 4.28 (d. J = 13.7, 1H), 4.23 (d, J = 13.7, 1H), 4.13-4.05 (m, 4H), 3.93 (dd, J = 12.3, 3.6, 1H), 3.72 (dd, J = 12.3, 4.6, 1H), 3.22 (m, 1H), 2.04-1.90 (m, 4H), 1.31 (dt, J = 7.0, 0.5, 6H). 13C NMR (125.7 MHz, CD3OD) 162.5 (C), 149.7 (C), 146.4 (C), 129.3 (CH), 119.4 (C), 107.7 (C), 63.5 (d, J = 6.4, CH2), 60.1 (CH2), 59.7 (d, J = 17.5, CH), 40.4 (CH2), 22.8 (d, J = 3.7, CH2), 22.2 (d, J = 142.3, CH2), 16.7 (d, J = 5.8, CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.3 MHz, CD3OD), 31.7 (s). ESI-HRMS for C15H25 (35) ClN4O5P [M+H]+ calcd 407.1251; found 407.1250.
Diethyl [(3R)-3-[({2-chloro-4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]-4-hydroxybutyl]phosphonate (56)
In the same way as that described for the synthesis of enantiomer (52 (Scheme 18), compound 55 (0.083 g, 0.317 mmol) is treated with triethylamine (0.030 mL, 0.211 mmol), aldehyde 49 (0.054 g, 0.21 mmol) and 2-picoline borane complex (0.029 g, 0.275 mmol) to give after treatment with a 1:1 mixture of 37% aq. HCl-MeOH, title compound 56 (0.039 g, 45%). (c 0.455, MeOH). ESI-HRMS f or C15H25(35) ClN4O5P [M+H]+ calcd 407.1251; found 407.1250. The1H, 13C and31P NMR spectra are identical to those o the enantiomer 52.
Diethyl [ (1R/S, 3S)-3-amino-1-fluoro-4-hydroxybutyl] phosphonate hydrochloride (59)
The α-fluoro-vinylphosphonate 5833 (0.222 g, 0.582 mmol) and 10% Pd/C (50 mg) are stirred in EtOH (10 mL) under a hydrogen atmosphere at ambient temperature and pressure for 5.5 h. The mixture is filtered through Celite and the solvent evaporated to a colourless gum (225 mg) which is dissolved in a mixture of EtOH (3 mL) and 37% aq. HCl (1 mL) and left at room temperature for 1.5 h. The solvent is evaporated to give title compound 59 as an ∼1:1 mixture of diastereomers as a dark yellow gum. 1H NMR (500 MHz, D2O), 5.38 (m, 0.5H), 5.28 (m, 0.5H), 4.38-4.30 (m, 4H), 3.96 (t, J = 3.4, 0.5H), 3.94 (t, J = 3.4, 0.5H), 3.81-3.70 (m, 2H), 2.46-2.24 (m, 2H), 1.43 (t, J = 7.1, 6H). Referenced to HOD at δ 4.79. 13C NMR (125.7 MHz, D2O) 86.9 (dd, J = 175.6, 173.7, CHF), 85.9 (dd, J = 177.2, 173.3, CHF), 66.0 (d, J = 7.0, CH2), 65.8 (d, J = 6.7, CH2), 61.6 (CH2), 61.0 (CH2), 51.3 (d, J = 16.0, CH), 50.6 (d, J = 14.7 CH), 29.9 (d, J = 19.8, CH2), 29.8 (d, J = 19.2, CH2), 16.3 (d, J = 4.9, CH3). Referenced to internal CH3CN at δ 1.47. 31P NMR (202. MHz, D2O), 18.0 (d, J = 76.0), 17.8 (d, J = 76.0). 19F NMR (470.5 MHz, D2O) -209.7 (d, J = 76.0). ES1-HRMS for C8H20FNO4P [M-HCl+H]+ calcd 244.1114; found 244.1111. The NMR spectra indicated 59 is a ∼1:1 mixture of diastereomers.
Diethyl [(1 R/S, 3S)-3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d ]pyrimidin-7-yl]methyl}amino)-1-fluoro-4-hydroxybutyl]phosphonate (60)
Compound 59 (0.157 g, 0.561 mmol), aldehyde 8 (0.113 g, 0.401 mmol), Et3N (0.034 mL, 0.241 mmol) and 2-picoline borane complex (0.056 g, 0.521 mmol) are stirred together in MeOH (15 mL) at 50 °C f or 24 h. The solvent is evaporated and the residue lash chromatographed (CHCl3-7M NH3 in MeOH, 99:1 then 99:2 v/v) to give title compound 60 as an ∼1:1 mixture of diastereomers as a colourless gum. 1H NMR (500 MHz, CD3OD) 8.42 (s, 1H), 7.52-7.49 (m, 3H), 7.39-7.29 (m, 3H), 5.61 (s, 2H), 5.13 (ddt, J = 46.4, 4.8, 2.4, 0.5H), 5.07 (ddt, J = 46.7, 6.9, 3.4, 0.5H), 4.23-4.12 (m, 4H), 4.08 (d, J = 13.5, 0.5H), 4.01 (s, 1H), 3.98 (d, J = 13.5, 0.5H), 3.72 (dt, J = 11.5, 4.7, 1H), 3.56 (dt, J = 10.7, 5.5, 1H), 2.97 (pent, J = 5.6, 0.5H), 2.91 (sext, J = 4.6, 0.5H), 2.14-1.81 (m, 2H), 1.36-1.30 (m, 6H). 13C NMR (125.7 MHz, CD3OD) 157.0 (C), 150.1 (CH), 150.0 (CH), 149.6 (C), 137.9 (C), 130.3 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 116.7 (C), 115.2 (C), 114.8 (C), 87.8 (dd, J = 177.5, 171.9 CHF), 87.5 (dd, J = 177.5, 171.9 CHF), 69.0 (CH2), 64.8 (t, J = 8.4, CH2), 64.5 (t, J = 7.5, CH2), 64.1 (CH2), 63.7 (CH2), 56.4 (d, J = 13.6, CH), 55.7 (d, J = 13.9, CH), 41.3 (CH2), 41.2 (CH2), 33.8 (d, J = 20.1, CH2), 32.7 (d, J = 19.7, CH2), 16.7 (CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.3 MHz, CD3OD) 19.0 (d, J = 77.6), 18.5 (d, J = 77.6). 19F NMR (470.5 MHz, CD3OD) -209.9 (d, J = 77.7), - 210.7 (d, J =77.4). ESI-HRMS for C22H31FN4O5P [M+H]+ calcd 481.2016; tound 481.2014. The NMR spectra indicated 60 is a ∼1:1 mixture of diastereomers.
Diethyl [(3S)-3-amino-1,1-difluoro-4-hydroxybutyl]phosphonate hydrochloride (63)
Compound 6234-35 (0.345 g, 0.860 mmol) is dissolved in a mixture of EtOH (3 mL) and 37% aq. HCl (1 mL) and left to stand at room temperature for 1.5 h. The solvent is evaporated to give title compound 63 as a yellow gum (0.256 g, 100%). 1H NMR (500 MHz, CDCl3) 8.24 (bs, 3H), 4.60 (bs, 1H), 4.35-4.28 (m, 4H), 4.02 (d, J = 9.1, 1H), 3.92- 3.81 (m, 2H), 2.91-2.74 (m, 1H), 2.63-2.47 (m, 1H), 1.39 (dt, J = 7.0, 1.0, 6H). 13C NMR (125.7 MHz, CDCl3) 119.3 (dt, J = 261.3, 216.5, CF2), 65.5 (d, J = 4.5, CH2), 61.5 (CH2), 48.2 (CH), 33.5 (m, CH2), 16.3 (d, J = 5.0, CH3). Referenced to the centre line of CDCl3 at δ 77.0. 31P NMR (202.3 MHz, CDCl3), 5.4 (t, J = 104.9). 19F NMR (470.5 MHz, CDCl3) -107.7 (dd, J = 301.1, 102.5), -111.2 (dd, J = 301.3, 105.9). ESI-HRMS for C8H19F2NO4P [M-HCl+H]+ calcd 262.1020; found 262.1017.
Diethyl [(3S)-3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d ]pyrimidin-7-yl]methyl}amino)-1,1-difluoro-4-hydroxybutyl]phosphonate (64)
Compound 63 (0.140 g, 0.470 mmol), aldehyde 8 (0.095 g, 0.336 mmol), Et3N (0.042 mL, 0.302 mmol) and 2-picoline borane complex (0.047 g, 0.437 mmol) are stirred in MeOH (15 mL) at 50 °C f or 24 h. The solvent is evaporated and the residue flash chromatographed (CHCl3-7M NH3 in MeOH, 99:1 then 98:2 v/v) to give title compound 64 as a colourless gum (0.088 g, 53%). (c 0.475, MeOH). 1H NMR (500 MHz, CD3OD) 8.42 (s, 1H), 7.53 (s, 1H), 7.51 (d, J = 7.3, 2H), 7.38-7.30 (m, 3H), 5.60 (s, 2H), 4.29-4.22 (m, 4H), 4.04 (d, J = 13.6, 1H), 3.99 (d, J = 13.6, 1H), 3.73 (dd, J = 11.3, 4.3 1H), 3.53 (dd, J = 11.3, 6.2, 1H), 3.18 (m, 1H), 2.34-2.24 (m, 2H), 1.34 (t, J = 7.0, 6H). 13C NMR (125.7 MHz, CD3OD) 157.0 (C), 150.1 (CH), 149.5 (C), 137.8 (C), 130.4 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 122.0 (dt, J = 259.1, 218.5, CF2), 116.7 (C), 114.5 (C), 69.0 (CH2), 66.2 (d, J = 6.7, CH2), 64.2 (CH2), 53.6 (CH), 41.1 (CH2), 36.3 (dt, J = 19.8, 14.3, CH2), 16.7 (CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.4 MHz, CD3OD) 6.7 (t, J = 109.9). 19F NMR (470.5 MHz, CD3OD) -110.3 (dd, J = 299.3, 109.9, -111.6 (dd, J = 299.3, 109.9). ESI-HRMS C22H30F2N4O5P [M+H]+ calcd 499.1922; found 499.1927.
[(3S)-4-Hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2- d ]pyrimidin-7-yl}methyl)amino]butyl]phosphonic acid hydrobromide (53)
Compound 52 (0.060 g, 0.147 mmol) is dissolved in EtOH (5 mL), 10% Pd-C (40 mg) added and the mixture stirred under a hydrogen atmosphere at ambient pressure and temperature for 3 h. The mixture is filtered through Celite and the solvent evaporated to give diethyl [(3S)-4-hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]butyl]phosphonate hydrochloride (0.055 g, 91%) as a colourless solid. (c 0.54, MeOH). 1H NMR (500 MHz, CD3OD) 7.97 (s, 1H), 7.67 (bs, 1H), 4.47 (d, J = 13.9, 1H), 4.43 (d, J = 13.9, 1H), 4.15-4.06 (m, 4H), 4.00 (dd, J = 12.4, 2.9, 1H), 3.76 (dd, J = 12.4, 4.1, 1H), 3.35 (m, partially hidden by residual CD3OD, 1H), 2.11-1.88 (m, 4H), 1.32 (t, J = 7.1, 6H). 13C NMR (125.7 MHz, CD 3OD) 155.7 (C), 145.2 (C), 143.8 (CH), 130.8 (CH), 119.6 (C), 108.0 (C), 63.6 (d, J = 4.3, CH2), 59.6 (d, J = 17.7, CH), 59.0 (CH2), 39.7 (CH2), 22.3 (d, J = 3.7, CH2), 22.2 (d, J = 142.7, CH2), 16.7 (d, J = 5.8, CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.3 MHz, CD3OD) 31.2 (s). ESI-HRMS or C15H26N4O5P [M-HC+H]+ calcd 373.1641; found 373.1642. This material (0.052 g, 0.127 mmol) is heated at 80 °C in hydrobromic acid (48% aq., 1.002 mL, 12.72 mmol) for 4 h. The solvent is evaporated and the residue eluted from a column of RP 18 silica gel (H2O) to afford title compound 53 as a colourless glass which slowly crystallized. (c 0.355, H2O). 1H NMR (500 MHz, D2O + DCl): δ 8.93 (s, 1H), 7.84 (s, 1H), 4.51 (d, J = 14.7 Hz, 1H), 4.48 (d, J = 14.7 Hz, 1H), 3.97 (dd, J = 13.1, 2.7 Hz, 1H), 3.81 (dd, J = 13.1, 4.7 Hz, 1H), 3.46 (m, 1H), 2.08-1.77 (m, 4H). Reerenced to HOD at 4.79 ppm. 13C NMR (125.7 MHz, D2O +DCl) 153.4 (C), 145.2 (CH), 133.8 (C) 133.7 (CH), 118.8 (C), 104.0 (C), 59.8 (d, J = 17.1 Hz, CH), 58.5 (CH2), 38.9 (CH2), 23.3 (d, J = 137.2 Hz, CH2), 21.5 (d, J = 3.2 Hz, CH2). Referenced to internal CH3CN at 1.47 ppm. 31P NMR (202.3 MHz, D2O + DCl) 28.5 (s). ES-HRMS for C11H16N4O5P [M-HBr-H]- calcd 315.0858; ound 315.0855.
[(3R)-4-Hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]butyl]phosphonic acid hydrobromide (57)
In the same way as that described for the synthesis of enantiomer 53 (Scheme 18), compound 56 (0.065 g, 0.160 mmol) is stirred under a hydrogen atmosphere in the presence of 10% Pd/C (40 mg) to give [(3R)-4-hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]butyl]phosphonate hydrochloride (0.063 g, 96%). (c 0.37, MeOH). ES-HRMS for C15H26N4O5P [M-HCl+H]+ calcd 373.1641; found 373.1647. This material (0.058 g, 0.142 mmol) is treated with 48% aq. hydrobromic acid (1.12 mL. 14.2 mmol) to give title compound 57 (0.04 g, 71%). (c 0.365, H2O). ESI- HRMS for C11H16N4O5P [M-HBr-H]- calcd 315.0858; found 315.0857. The1H, 13C and31P NMR spectra are identical to those of the enantiomer 53.
[(1R/SV, 3S)-1-Fluoro-4-hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]butyl]phosphonic acid; triethylamine (61)
Compound 60 (0.1 g, 0.208 mmol) and 10% Pd/C (30 mg) are stirred in EtOH (6 mL) under a hydrogen atmosphere at ambient temperature and pressure for 3 h. The mixture is filtered and the solvent evaporated. The residue is dissolved hydrogen bromide (48% aq. 1.639 mL, 20.81 mmol) and heated to 80 °C f or 6 h. he solvent is evaporated several times rom H2O then the residue dissolved in hot water and chromatographed on RP 18 silica gel (H2O) to give the sparingly soluble product hydrobromide salt. It is dissolved in 5% aq. Et3N and the solvent evaporated then the residue flash chromatographed (1,4-dioxane-H2O-Et3N, 80:20:3 then 70:30:3 v/v/v) to give title compound 61 as an ∼1:1 mixture of diastereomers as a colourless amorphous solid. 1H NMR (500 MHz, D2O) 7.97 (s, 1H), 7.69 (s, 1H), 4.79 (HOD + 0.5H), 4.69 (bs, 0.5H), 4.46 (dd, J = 13.8, 1.9, 1H), 4.36 (dd, J = 13.8, 2.7, 1H), 4.08 (dd, J = 12.8, 3.6, 1H), 3.91 (dt, J = 12.8, 5.0, 1H), 3.66-3.58 (m, 1H), 3.22 (q, J = 7.4, 6H), 2.44-2.24 (m, 2H), 1.30 (t, J = 7.4, 9H). Referenced to HOD at 4.79 ppm. Approximately 1 eq. of Et3N present. 13C NMR (125.7 MHz, D2O) 155.5 (C), 143.8 (C), 143.3 (CH), 131.3 (CH), 131.2 (CH), 118.0 (C), 107.0 (C), 106.9 (C), 89.7 (bm, CHF), 60.4 (CH2), 59.5 (CH2), 57.6 (CH), 56.1 (CH), 47.3 (CH2), 38.7 (CH2), 38.5 (CH2), 31.1 (d, J = 20.9, CH2), 30.6 (d, J = 19.7, CH2), 8.9 (CH3). Referenced to internal CH3CN at δ 1.47. 31P NMR (202.3 MHz, CD3OD + drop Et3N) 11.0 (d, J = 61.6), 10.9 (d, J = 63.1). 19F NMR (470.5 MHz, D2O) -197.9 (d, J = 63.4), -203.9 (d, J = 62.1). ESI- HRMS for C11H15FN4O5P [M-Et3N-H]- calcd. 333.0764; ound 333.0754. The NMR spectra indicated compound 61 is an ∼1:1 mixture of diastereomers.
[(3S)-1,1-Difluoro-4-hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]butyl]phosphonic acid; triethylamine (65)
Compound 64 (0.088 g, 0.177 mmol) and 10% Pd/C (30 mg) are stirred in EtOH (5 mL) under a hydrogen atmosphere at ambient temperature and pressure for 3 h. The mixture is filtered through Celite and the solvent evaporated. The residue is heated at 80 °C in hydrobromic acid (48% aq., 1.39 mL, 17.65 mmol) for 10 h. The solvent is evaporated and the residue dissolved in 5% aq. Et3N, evaporated, dissolved in H2O and loaded on to a column of Dowex 50WX8 (H +) resin (4 cm × 1 cm) and eluted with water (50 mL, discarded) then 5% aq. Et3N (250 mL). After leaving the column standing in 5% aq. Et3N overnight it is eluted with more 5% aq. Et3N (100 mL). The combined basic fractions are evaporated and the residue flash chromatographed (1,4-dioxane-H2O-Et3N, 85:15:3 then 80:20:3 v/v/v)to give title compound 65 as a cream coloured solid (0.058 g, 73%). (c 0.55, 3% aq. Et3N). 1H NMR (500 MHz, D2O) 7.94 (s, 1H), 7.68 (s, 1H), 4.46 (d, J = 13.8, 1H), 4.32 (d, J = 13.8, 1H), 4.10 (dd, J = 12.8, 3.8, 1H), 3.91 (dd, J = 12.8, 5.0, 1H), 3.72 (m, 1H), 3.22 (q, J = 7.3, 6.5H), 2.62 (m, 1H), 2.43 (m, 1H), 1.3 (t, J = 7.3, 9.75H). Referenced to HOD at δ 4.79. Approximately 1.1 eq. of Et3N present. 13C NMR (125.7 MHz, D2O) 155.4 (C), 143.6 (C), 143.2 (CH), 131.3 (CH), 123.0 (dt, J = 260.5, 179.1, CF2), 117.9 (C), 106.9 (C), 60.4 (CH2), 54.6 (CH), 47.3 (CH2), 38.4 (CH2), 34.9 (dt, J = 23.1, 13.8, CH2), 8.9 (CH3). Referenced to internal CH3CN at δ 1.47. 31P NMR (202.4 MHz, D2O) 4.6 (t, J = 83.1). 19F NMR (470.5 MHz, D2O) -104.1 (dd, J = 286.6, 83.5, -110.6 (dd, J = 286.4, 82.8). ESI-HRMS for C11H14F2N4O5P [M-Et3N-H]- calcd 351.0670; found 351.0671.
di-tert-Butyl [(3R)-4-(benzyloxy)-3-hydroxybutyl]phosphonate (68)
In a modification of a known method, 47 sodium hydride (60% in oil, 2.039 g, 51.0 mmol) is added in one portion to a solution of di-tert-butyl phosphonate (6.6 g, 34.0 mmol) in CH3CN (30 mL) at room temperature and the mixture stirred 30 min. Iodomethane (4.25 mL, 68.0 mmol) is added in 0.5 mL portions controlling the resulting exotherm with an ice-bath so that the temperature did not go above 50 °C. After the temperature has dropped back to room temperature, H2O (1 mL) is added and the solvent evaporated. The residue is suspended in DCM and filtered through Celite then the solvent evaporated. The residue is flash chromatographed (EtOAc-hexanes, 4:6 then 6:4 v/v) to give di-tert-butyl methylphosphonate (66) (5.4 g, 76%) as a colourless oil. The1H and 31P NMR are in agreement with that reported in the literature47 except for the sign in the 31P NMR which is +ve and opposite to that reported. 13C NMR (500 MHz, CDCl3) δ 81.4 (d, J = 89.6 Hz, C), 30.4 (d, J = 3.7 Hz, CH3), 17.0 (d, J = 148.5 Hz, CH3). Referenced to the centre line of CDCl3 at 77.0 ppm. ES-HRMS or C9H21Na3P [M+Na]+ calcd 231.1126; found 231.1132. n-Butyllithium (1.5M, 17.05 mL, 25.6 mmol) is added dropwise to a solution of di- tert butyl methylphosphonate (66) (5.33 g, 25.6 mmol) in THF (25 mL) keeping the temperature below -70 °C throughout the addition. After 15 m, boron trifluoride diethyl etherate (3.35 mL, 26.4 mmol) is added dropwise to the dark coloured mixture and after 10 min (2R)-2-[(benzyloxy)methyl]oxirane (67)48 (1.4 g, 8.53 mmol) in THF (0.5 mL) added dropwise. After 30 min, sat. aq. NaHCO3 (5 mL) is added then triethylamine (23.97 mL, 171 mmol) and the mixture warmed to room temperature. The solvent is evaporated and the residue dissolved in Et2O (150 mL) and washed with sat. aq. NaHCO3, dried and the solvent evaporated. The residue is flash chromatographed (CHCl3-MeOH, 99:1 v/v) to give 68 as a yellow oil (2.53 g, 80%). (c, 0.82, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.36-7.27 (m, 5H), 4.55 (s, 2H), 3.85 (m, 1H), 3.48 (dd, J = 9.5 Hz, 4.1, 1H), 3.40 (dd, J = 9.5, 7.0 Hz, 1H), 3.07 (d, J = 3.0, exchanged to D2O, 1H), 1.86- 1.67 (m, 4H), 1.49 (s, 18H). 13C NMR (125.7 MHz, CDCl3) 138.0 (C), 128.4 (CH), 127.7 (CH), 81.6 (d, J = 8.5 Hz, C), 74.1 (CH2), 73.3 (CH2), 70.4 (d, J = 13.7 Hz, CH), 30.4 (CH3), 27.3 (d, J = 5.1 Hz, CH2), 26.5 (d, J = 146.5 Hz, CH2). Referenced to the centre line of CDCl3 at δ 77.0. 31P NMR (202.4 MHz, CDCl3) 24.4 (s). ESI-HRMS for C19H33NaO5P [M+Na]+ calcd 395.1963, found 395.1969.
di-tert-Butyl [(3S)-3-amino-4-(benzyloxy)butyl]phosphonate (69)
A mixture of diisopropyl azodicarboxylate (DIAD, 1.78 mL, 9.13 mmol) and diphenylphosphoryl azide (DPPA,1.97 mL, 9.13 mmol) in dry toluene (10 mL) is added dropwise to a solution of 68 (2 g, 5.37 mmol) and triphenyl phosphine (2.39 g, 9.13 mmol) in toluene (20 mL) at 0 °C. After 10 mins, the mixture is allowed to warm to room temperature and then stirred overnight, filtered through Celite and the solvent evaporated. The residue is flash chromatographed (EtOAc-hexanes, 4:6 v/v) to give the crude azide as a yellow oil (1.6 g) which is dissolved in dry Et2O (30 mL), cooled in an ice-bath and LiAlH4 (1M in THF, 8.06 mL, 8.06 mmol) added dropwise. The mixture is warmed to room temperature and stirred for 30 min then cooled in an ice-bath and H2O (0.3 mL), 15% aq. NaOH (0.3 mL) then H2O (0.9 mL) added successively. After diluting with EtOAc, the mixture is filtered through Celite and the solvent evaporated. The residue is flash chromatographed (CHCl3-7M NH3 in MeOH, 98:2 then 96:4 v/v) to give 69 as a colourless oil (0.688 g, 35%). (c, 1.09, CHCl3). 1H NMR δ (500 MHz, CDCl3) 7.36-7.26 (m, 5H), 4.54 (d, J = 12.1 Hz, 1H), 4.51 (d, J = 12.1 Hz, 1H), 3.45 (dd, J = 9.1, 4.0 Hz, 1H), 3.27 (dd, J = 9.1, 7.3 Hz, 1H), 3.01 (m, 1H), 1.8-1.49 (m, 6H, after D2O exchange, became 4H), 1.49 (d, J = 1.5 Hz, 18H). 13C NMR (125.7 MHz, CDCl3) δ 138.2 (C), 128.3 (CH), 127.6 (CH), 81.4 (d, J = 8.6, C), 75.3 (CH2), 73.2 (CH2), 51.4 (d, J = 16.2 Hz, CH), 30.4 (d, J = 3.5 Hz, CH3), 28.1 (d, J = 5.5 Hz, CH2), 26.9 (d, J = 146.5 Hz, CH2). Referenced to the centre line of CDCl3 at δ 77.0. 31P NMR (202.4 MHz, CDCl3) 23.7 (s). ESI-HRMS for C19H35NO4P [M+H]+ calcd 372.2304; found 372.2311. The % d.e. of compound 69 is determined in two ways, as follows. The (S, R) Mosher amide49 is prepared by dissolution of di-tert-butyl [(3S)-3-amino-4-(benzyloxy)butyl]phosphonate (69) (6.3 mg, 0.017 mmol) in a mixture of CDCl3 and Et3N (3 eq.) and a solution of (S)-MTPACl (1.2 eq.) in CDCl3 (prepared from (R)-MTPA, 99% e.e.) is added to give a total volume of 0.6 mL. The mixture is spiked with CFCl3 and left for 30 min. 19F NMR (470.5 MHz, CDCl3, ref. CFCl3 δ 0): δ -69.3 (s, 2%), -69.4 (s, 98%). The % d.e. is 96%. The (S, S) Mosher amide is prepared by dissolution of di-tert-butyl [(3S)-3-amino-4-(benzyloxy)butyl]phosphonate (69) (5.0 mg, 0.019 mmol) in a mixture of CDCl3 and Et3N (3.6 eq.) and a solution of (R)-MTPACl (1.2 eq.) in CDCl3 (prepared from (S)-MTPA, 99% e.e.) added to give a total volume o 0.6 mL. he mixture is spiked with CFCl3 and left or 30 m. 19F NMR (470.5 MHz, CDCl3, ref. CFCl3 δ 0): δ -69.3 (s, 97%), -69.4 (s, 3%). The % d.e. is 94%. The average % d.e. from these two assessments is 95%.
[(3S)-4-Hydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]butyl]phosphonic acid hydrobromide (53)
Compound 69 (0.15 g, 0.404 mmol), 2-picoline borane complex (0.043 g, 0.404 mmol), 4-benzyloxy-5H-pyrrolo[3,2-d]pyrimidine-7-carbaldehyde (8) (0.087 g, 0.311 mmol) and Et3N (0.043 mL, 0.311 mmol) are stirred in MeOH (15 mL) at 40 °C overnight to give a clear colourless solution. The solvent is evaporated and the residue is chromatographed (0 → 10% continuous gradient of 0.5% v/v Et3N/MeOH in EtOAc) to give di-tert-butyl [(3S)-4-(benzyloxy)-3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)butyl]phosphonate as a colourless gum (0.135 g, 71%). (c, 0.6, CHCl3). 1H NMR (500 MHz, CDCl3): δ 9.13 (bs, exchanged D2O, 1H), 8.53 (s, 1H), 7.48-7.46 (m, 2H), 7.40-7.23 (m, 9H), 5.57 (s, 2H), 4.51 (d, J = 11.9, 1H), 4.48 (d, J =11.9, 1H), 4.05 (s, 2H), 3.53 (dd, J = 9.5, 4.6, 1H), 3.46 (dd, J = 9.5, 6.0, 1H), 2.94-2.89 (m, 1H), 2.21 (bs, exchanged D2O, 1H), 1.84-1.76 (m, 2H), 1.72-1.63 (m, 2H), 1.443, 1.442 (2s, 18H). 13C NMR (125.7 MHz, CDCl3) 155.3 (C), 149.6 (CH), 149.2 (C), 138.3 (C), 136.4 (C), 128.6 (CH), 128.4 (CH), 128.3 (CH), 127.6 (CH), 127.5 (CH), 127.1 (CH), 115.4 (C) 115.3 (C), 81.5 (d, J = 3.0, C), 81.4 (d, J = 2.6, C), 73.2 (CH2), 71.7 (CH2), 67.7 (CH2), 56.8 (d, J = 16.5, CH), 40.9 (CH2), 30.40 (CH3), 30.37 (CH3), 26.4 (d, J = 146.2, 2H), 25.2 (CH2). Referenced to the centre line of CDCl3 at δ 77.0. 31P NMR (202.4 MHz, CDCl3) δ 24.1 (s). ESI-HRMS for C33H46N4O5P [M+H]+ calcd 609.3206; found 609.3203. This material (0.120 g, 0.197 mmol) is heated at 80 °C in 48% aq. HBr (3 mL) for 16 h. After cooling, the solution is washed with CHCl3 (2x) then the aqueous phase evaporated and the residue chromatographed on RP 18 silica gel (H2O) to give 53 as a colourless glass. (c2.03, H2O). ESI-HRMS for C11H18N4O5P [M+H]+ calcd 317.1015; ound 317.1013. The1H, 13C and31P NMR of compound 53 made by this route are the same as those reported above in an alternative synthesis.
[7-({[(2S)-4-(Diethoxyphosphoryl)-4,4-difluoro-1-hydroxybutan-2-yl]amino}methyl)-2-[(E)- [(dimethylamino)methylidene]amino]-4-oxo-3H,4H,5H-pyrrolo[3,2- d ]pyrimidin-3-yl]methyl 2,2-dimethylpropanoate (71)
The phosphonate 63 (0.135 g, 0.454 mmol), deazaguanine derivative 7036 (0.113 g, 0.324 mmol), Et3N (0.041 mL, 0.292 mmol) and 2-picoline borane complex (0.045 g, 0.421 mmol) are stirred in MeOH (5 mL) at 50 °C for 1 h then at room temperature for 16 h. The solvent is evaporated and the residue flash chromatographed (CHCl3-7M NH3 in MeOH, 99:1 then 98:2 v/v) to give 71 as a yellow amorphous solid together with a mixture of 70 + 71. The latter is chromatographed twice more to give a total amount of 71, 0.109 g, 57%. (c 1.44, MeOH). 1H NMR (500 MHz, CD3OD) 8.58 (s, 1H), 7.25 (s, 1H), 6.31 (s, 2H), 4.31-4.24 (m, 4H), 3.90 (d, J = 13.2, 1H), 3.83 (d, J = 13.2, 1H), 3.73 (dd, J = 11.2, 4.6, 1H), 3.52 (dd, J = 11.2, 6.7, 1H), 3.22 (m, 1H), 3.18 (s, 3H), 3.06 (s, 3H), 2.37-2.19 (m, 2H), 1.37 (t, J = 7.0, 6H), 1.15 (s, 9H). 13C NMR (125.7 MHz, CD3OD) 179.0 (C), 158.7 (CH), 156.5 (C), 155.3 (C), 144.9 (C), 128.6 (CH), 121.1 (dt, J = 259.0, 218.3, CF2), 115.3 (C), 114.7 (C), 67.0 (CH2), 66.3 (d, J = 6.9, CH2), 64.6 (CH2), 53.8 (CH), 41.7 (CH2), 41.1 (CH3), 39.8 (C), 36.2 (dt, J = 20.0, 14.1, CH2), 35.2 (CH3), 27.4 (CH3), 16.7 (d, J = 5.0, CH3). Referenced to the centre line of CD3OD at 49.0 ppm. 31P NMR (202.4 MHz, CD3OD) 6.6 (t, J = 109.6). 19F NMR (470.5 MHz, CD3OD) -110.3 (dd, J = 298.9, 109.4), -111.4 (dd, J = 298.9. 109.8). ES1-HRMS for C24H40F2N6O7P [M+H]+ calcd 593.2664; found 593.2667.
[(3S-3-[({2-Amino-4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]-1,1-difluoro-4-hydroxybutyl]phosphonic acid; triethylamine (72)
Compound 71 (0.1 g, 0.169 mmol) is heated at 80 °C in 48% aq. hydrobromic acid for 16 h then evaporated to the sparingly soluble hydrobromide salt form of 72. It is dissolved in 5% aq. Et3N, evaporated and the residue dissolved in H2O and loaded on to a column of Dowex 50WX8 (H +) resin (4 cm × 1 cm) and eluted with water (50 mL, discarded) then 5% aq. Et3N (250 mL). After leaving the column standing in 5% aq. Et3N overnight it is eluted with more 5% aq. Et3N (100 mL). The combined basic fractions are evaporated and the residue flash chromatographed (1,4-dioxane-H2O-Et3N, 80:20:3 then 70:30:3 v/v/v) to give a yellow glassy solid which is dissolved in 3% aq. Et3N and chromatographed on RP 18 silica gel (3% aq. Et3N) to give 72 as a cream coloured amorphous solid (0.051 g, 65%). (c 0.62, 3% aq. Et3N). 1H NMR (500 MHz, D2O) 7.58 (s, 1H), 4.43 (d, J = 13.7, 1H), 4.32 (d, J = 13.7, 1H), 4.08 (dd, J = 12.8, 4.0, 1H), 3.92 (dd, J = 12.8, 6.0, 1H), 3.75 (m, 1H), 3.27 (q, J = 7.3, 7.3H), 2.65 (m, 1H), 2.44 (m, 1H), 1.35 (t, J = 7.3, 10.9H). Referenced to HOD at δ 4.79. Approximately 1.2 eq. of Et3N present. 13C NMR (125.7 MHz, D2O) 158.0 (C), 152.6 (C), 141.8 (C), 130.6 (CH), 122.8 (dt, J = 260.6, 180.2, CF2), 112.9 (C), 103.1 (C), 60.4 (CH2), 54.6 (CH), 47.2 (CH2), 38.3 (CH2), 34.4 (dt, J = 23.9, 14.5, CH2), 8.8 (CH3). 31P NMR (202.4 MHz, D2O) 4.8 (t, J = 83.71 19F NMR (470.5 MHz, D2O) -103.8 (dd, J = 287.8, 84.2, -110.6 (dd, J = 287.6, 83.4). ESI- HRMS for C11H15F2N5O5P [M-Et3N-H]- calcd 366.0779; found 366.0785.
2-({[4-(Benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)ethan-1-ol (73)
Ethanolamine (120 μL, 5 eq.) is added to acetyl chloride (142 μL, 5 eq.) in methanol (8 mL) and a few microdrops of acetyl chloride added to adjust the pH to 5. To this solution is added aldehyde 8 (100 mg, 1 eq.) and the mixture is stirred at 40 °C for 30 min. Picoline borane (64 mg, 1.5 eq.) is added and the mixture stirred at 40 °C for 4 h. The product is evaporated onto silica gel and subjected to flash chromatography (9/1/0.1 v/v/v DCM/MeOH/conc. NH3) to give title compound 73 (47 mg). 1H NMR (500 MHz, MeOD) δ 8.35 (s, 1H), 7.51 (s, 1H), 7.43-7.23 (m, 5H), 5.53 (s, 2H), 4.04 (s, 2H), 3.63 (m, 2H), 2.80 (m, 2H). 13C NMR (125 MHz, MeOD) δ 157.2, 150.5, 149.6, 137.0, 131.4, 129.5, 116.8, 111.8, 69.2, 60.2, 51.0, 42.9. ESI-HRMS or C16H19N4O2 [M+H]+ calcd 99.1508; found 299.1514.
tert-Butyl N-{[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}-N-(2-[bis(benzyloxy)phosphoryl]oxy} ethyl)carbamate (74)
To a solution of 73 (47 mg, 1 eq.) in MeOH (5 mL) and Et3N (22.5 μL) is added di-tert-butyl dicarbonate (35 mg, 1 eq.) and the mixture is stirred for 30 min. Evaporation and flash chromatography (EtOAc) gives tert-butyl N-{[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}-N-(2-hydroxyethyl)carbamate (32 mg). To this carbamate and tetrazole (28 mg, 5 eq.) in DCM (2 mL) is added dibenzyl diisopropylphosphoramidite (52.5 μL, 1.5 eq.) and the mixture is stirred under argon for 30 min. tert-Butyl hydroperoxide (0.08 mL) is then added followed by Na2SO3 (10% aq., 0.08 mL) and the product is extracted with DCM, washed with brine, dried and evaporated. Flash chromatography (50%, 70%, then 100% v/v EtOAc in hexanes) gives 74 (32 mg). 1H NMR (500 MHz, CDCl3) δ 8.52 (s, 1H), 7.46-7.26 (m, 6H), 5.56 (s, 2H), 5.03-4.97 (m, 4H), 4.60 (s, 2H), 4.14-4.08 (bm, 2H), 3.60 (bs, 2H), 1.42 (s, 9H). 13C NMR (125 MHz, CDCl 3) δ 155.5, 155.3, 149.9, 136.3, 135.9, 128.4, 127.9, 114.9, 114.4, 113.9, 80.0, 69.3, 67.8, 65.7, 47.3, 41.9, 41.1, 28.4. 31P NMR (202 MHz, CDCl3) δ -1.0. ESI-HRMS for C35H40N4O7P [M+H]+ calcd 659.2635; found 659.2640.
({2- [({4-Hydroxy-5H-pyrrolo[3,2-]pyrimidin-7-yl}methyl)amino]ethoxy}phosphonic acid (75)
The carbamate 74 (23 mg) is hydrogenated at atmospheric pressure with 10% Pd/C (20 mg) in ethanol (5 mL) for 2 h, filtered and evaporated. The crude reduced product is hydrolysed with 80% aqueous TFA (5 mL) for 2 h and then evaporated twice from dioxane followed by evaporation from 1 N HCl to give 75 (14 mg). 1H NMR (500 MHz, D2O) δ 8.05 (s, 1H), 7.71 (s, 1H), 4.42 (s, 2H), 4.05-4.02 (m, 2H), 3.32-3.30 (m, 2H). 13C NMR (125 MHz, D2O) δ 153.2,144.4,134.7, 132.3,118.2,103.6,60.5, 46.9, 40.1. 31P NMR (202 MHz, D2O) δ 2.3. ESI-HRMS for C9H14N4O5P [M+H]+ calcd 289.0702; found 289.0699.
9H-Fluoren-9-ylmethyl N-{(±)-2(R/S)-1-[(dimethoxyphosphoryl)oxy]-3-hydroxypropan-2-yl}carbamate (±)-(77)
Iodine (1.94 g, 7.66 mmol) is added in one portion to a solution of trimethyl phosphite (0.943 mL, 7.98 mmol) in CH2Cl2 (25 mL) at 0 °C, which is then warmed to room temperature and stirred for 5 min to give a colourless solution. A portion (6.7 mL, 1.3 eq) of this is added dropwise to a solution of compound 7637 (0.5 g, 1.596 mmol) in a mixture of CH2Cl2 (6 mL) and pyridine (3 mL) at 0 °C and stirred for 1 h. The mixture is warmed to room temperature and stirred for 30 min then the solvent is evaporated and the residue subjected to flash chromatography (EtOAc-hexanes, 9:1 v/v then EtOAc) to give first unreacted 76 (190 mg) then (±)-77 (0.24 g, 36%) as a colourless gum. 1H NMR (500 MHz, CD3OD) 7.76 (d, J = 7.5, 2H), 7.63 (d, J = 73, 2H), 7.37 (t, J = 7.4, 2H), 7.29 (t, J = 7.4, 2H), 4.40-4.33 (m, 2H), 4.19-4.13 (m, 2H), 4.08 (m, 1H), 3.87 (m, 1H), 3.74 (d, J = 11.1, 3H), 3.72 (d, J = 11.2, 3H), 3.65-3.56 (m, 2H). 13C NMR (125.7 MHz, CD3OD) 158.4 (C), 145.3 (d, J = 4.8, C), 142.6 (C), 128.7 (CH), 128.1 (CH), 126.1 (CH), 120.9 (CH), 67.74 (CH2), 67.70 (CH2), 61.5 (CH2), 55.3 (d, J = 6.1, CH3), 54.2 (d, J = 7.0, CH), 48.4 (CH). Referenced to the centre line of CD3OD at 49.0 ppm. 31P (121.5 MHz, CD3OD) 2.2 (s). ESI-HRMS f or C20H24NNaO7P [M+Na]+ calcd 444.1188; found 444.1179.
[(±)-2-(R/S)-2-({[4-(Benzyloxy)-5H-pyrrolo[3,2-d ]pyrimidin-7-yl]methyl}amino)-3-hydroxypropoxy]phosphonic acid; triethylamine (±)-(79)
Compound (±)-77 (0.4 g, 0.949 mmol) is dissolved in CH2Cl2 (10 mL) and bromotrimethylsilane (1.232 mL, 9.49 mmol) added. The clear solution is left at room temperature for 16 h then the solvent is evaporated. 10% aq. Et3N (2 mL) is added and the solution evaporated again. The residue is dissolved in DMF (4 mL) and piperidine (0.8 mL) added. After 30 min at room temperature the solvent is evaporated. The residue is dissolved in H2O (10 mL) and washed with EtOAc (10 mL). After evaporating the aqueous solution, the residue is repeatedly evaporated from 1:1 v/v MeOH-Et3N (6 mL) to exchange most of the piperidine with Et3N. The residue is chromatographed on silica gel (1,4-dioxane-H2O-Et3N, 60:40:1 v/v/v) then on RP-18 silica gel (H2O) to give (±)-78 (160 mg, 0.428 mmol, ∼ 80-85% pure) which is suspended in a mixture of MeOH (15 mL) and Et3N (0.040 mL, 0.286 mmol). Aldehyde 8 (0.072 g, 0.286 mmol) and sodium cyanoborohydride (0.023 g, 0.371 mmol) are added and the mixture heated at 50 °C f or 16 h. he solvent is evaporated and the residue flash chromatographed (THF-H2O-Et3N, 85:15:1 then 80:20:1 v/v/v) then on RP-18 silica gel (H2O then MeOH-THF-Et3N, 70:30:1 v/v/v) to give (±)-79 as a colourless amorphous solid (71 mg, 49%). 1H NMR (500 MHz, CD3OD) 8.48 (s, 1H), 7.85 (s, 1H), 7.52 (d, J = 7.2, 2H), 7.39-7.30 (m, 3H), 5.63 (s, 2H), 4.44 (s, 2H), 4.18 (ddd, J = 12.4, 8.8, 3.4, 1H), 4.04 (ddd, J = 12.4, 9.7, 5.6 1H), 3.85 (dd, J = 12.0, 5.1, 1H), 3.79 (dd, J = 12.0, 6.1, 1H), 3.31 (m, residual CD3OD + 1H), 3.07 (q, J = 7.3, 5.2H), 1.26 (t, J = 7.3, 8H). Approximately 0.9 eq of Et3N present. 13C NMR (125.7 MHz, CD3OD) 157.1 (C), 150.8 (CH), 149.6 (C), 137.7 (C), 133.3 (CH), 129.5 (CH), 129.4 (CH), 129.3 (CH), 116.5 (C), 107.9 (C), 69.1 (CH2), 62.6 (d, J = 3.8, CH2), 60.7 (d, J = 3.6, CH), 59.9 (CH2), 47.1 (CH2), 40.2 (CH2), 9.1 (CH3). Referenced to the centre line of CD3OD at δ 40.0. 31P (202.3 MHz CD3OD), 3.2 (s). ESI-HRMS for C17H20N4O6P [M-Et3N-H]- calcd 407.1120; ound 407.1128.
{(±)-2(R/S)-3-Hydroxy-2-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]propoxy}phosphonic acid; triethylamine (±)-(80)
Compound (±)-79 (0.07 g, 0.137 mmol) is dissolved in a 1:1 mixture of MeOH-H2O (10 mL), 20% Pd(OH)2/C (40 mg) added and the mixture stirred under a hydrogen atmosphere at ambient pressure and temperature for 4 h. After filtering through Celite the solvent is evaporated to give (±)-80 as a colourless solid (54 mg, 94%). 1H NMR (500 MHz, D2O) 8.14 (s, 1H), 7.82 (s, 1H), 4.59 (s, 2H), 4.23 (ddd, J = 12.4, 6.7, 3.5, 1H), 4.11 (ddd, J = 12.4, 7.5, 5.5, 1H), 4.02 (dd, J = 12.6, 5.0, 1H), 3.96 (dd, J = 12.6, 5.9, 1H), 3.60 (m, 1H), 3.27 (q, J = 7.4, 2.6H), 1.35 (t, J = 7.4, 3.9H). Referenced to HOD at 4.79 ppm. Approximately 0.4 eq. of Et3N present. 13C NMR (75.5 MHz, D2O-CD3OD, 3:1) 156.0 (C), 144.5 (C), 143.8 (CH), 131.7 (CH), 118.6 (C), 107.8 (C), 61.6 (CH2), 60.2 (CH), 59.1 (CH2), 47.6 (CH2), 39.9 (CH2), 9.2 (CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (121.5 MHz, D2O) 3.0 (s). ESI-HRMS for C10H14N4O6P [M-Et3N-H]-calcd 317.0651; found 317.0644.
{(±)-2(R/S)-[({2-Chloro-4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]-3-hydroxypropoxy}phosphonic acid; triethylamine (±)-(81)
A mixture of (±)-78 (0.19 g, 0.509 mmol), Et3N (0.048 mL, 0.339 mmol), the aldehyde 49 (0.086 g, 0.339 mmol) and 2-picoline borane complex (0.047 g, 0.441 mmol) are stirred together in MeOH (15 mL) at 50 °C f or 16 h then the solvent evaporated and the residue chromatographed on silica gel (1,4-dioxane-H2O-1% aq. Et3N, 75:25:1 v/v/v). The crude product is further chromatographed on silica gel (THF-H2O-Et3N, 85:15:1 then 80:20:1 v/v/v) to give [(±)-2(R/S)-2-({[4-(tert-butoxy)-2-chloro-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)-3-hydroxypropoxy]phosphonic acid; triethylamine as a colourless amorphous solid (105 mg, 51%). 1H NMR (500 MHz, CD3OD) 7.80 (s, 1H), 4.32 (d, J = 13.9, 1H), 4.29 (d, J = 13.9, 1H), 4.13 (ddd, J = 12.4, 9.2, 3.6, 1H), 4.00 (ddd, J = 12.4, 10.3, 5.6, 1H), 3.82 (dd, J = 11.9, 5.2, 1H), 3.77 (dd, J = 11.9, 5.9, 1H), 3.16 (m, 1H), 2.89 (q, J = 7.3, 12H), 1.72 (s, 9H), 1.19 (t, J = 7.3, 18H). Approximately 2 eq. of Et3N present. 13C NMR (125.7 MHz, CD3OD) 157.4 (C), 151.2 (C), 150.9 (C), 133.2 (CH), 116.5 (C), 109.4 (C), 85.2 (C), 62.8 (d, J = 4.5, CH2), 60.9 (d, J = 3.5, CH), 60.5 (CH2), 47.0 (CH2), 40.3 (CH2), 28.7 (CH3), 9.9 (CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.3 MHz, CD3OD) 3.0 (s). ESI-HRMS for C14H21(35) ClN4O6P [M-Et3N-H]- calcd 407.0887; found 407.0895. This material (0.095 g, 0.155 mmol) is dissolved in 80% aq. TFA (2 mL) and left at room temperature. After 15 min the solvent is evaporated to leave a colourless solid which is chromatographed on silica gel (1,4-dioxane-H2O-Et3N 75:25:1 v/v/v) to give (±)-81 as a colourless gum (84 mg, 97%). 1H NMR (500 MHz, CD3OD) 7.53 (s, 1H), 4.36 (s, 2H), 4.18 (ddd, J = 12.4, 8.7, 3.4, 1H), 4.05 (ddd, J = 12.4, 9.7, 5.4, 1H), 3.86 (dd, J = 12.0, 5.2, 1H), 3.81 (dd, J = 12.0, 6.0, 1H), 3.31 (residual CD3OD + 1H), 3.08 (q, J = 7.3, 10.8H), 1.26 (t, J = 7.3, 16.2H). Approximately 1.8 eq. of Et3N present. 13C NMR (125.7 MHz, CD3OD) 164.7 (C), 152.5 (C), 147.2 (C), 129.5 (CH), 119.8 (C), 106.7 (C), 62.2 (CH2), 60.8 (CH), 59.9 (CH2), 47.1 (CH2), 40.7 (CH2), 9.5 (CH3). Referenced to the centre line of CD3OD at 49.0 ppm. 31P NMR (202.3 MHz, CD3OD), 4.1 (s). ESI-HRMS for C10H13 (35) ClN4O6P [M-Et3N-H]- calcd 351.0261; found 351.0264.
[2-Amino-3-hydroxy-2-(hydroxymethyl)propoxy]phosphonic acid (83)
Dibenzyl diisopropyl phoshoramidite (3.64 mL, 10.83 mmol) is added to an ice cold solution of compound 8239 (1.35g, 5.42 mmol) and 1 H-tetrazole (1.52 g, 21.7 mmol) in CH2Cl2 (50 mL). The solution is stirred at room temperature for 1 h and then cooled to -30 °C. A solution of MCPBA (4.67g, 16.3 mmol) in CH2Cl2 (25 mL) is added and the solution bought slowly to room temperature. The solution is partitioned between CH2Cl2 and 10% aqueous sodium bicarbonate. The organic phase is dried and concentrated under reduced pressure. The residue is flash chromatographed (CH2Cl2-MeOH, 9:1) to give crude tert-butyl N-[5-(hydroxymethyl)-2,2-dimethyl-1,3-dioxan-5-yl]carbamate (2.81 g, 99%). A portion of this material (0.2 g) is further flash chromatographed (EtOAc-hexanes) and the purified product is then dissolved in methanol (40 mL) and stirred with palladium on charcoal (5%, 22 mg) under a balloon of hydrogen. After 2 h the catalyst is removed by filtration. The solution is concentrated to dryness and the residue dissolved in 90% aqueous TFA. After 30 min the solution is concentrated under reduced pressure and the residue dissolved in H2O. Lyophilization gives 83 (90 mg, 0.29 mmol, 76%). 1H NMR (500 MHz, D2O, HOD 4.70 ppm) δ 3.99 (s, 2H), 3.75 (s, 4H). 13C NMR (125.7 MHz, D2O, referenced to internal acetone at δ 30.3) δ 62.8 (d, JC,P = 4.6 Hz), 60.8 (d, JC,P = 8.1 Hz), 59.3. 31P NMR (202.4, MHz, D2O) δ 0.0. ES-HRMS C4H13NO6P [M+H]+ calcd. 202.0481, found 202.0484.
{3-Hydroxy-2-[({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]-2-(hydroxymethyl)propoxy}phosphonic acid (84)
Compound 83 (55 mg, 0.18 mmol), aldehyde 8 (93 mg, 0.26 mmol) and sodium cyanoborohydride (7.8 mg, 0.48 mmol) are stirred in MeOH (5 mL) at 40 °C for 16 h and then at room temperature for 48 h. The solution is concentrated onto silica gel and then flash chromatographed on silica (MeCN-H2O, 4:1 v/v), Further purification by flash chromatography (CH2Cl2–MeOH-28% aq. NH4OH, 25:25:10 v/v/v then 50:80:20 v/v/v) gives [2-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)-3-hydroxy-2-(hydroxymethyl)propoxy]phosphonic acid (7.0 mg, 0.015 mmol, 9%). 1H NMR (500 MHz, D2O, HOD 4.70 ppm) δ 8.35 (s, 1H), 7.79 (s, 1H), 7.46 (m, 2H), 7.41 (m, 3H), 5.45 (s, 2H), 4.47 (s, 2H), 4.09 (m, 2H), 3.86 (m, 4H). 13C NMR (125.7 MHz, D2O) δ 155.9, 149.3, 146.6, 135.6, 133.0, 128.8, 128.6, 127.8, 115.2, 104.6, 68.6, 65.7(d, Jc,p = 7 Hz), 60.9, 57.9, 35.4. 31P NMR (202.4 MHz, D2O) δ 1.2. ESI-HRMSMS C18H24N4O7P [M+H]+ calcd. 439.1383, found 439.1385. This material in methanol (5 mL) and water (5 mL) is stirred with Pearlmann's catalyst (2 mg) under a balloon of hydrogen for 16 h. The solution is filtered and the solvent evaporated to give 84 (6.0 mg, 0.014 mmol) and crystallised from MeCN-H2O. (2.0 mg, 41%) H NMR (500 MHz, D2O, HOD 4.70 ppm) δ 7.98 (s, 1H), 7.61 (s, 1H), 4.22 (s, 2H), 3.90 (d, J = 6.4 Hz, 2H,), 3.77 (m, 4H). 13C NMR (125.7 MHz, D2O) δ 155.6, 143.1, 142.7, 130.0, 117.4, 109.8, 63.7, 61.5, 59.0, 35.0. 31P NMR (202.4 MHz, D2O) δ 4.5. ESI-HRMS C11H18N4O7P [M+H]+ calcd. 349.0913, found 349.0911.
[(2R)-2-Amino-3-hydroxypropoxy]phosphonic acid (86)
Dibenzyl diisopropylphosphoramidite (1.067 mL, 3.24 mmol) is added to compound 8540,41 (0.5 g, 2.162 mmol) and 1H-tetrazole (0.454 g, 6.49 mmol) in CH3CN (10 mL) and the mixture stirred for 1 h. The solvent is evaporated and the residue flash chromatographed (EtOAc-hexanes, 1:9 v/v) to give tert-butyl (4R)-4-({[bis(benzyloxy)phosphanyl]oxy}methyl)-2,2-dimethyl-1,3-oxazolidine-3-carboxylate as a colourless oil. This is dissolved in CH2Cl2 (10 mL) cooled in ice-water and MCPBA (m-chloroperoxybenzoic acid) (0.995 g, 4.32 mmol) added and stirred for 30 min. The mixture is diluted with CH2Cl2 (50 mL) and washed with sat. aq. Na2SO3, sat. aq. NaHCO3 (3x) then brine, dried and the solvent evaporated. The residue is flash chromatographed (DCM-hexanes-EtOAc, 4:3:1 v/v/v) to give tert-butyl (4R)-4-({[bis(benzyloxy)phosphoryl]oxy}methyl)-2,2-dimethyl-1,3- oxazolidine-3-carboxylate as a colourless oil (0.816 g, 77%). (c 0.57, CHCl3). 1H NMR (500 MHz, CDCl3) 7.34 (s, 10H), 5.08-5.00 (m, 4H), 4.22-4.16 (m, 0.5H), 4.13-4.06 (m, 1H), 3.97-3.83 (m, 3H), 3.77 (q, J =8.6, 0.5H), 1.52-1.41 (m, 15H). 13C NMR (125.7 MHz) 152.1, 151.4 (C), 135.7 (C), 128.6 (CH), 127.9 (CH), 94.1, 93.6 (C), 80.6, 80.3 (C), 69.4 (CH2), 65.7, 65.3 (CH2), 64.8, 64.6 (CH2), 56.6, 56.5, 56.32, 56.26 (CH), 28.3 (CH3), 27.4, 24.3 (CH3), 26.6, 23.0 (CH3). Referenced to the centre line of CDCl3 at δ 77.0. 31P NMR (202.3 MHz, CDCl3) -1.0 (s), -1.1 (s). ESI-HRMS for C25H34NNaO7P [M+Na]+ calcd 514.1971; found 514.1968. his material (0.77 g, 1.567 mmol) and 10% Pd-C (100 mg) are stirred in EtOH (15 mL) under a hydrogen atmosphere at ambient temperature and pressure for 16 h. The mixture is filtered through cellulose paper and the solvent is evaporated to give {[(4R)-3-[(tert-butoxy)carbonyl]-2,2-dimethyl-1,3-oxazolidin-4-yl]methoxy}phosphonic acid as a colourless gum (480 mg). This is dissolved in 80% aq. TFA (10 mL) and left at room temperature for 2 h. The solvent is evaporated and the residue dissolved in H2O (10 mL) and washed with CH2Cl2 (2 ×10 mL) then evaporated. The residue is dissolved in H2O and chromatographed on RP 18 silica gel (H2O) to give 86 as a colourless gum which solidified (0.26 g, 97%). 0 (c0.565, H2O). No measurable rotation observed. 1H NMR (500 MHz, D2O) 4.19-4.12 (m, 1H), 4.11-4.03 (m, 1H), 3.90 (dd, J = 12.3, 4.7, 1H), 3.81 (dd, J = 12.3, 6.7, 1H), 3.63 (m, 1H). Reference to HOD at δ 4.79. 13C NMR (125.7 MHz, D2O) 62.9 (d, J = 3.2, CH2), 59.1 (CH2), 53.5 (d, J = 7.4, CH). Referenced to internal CH3CN at δ 1.47. 13P NMR (202.3 MHz, D2O) 0.0 (s). ESI-HRMS or C3H9NO5P [M-H]- calcd 170.0218; found 170.0211.
[(2R)-3-Hydroxy-2-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]propoxy]phosphonic acid; triethylamine (87)
Compound 86 (25 mg, 0.146 mmol) is suspended in methanol (50 mL) and brought to pH7 with Et3N. Aldehyde 8 (22 mg, 0.088 mmol) and then picoline borane complex (20.3 mg, 0.19 mmol) are added and the suspension stirred at 50 °C for 60 h. The solution is evaporated onto silica gel. Flash chromatography (1,4-dioxane-H2O-Et3N, 30:10:0.4 v/v/v then 20:10:0.3 v/v/v) gives [(2R)-3-hydroxy-2-[({4-benzyloxy-5H-pyrrolo[3,2-d]pyrimidin-7-ylmethyl)amino]propoxy]phosphonic acid; triethylamine (18 mg, 40%). 1H NMR (500 MHz, CD3OD) 8.39 (s, 1H), 7.75 (s, 1H), 7.43 (m, 2H), 7.28 (m, 3H), 5.55 (s, 2H), 4.37 (m, 2H), 4.09 (m, 1H), 3.95 (m, 1H), 3.76 (dd, J = 12.1, 5.0 Hz, 1H), 3.69 (dd, J = 12.1, 6.2 Hz, 1H), 3.23 (m, 1H), 3.01 (q, J = 7.1 Hz, 7H), 1.18 (t, J = 7.1 Hz, 11H). Approximately 1.1 eq. of Et3N present. 13C NMR (125.7 MHz, CD3OD, centre line 49.0 ppm) 157.3, 150.8, 149.6, 137.8, 133.1, 129.7, 116.8, 108.2, 69.4, 62.7 (d, Jc,p = 5 Hz), 60.7 (d, Jc,p = 5 Hz), 60.0, 47.6, 40.4, 9.4. 31P NMR (202.4 MHz, CD3OD) δ 2.7 ESI-HRMS for C17H20N4O6P [M-H]- calcd. 407.1120, found 407.1129. A solution of this material (14 mg, 0.028 mmol) in methanol (5 mL) and water (5 mL) is stirred with Pearlmann's catalyst (2 mg) under a balloon of hydrogen for 16 h. The solution is filtered and the solvent evaporated to give 87 (4 mg, 46%). 1H NMR (500 MHz, D2O-CD3OD, 2:1) δ 7.91 (s, 1H), 7.61 (s, 1H), 4.39 (m, 2H), 4.08 (m, 1H), 3.96 (m, 1H), 3.81 (dd, J = 12.1, 5.5 Hz, 1H), 3.74 (dd, J = 12.1, 6.1, Hz, 1H), 3.41 (m, 1H), 3.11 (q, J = 7.1 Hz, 4H), 1.14 (t, J = 7.1 Hz, 11H). Approximately 0.7 eq. of Et3N present. 13C NMR (125.7 MHz, D2O-CD3OD, 2:1 centre line of CD3OD 49.0 ppm) δ 155.0, 143.6, 142.8, 130.8, 117.7, 106.0, 60.9 (d, Jc,p = 5 Hz), 58.3 (d, Jc,p = 7 Hz), 57.6, 46.6, 38.7, 8.1. 31P NMR (202.4 MHz, D2O-CD3OD, 2:1) δ 1.0. ESI-HRMS for C10H14N4O6P [M-H]- calcd. 317.0651, found 317.0644.
[(2R)-2-[({2-Amino-4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]-3-hydroxypropoxy]phosphonic acid hydrochloride (88)
A mixture of 86 (0.15 g, 0.877 mmol), triethylamine (0.123 mL, 0.877 mmol), 7036 (0.122 g, 0.351 mmol) and 2-picoline borane complex (0.049 g, 0.456 mmol) are stirred together in MeOH (15 mL) at 50 °C f or 17 h then the solvent is evaporated and the residue ash chromatographed (3% aq. Et3N in 1,4-dioxane-H2O, 8:2 v/v) to give [(2R)-2-[({2-[(E)-[(dimethylamino)methylidene]amino]-3-{[(2,2-dimethylpropanoyl)oxy]methyl}-4-oxo-3H,4H,5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]-3-hydroxypropoxy]phosphonic acid; triethylamine as a yellow solid (0.144 g, 68.0%). (c 0.51, MeOH). 1H NMR (500 MHz, CD3OD, 4 mg in 0.6 mL) 8.70 (s, 1H), 7.47 (s, 1H), 6.31 (s, 2H), 4.38 (d, J = 13.6, 1H), 4.32 (d. J = 13.6, 1H), 4.25 (ddd, J = 12.2, 8.6, 3.2, 1H), 4.03 (ddd, J =12.2, 10.1, 5.1, 1H), 3.89 (dd, J = 12.0, 5.1, 1H), 3.84 (dd, J = 12.0, 6.7, 1H), 3.34 (m, 1H), 3.22 (s, 3H), 3.08 (s, 3H), 3.02 (q, J = 7.3, 5.4H), 1.24 (t, J = 7.3, 8H), 1.15 (s, 9H). Approximately 0.9 eq. of Et3N present. 1H NMR (500 MHz, CD3OD, 25 mg in 0.6 mL) 8.69 (s, 1H), 7.47 (s, 1H), 6.27 (d, J =9.0, 1H), 6.22 (d, J = 9.1, 1H), 4.37 (d, J = 13.6, 1H), 4.32 (d. J = 13.6, partly overlapped with ddd at 4.28, 1H), 4.28 (ddd, J = 12.3, 9.0, 3.0, 1H, partly overlapped with d at 4.32), 4.08 (ddd, J = 12.6, 10.6, 5.3, 1H), 3.96-3.86 (m, 2H), 3.38 (m, 1H), 3.23 (s, 3H), 3.09 (s, 3H), 3.05 (q, J =73, 5.4H), 1.25 (t, J = 7.3, 8H), 1.13 (s, 9H). 13C NMR (125.7 MHz, CD3OD, 25 mg in 0.6 mL) 179.0 (C), 159.0 (CH), 156.2 (C), 155.6 (C), 144.9 (C), 130.5 (CH), 115.1 (C), 108.4 (C), 67.1 (CH2), 62.1 (d, J = 4.7, CH2), 61.4 (d, J = 2.9, CH), 60.3 (CH2), 46.9 (CH2), 41.3 (CH3), 39.8 (C), 35.3 (CH3), 27.4 (CH3), 9.9 (CH3). Referenced to the centre line of CD3OD at δ 49.0. 31P NMR (202.3 MHz, CD3OD) 3.4 (s). ESI-HRMS or C19H30N6O8P [M-Et3N-H]- calcd 501.1863; found 501.1857. The1H and13C NMR spectra displayed significant concentration effects. This material (0.138 g, 0.229 mmol) is heated at 100 °C in 6M aq. HCl (6 mL) for 1.5 h. The solvent is evaporated and the residue flash chromatographed (1,4-dioxane-H2O-Et3N, 75:25:3 then 70:30:3 v/v/v) to give the triethylammonium form of the product as a light brown solid. This is dissolved in 5% HCl (10 mL), the solvent evaporated and the residue dissolved in hot H2O and loaded on to an Amberlyst A21 resin column. The column is eluted first with H2O then the product is eluted off with 1M HCl. The residue after evaporation of the solvent is chromatographed on RP 18 silica gel (H2O) to give 88 as a colourless solid after freeze drying (56 mg, 66%). (c 0.45, 1.5M HCl). 1H NMR (500 MHz, D2O + drop of DCl) 7.62 (s, 1H), 4.49 (d, J = 14.2, 1H), 4.43 (d, J = 14.2. 1H), 4.31 (ddd, J = 12.1, 6.1, 3.6, 1H), 4.19 (ddd, J = 12.1, 6.0, 6.0, 1H), 3.94 (dd, J = 12.5, 4.9, 1H), 3.88 (dd, J = 12.5, 6.3, 1H), 3.65 (m, 1H). Referenced to HOD at 4.79 ppm. 13C NMR (125.7 MHz, D2O + drop of DCl) 154.2 (C), 151.2 (C), 133.1 (C), 132.3 (CH), 112.4 (C), 101.5 (C), 61.7 (d, J = 3.6, CH2), 59.2 (d, J = 6.8, CH), 58.5 (CH2), 39.3 (CH2). Referenced to internal CH3CN at 1.47 ppm. 31P NMR (202.3 MHz, D2O + DCl) -0.2 (s). ESI-HRMS for C10H15N5O6P [M-HCl-H]- calcd 332.0760; found 332.0755. Found 10.1% Cl, C10H16N5O6P•HCl required 9.6% Cl indicating the product is a mono hydrochloride salt.
[(2S)-3-hydroxy-2- [({4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-l}methyl)amino]propoxy]phosphonic acid; triethylamine (89) is prepared, in the same manner as that of the enantiomer 87 and has the same1H, 13C and31P NMR as the enantiomer 87. ES-HRMS or C10H15N4NaO6P [M+Na]+ calcd. 341.0627, found 341.0626.
[(2S)-2-[({2-amino-4-hydroxy-5H-pyrrolo[3,2-d]pyrimidin-7-yl}methyl)amino]-3-hydroxypropoxy]phosphonic acid hydrochloride (90) is prepared, in the same manner as that of the enantiomer 88 as a colourless amorphous solid. (c 0.42, 1.5M HCl). ESI-HRMS or C10H15N5O6P [M-HCl-H]- calcd 332.0760; found 332.0756. The1H, 13C and31P NMR spectra are identical to those of the enantiomer 88.
Benzyl N-[(1 R)-2-[(tert-butyldiphenylsilyl)oxy]-1-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]ethyl]carbamate (92)
A solution of compound 91 (2.0g, 12.4 mmol) and imidazole (3.38 g, 49.6 mmol) in DMF (50 mL) is cooled in an ice-water bath whilst tert-butylchlorodiphenylsilane(11.5 mL, 43.4 mmol) is added from a syringe. The mixture is stirred at room temperature for 2 h and then diluted with water and extracted three times with EtOAc. The combined extracts are washed with saturated aqueous NaCl, dried and concentrated under reduced pressure. The residue is flash chromatographed EtOAc-CH2Cl2-Et3N, 10:90:0.1 then stepwise to 40:60:0.1) to give O-silylated hydroxylamine (3.9 g, 79%). This material is dissolved in THF-water (2:1, 90 mL) and benzylchloroformate (1.53 mL, 10.7 mmol) and NaHCO3 (2.46g, 29.3 mmol) are added. After stirring for 48 h the mixture is partitioned between EtOAc and water. The organic phase is washed with saturated aqueous NaCl, dried and concentrated under reduced pressure. The residue is flash chromatographed (EtOAc-hexanes, 1:9 v/v then stepwise to 2:8 v/v) to give 92 (4.0 g, 77%). 1H NMR (500 MHz, CDCl3) δ 7.65 (m, 4H), 7.37 (m, 11H), 5.11 (d, J = 12.3 Hz, 1H), 5.05 (d, J = 12.3 Hz, 1H), 4.95 (bd, J = 9.4 Hz, 1H), 4.44 (m, 1H), 4.02 (m, 1H), 3.91 (m, 1H), 3.70 (m, 3H), 1.38 (s, 3H), 1.34 (s, 3H), 1.05 (s, 9H). 13C NMR (125 MHz, CDCl3, centre line δ 77.0) δ 156.4, 136.5, 135.6, 133.2, 129.8, 128.5, 128.1, 127.7, 109.2, 73.9, 66.9, 66.2, 63.8, 52.7, 26.8, 26.3, 25.1, 19.2. ESI-HRMS for C31H39NO5SiNa [M+Na]+ calcd. 556.2496, found 556.2486.
Benzyl (4R,5S)-5-({[bis(benzyloxy)phosphoryl]oxy}methyl)-4-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,2-dimethyl-1,3-oxazolidine-3-carboxylate (93)
A solution of 92 (0.75g, 1.41 mmol) in AcOH- THF-water (3:1:1, 35 mL) is heated at 50 °C for 5 h. The solvents are removed under reduced pressure. The residue is dissolved in, and evaporated from, ethanol and toluene to give benzyl N-[(2R,3S)-1-[(tert-butyldiphenylsilyl)oxy]-3,4-dihydroxybutan-2-yl]carbamate (0.71g, >100%) which is used without further purification. A portion of this material (0.33g, 0.67 mmol) and pyridine (0.16 mL, 2.0 mmol) are dissolved in CH2Cl2 (10 mL) and cooled in a dry ice-acetone bath then benzoyl chloride (0.135 mL, 1.15 mmol) is added. The solution is stirred for 30 min and then quenched with water, brought to room temperature and washed with water, 10% aqueous NaHCO3 and saturated aqueous NaCl, dried and concentrated under reduced pressure. The resulting (2S,3R)-3- {[(benzyloxy)carbonyl]amino}-4-[(tert-butyldiphenylsilyl)oxy]-2-hydroxybutyl benzoate is taken up in acetone (16 mL) and 2,2-dimethoxypropane (4 mL), with a trace of toluenesulfonic acid, and stirred at 50 °C for 3 h. The solution is cooled and concentrated under reduced pressure. The residue is taken up in EtOAc, washed with 10% aqueous NaHCO3, dried and concentrated under reduced pressure. The resulting benzyl (4R,5S)-5-[(benzoyloxy)methyl]-4-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,2-dimethyl-1,3-oxazolidine-3-carboxylate is dissolved in methanol (20 mL) and basified with sodium methoxide. After 1 h saturated aqueous ammonium chloride (0.5 mL) is added, the mixture is concentrated under reduced pressure, taken up in EtOAc, washed with water, dried (MgSO4) and evaporated to dryness. Flash chromatography (EtOAc-hexanes 20:80 v/v, then stepwise to 25:75 v/v) gives benzyl (4R,5S)-4-{[(tert-butyldiphenylsilyl)oxy]methyl}-5-(hydroxymethyl)-2,2-dimethyl-1,3-oxazolidine-3-carboxylate (0.16 g, 43%). 1H NMR (500 MHz, CDCl3) δ 7.60 (m, 4H), 7.47-7.12 (m, 11H), 5.20-4.84 (m, 2H), 4.36 (m, 1H), 4.05-3.60 (m, 5H), 2.10 (m, 1H), 1.67 (s, 3H), 1.52 (s, 3H), 1.05 (s, 9H). 13C NMR (125.7 MHz, CDCl3 centre line δ 77.0, signals split due to hindered rotation) δ 152.4, 136.3, 135.7, 133.1, 129.8, 128.4, 128.0, 127.8, 95.4, (78.5, 77.6), 66.9, (63.5, 61.7), (59.8, 58.9), 28.1, 26.9, 26.1, 19.2. ESI-HRMS C31H39NO5SiNa [M+Na]+ calcd. 556.2495, found 556.2495. A solution of this material(160 mg, 0.30 mmol) and 1H-tetrazole (42 mg, 0.60 mmol) in MeCN (9 mL) is cooled in an ice-H2O bath and dibenzyl diisopropylphosphoramidite (0.168 mL, 0.51 mmol) is added. The mixture is briefly warmed to room temperature, then cooled again in the ice-water bath whilst MCPBA (138 mg, 0.60 mmol) is added. The solution is warmed to room temperature and partitioned between EtOAc and 10% aqueous NaHCO3. The organic phase is concentrated under reduced pressure and the residue flash chromatographed (EtOAc-hexanes, 25:75) to give crude 93 (200 mg, 0.25 mmol, 83%). A portion of this is further purified by flash chromatography (EtOAc-CH2Cl2, 4:96) to give clean 93. 1H NMR (500 MHz, CDCl3) δ 7.58 (bs, 4H), 7.30(m, 20H), 7.11 (bs, 1H), 5.20-4.82 (bm, 2H), 5.02(m, 4H), 4.46(m, 1H), 4.11 (m, 1H), 4.05(m, 1H), 3.85(bs, 1H), 3.71(bs, 2H), 1.61(bs, 3H), 1.50(bs, 3H), 1.09(s, 9H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0, signals split due to hindered rotation) δ 152.2, 135.4, 129.9, 128.4, 128.1, 128.0, 127.8, (95.7, 95.1), (76.4, 75.6), 69.4, 67.7, (67.1, 66.8), (63.0, 61.5), (60.2, 59.2), 28.3, 26.9, 26.5, 14.1. 31P NMR (202,4 MHz, CDCl3) δ -1.0 ES-HRMS for C45H52NO8PSiNa [M+Na]+ calcd. 816.3098, found 816.3095.
Benzyl N-[(2R,3S)-4-{[bis(benzyloxy)phosphoryl]oxy}-1,3-dihydroxybutan-2-yl]carbamate (94)
Compound 93 (200 mg, 0.25 mmol) is dissolved in THF (5 mL) and TBAF (1M, 0.44 mL, 0.44 mmol) is added. After a few minutes the solution is filtered through a pad of silica gel and concentrated under reduced pressure to give crude benzyl (4R,5S)-5-({[bis(benzyloxy)phosphoryl]oxy} methyl)-4-(hydroxymethyl)-2,2-dimethyl-1,3-oxazolidine-3-carboxylate. The residue is stirred in THF-TFA-H2O(1:1:1) for 4 h, concentrated under reduced pressure and flash chromatographed (EtOAc) to give 94 (46 mg, mmol, 36%). 1H NMR (500 MHz, CDCl3) δ 7.34 (m, 15H), 5.48 (m, 1H), 5.06 (m, 6H), 4.08 (m, 1H), 3.99 (m, 2H), 3.79 (m, 1H), 3.71 (m, 2H), 2.10 (bs, 2H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0) δ 156.7, 136.3, 135.4, 133.1, 128.8, 128.7, 128.2, 128.1, 128.1, 71.7, 70.2, 69.9, 67.1, 64.2, 52.6. 31NMR (202.4 MHz, CDCl3) δ 0.5 ES-HRMS for C26H30NO8PNa [M+Na]+ calcd. 538.1067, found 538.1605.
[(2S,3R)-2,4-Dihydroxy-3-[({4-hydroxy-5H-pyrrolo[3,2-d ]pyrimidin-7-yl}methyl)amino]butoxy]phosphonic acid (95)
A solution of compound 94 (100 mg, 0.19 mmol) in methanol (10 mL) is stirred with Pd black (5 mg) under a balloon of hydrogen. After 3 h the catalyst is removed by filtration, 7M NH3 in MeOH (1 drop) added, and the solution concentrated to dryness to give [(2S,3R)-3-amino-2,4-dihydroxybutoxy]phosphonic acid; ammonia (33 mg, 86%). 1H NMR (500 MHz, D2O, HOD 4.70 ppm) δ 4.00 (m, 3H), 3.85 (dd, J = 12.4, 4.2 Hz, 1H), 3.74 (dd, J = 12.4, 7.1 Hz, 1H), 3.49 (m, 1H), 3.31 (s, 1H). 13C NMR (125.7 MHz, D2O) δ 66.9 (d, Jc,p = 8 Hz), 65.1(d, Jc,p = 6 Hz), 59.0, 54.6. 31P NMR (202.4 MHz, D2O) δ 4.8. ESI-HRMS for C4H12NNaO6P [M+Na]+ calcd. 224.0300, ound 224.0299. his material (15 mg, 0.075 mmol) is suspended in methanol and brought to pH 6 with triethylamine, aldehyde 8 (20 mg, 0.060 mmol) and sodium cyanoborohydride (7.0 mg, 0.11 mmol) are added and the mixture stirred at 50 °C for 48 h. The solution is concentrated onto silica gel and the residue flash chromatographed (i-PrOH-H2O, 8:2 v/v) to give crude give [(2S,3R)-3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-yl]methyl}amino)-2,4-dihydroxybutoxy]phosphonic acid which is dissolved in H2O, and chromatographed on a Strata-X cartridge (Phenomenex) eluting with H2O-methanol-28% aq. NH4OH (5:4:1 v/v/v) to give [(2S,3R)-3-({[4-(benzyloxy)-5H-pyrrolo[3,2-d]pyrimidin-7-ylmethyl}amino)-2,4-dihydroxybutoxy]phosphonic acid (12 mg, 38%). 1H NMR (500 MHz, D2O-MeOD, 9:1, HOD 4.70 ppm) δ 8.38 (s, 2H) 8.34 (s, 1H), 7.81 (s, 1H), 7.46 (m, 2H), 7.37 (m, 3H), 5.50 (s, 2H), 4.54 (d, J = 14.1 Hz, 1H), 4.48 (d, J = 14.1 Hz, 1H), 4.02-3.92 (m, 3H), 3.86 (m, 2H), 3.40 (m, 1H). 13C NMR (125.7 MHz, D2O-MeOD, 9:1) δ 170.6 155.8, 149.6, 147.6, 135.9, 132.7, 128.7, 128.5, 127.9, 115.3, 104.6, 68.5, 67.1, 65.6, 59.9, 56.8, 39.0. 31P NMR (202.4 MHz, D2O) δ 1.3. ESI-HRMS for C18H24N4O7P [M+HJ]+ calcd. 439.1383, found 439.1378. A solution of this material (11 mg, 0.022 mmol) in 20% aqueous methanol (7mL) is stirred with Pd black (1 mg) under a balloon of hydrogen. After 14 h the catalyst is removed by filtration and the residue after evaporation purified by flash chromatography (i-PrOH-H2O, 7:3 v/v) to give 95 (3.2 mg, 42%)1H NMR (500 MHz, D2O, HOD 4.70 ppm) δ 8.05 (s, 1H), 7.73 (s, 1H), 4.56 (d, J = 14.1 Hz, 1H), 4.49 (d, J = 14.1 Hz, 1H), 4.00(m, 3H), 3.90 (m, 2H), 3.48 (m, 1H). 13C NMR (125.7 MHz, D2O) δ 155.2, 150.6, 143.1, 130.9, 117.7, 106.2, 67.3 (d, JC,P = 6 Hz), 65.4, 60.0, 56.8, 39.0. 31P NMR (202.4 MHz, D2O) δ 2.9. ESI-HRMS for C11H17N4NaO7P [M+Na]+ calcd. 371.0733, found 371.0733.
Enzyme Inhibition Assays
PfHGXPRT activity was measured using spectrophotometric assays observing the conversion of xanthine and 5-phospho-α-D-ribose-1-pyrophosphate (PRPP) to xanthosine-5′-monophosphate and inorganic pyrophosphate (PPi) at 247 nm (ε257 – 6.8 mM -1 cm -1) or the conversion of guanine and PRPP to guanosine-5′-monophosphate and PPi (ε257 – 5.8 mM -1 cm -1) on a Varian Cary 100 spectrophotometer (Palo Alto, CA) at 37°C. All assays were conducted in 10 mM potassium phosphate (pH 7.6), 10 mM MgCl2, 1 mM PRPP, 150 mM xanthine or 50 mM guanine, 0.5 mM DTT with varying amounts of inhibitor and concentrations of PfHGXPRT between 2 and 10 nM or human HGPRT of 1 nM. As the dissociation constants of many of the inhibitors were near or below that of the enzyme concentration, the data were fit to the Morrison equation for tight-binding inhibitors.50
Scheme 8.

(a) i, (BnO)2PN(iPr)2, ii, MCPBA, iii, H2, Pd/C, iv, aq TFA; (b) i, 2-picoline borane, 8, ii, H2, Pd/C; (c) i, 2-picoline borane, 70, ii, 6M aq HCl.
Table 1. a Activity of synthesized acyclic aza-C-nucleoside phosph(on)ates as inhibitors of Pf and human (Hs) HGXPRT.
| Pf HGXPRT Ki (nM) | Hs HGPRT Ki (nM) | Pf HGXPRT Ki (nM) | Hs HGPRT Ki (nM) | ||
|---|---|---|---|---|---|
| 19 | > 20,000 | > 10,000 | 61 | 1.2 ± 0.1 | 420 ± 10 > 10,000 |
| 20 | 1,900 ± 100 | > 10,000 | 65 | 87 ± 8 | |
| 4 | 10.6 ± 1.324 | 4,900 ± 30024 | 72 | 100 ± 11 | > 10,00 0 |
| 21 | 490 ± 35 | > 10,000 | 75 | 14,400 | > 10,000 |
| 22 | 8,600 ± 900 | > 10,000 | 80 | 19 ± 2 | > 10,000 |
| 24 | 27.6 ± 1.9 | > 10,000 | 81 | 9,400 | > 10,000 |
| 31 | 620 ± 61 | > 10,000 | 84 | 15,300 | > 10,000 |
| 32 | > 20,000 | 11,320 ± 1,300 | 87 | 2.4 ± 0.2 | > 10,000 |
| 44 | 9.1 ± 0.1 | > 10,000 | 88 | 14.3 ± 1.7 | > 10,000 |
| 45 | 490 ± 60 | > 10,000 | 90 | 650 ± 150 | > 10,000 |
| 53 | 0.65 ± 0.0424 | 380 ± 4024 | 95 | 980 ± 70 | > 10,000 |
| 57 | 23.4 ± 2.3 | 16,000 ± 1,000 |
The Ki values were determined from analyses assuming competitive inhibition as in ref. 24.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. World Malaria Report 2011. WHO; 2011. [Google Scholar]
- 2.Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, Tan JC, Al Saai S, Phyo AP, Moo CL, Lwin KM, McGready R, Ashley E, Imwong M, Stepniewska K, Yi P, Dondorp AM, Mayxay M, Newton PN, white NJ, Nosten F, Ferdig MT, Anderson TJC. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bejon P, Lusingu J, Olotu A, Leach A, Lievens M, Vekemans J, Mshamu S, Lang T, Gould J, Dubois MC, Demoiti MA, Stallaert JF, Vansadia P, Carter T, Njuguna P, Awuondo KO, Malabeja A, Abdul O, Gesase S, Mturi N, Drakeley CJ, Savarese B, Villafana T, Ballou WR, Cohen J, Riley EM, Lemnge MM, Marsh K, von Seidlein L. New Engl J Med. 2008;359:2521–2532. doi: 10.1056/NEJMoa0807381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyde JE. Curr Drug Targets. 2007;8:31–47. doi: 10.2174/138945007779315524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reyes P, Rathod PK, Sanchez DJ, Mrema JEK, Rieckmann KH, Heidrich HG. Mol Biochem Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 6.Wang CC. J Med Chem. 1984;27:1–9. doi: 10.1021/jm00367a001. [DOI] [PubMed] [Google Scholar]
- 7.Cassera MB, Hazleton KZ, Riegelhaupt PM, Merino EF, Luo M, Akabas MH, Schramm VL. J Biol Chem. 2008;283:32889–32899. doi: 10.1074/jbc.M804497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kicska GA, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K. J Biol Chem. 2002;277:3226–3231. doi: 10.1074/jbc.M105906200. [DOI] [PubMed] [Google Scholar]
- 9.Cassera MB, Hazleton KZ, Merino EF, Obaldia N, III, Ho MC, Murkin AS, DePinto R, Gutierrez JA, Almo SC, Evans GB, Babu YS, Schramm VL. PLoS ONE. 2011;6:e26916. doi: 10.1371/journal.pone.0026916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piper JR, Laseter AG, Montgomery JA. J Med Chem. 1980;23:357–364. doi: 10.1021/jm00178a002. [DOI] [PubMed] [Google Scholar]
- 11.Piper JR, Laseter AG, Johnston TP, Montgomery JA. J Med Chem. 1980;23:1136–1139. doi: 10.1021/jm00184a015. [DOI] [PubMed] [Google Scholar]
- 12.Schramm VL. J Biol Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Luo M, Schramm VL. J Am Chem Soc. 2009;131:4685–4694. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinch K, Evans GB, Frohlich RFG, Furneaux RH, Kelly PM, Legentil L, Murkin AS, Li L, Schramm VL, Tyler PC, Woolhouse AD. J Med Chem. 2009;52:1126–1143. doi: 10.1021/jm801421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewandowicz A, Ringia EAT, Ting LM, Kim K, Tyler PC, Evans GB, Zubkova OV, Mee S, Painter GF, Lenz DH, Furneaux RH, Schramm VL. J Biol Chem. 2005;280:30320–30328. doi: 10.1074/jbc.M505033200. [DOI] [PubMed] [Google Scholar]
- 16.Schramm VL, Gutierrez JA, Cordovano G, Basu I, Guha C, Belbin TJ, Evans GB, Tyler PC, Furneaux RH. Nucl Acids Symp Ser. 2008;52:75–76. doi: 10.1093/nass/nrn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schramm VL, Tyler PC. Iminosugars. John Wiley & Sons, Ltd; 2008. Transition State Analogue Inhibitors of N-Ribosyltransferases; pp. 177–208. [Google Scholar]
- 18.Schwartz SD, Schramm VL. Nat Chem Biol. 2009;5:551–558. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CM, Tyler PC, Furneaux RH, Kicska G, Xu Y, Grubmeyer C, Girvin ME, Schramm VL. Nat Struct Mol Biol. 1999;6:582–587. doi: 10.1038/9367. [DOI] [PubMed] [Google Scholar]
- 20.Shi W, Li CM, Tyler PC, Furneaux RH, Cahill SM, Girvin ME, Grubmeyer C, Schramm VL, Almo SC. Biochemistry. 1999;38:9872–9880. doi: 10.1021/bi990664p. [DOI] [PubMed] [Google Scholar]
- 21.Shi W, Li CM, Tyler PC, Furneaux RH, Grubmeyer C, Schramm VL, Almo SC. Nat StructMol Biol. 1999;6:588–593. doi: 10.1038/9376. [DOI] [PubMed] [Google Scholar]
- 22.Keough DA, Naesens LMJ, Skinner-Adams TS, de Jersey J, Guddat LW. J Med Chem. 2009;52:4391–4399. doi: 10.1021/jm900267n. [DOI] [PubMed] [Google Scholar]
- 23.Hocková D, Keough DT, Janeba Z, Wang TH, de Jersey J, Guddat LW. J Med Chem. 2012;55:6209–6223. doi: 10.1021/jm300662d. [DOI] [PubMed] [Google Scholar]
- 24.Hazleton KZ, Ho MC, Cassera MB, Clinch K, Crump DR, Rosario I, Merino EF, Almo SC, Tyler PC, Schramm VL. Chem Biol. 2012;19:721–730. doi: 10.1016/j.chembiol.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirschmann R, Yager KM, Taylor CM, Witherington J, Sprengeler PA, Phillips BW, Moore W, Smith AB. J Am Chem Soc. 1997;119:8177–8190. [Google Scholar]
- 26.Bak P, Novák T, Ludányi K, Pete B, Lászl T, Keglevich G. Tetrahedron: Asymm. 1999;10:2373–2380. [Google Scholar]
- 27.Clinch K, Greatrex BW, Mason JM, Tyler PC. WO 2009/082247 2009 [Google Scholar]
- 28.Kamath VP, Juarez-Brambila JJ, Morris CB, Winslow CD, Morris PE. Org Process Res Dev. 2009;13:928–932. [Google Scholar]
- 29.Ganesan M, Muraleedharan KM. Nucleosides, Nucleotides Nucleic Acids. 2010;29:91–96. doi: 10.1080/15257771003597709. [DOI] [PubMed] [Google Scholar]
- 30.Farhanullah, Kang T, Yoon EJ, Choi EC, Kim S, Lee J. Eur J Med Chem. 2009;44:239–250. doi: 10.1016/j.ejmech.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Ho MC, Shi W, Rinaldo-Matthis A, Tyler PC, Evans GB, Clinch K, Almo SC, Schramm VL. Proc Natl Acad Sci. 2010;707:4805–4812. doi: 10.1073/pnas.0913439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clinch K, Evans GB, Fr hlich RFG, Gulab SA, Gutierrez JA, Mason JM, Schramm VL, Tyler PC, Woolhouse AD. Bioorg Med Chem. 2012;20:5181–5187. doi: 10.1016/j.bmc.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foss FW, Jr, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Bioorg Med Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berkowitz DB, Eggen M, Shen Q, Shoemaker RK. J Org Chem. 1996;61:4666–4675. doi: 10.1021/jo9604752. [DOI] [PubMed] [Google Scholar]
- 35.Otaka A, Miyoshi K, Burke TR, Jr, Roller PP, Kubota H, Tamamura H, Fujii N. Tetrahedron Lett. 1995;36:927–930. [Google Scholar]
- 36.Semeraro T, Lossani A, Botta M, Ghiron C, Alvarez R, Manetti F, Mugnaini C, Valensin S, Focher F, Corelli F. J Med Chem. 2006;49:6037–6045. doi: 10.1021/jm060547+. [DOI] [PubMed] [Google Scholar]
- 37.Choi JY, Borch RF. Org Lett. 2006;9:215–218. doi: 10.1021/ol062643a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stowell JK, Widlanski TS. Tetrahedron Lett. 1995;36:1825–1826. [Google Scholar]
- 39.Ooi H, Ishibashi N, Iwabuchi Y, Ishihara J, Hatakeyama S. J Org Chem. 2004;69:7765–7768. doi: 10.1021/jo048817o. [DOI] [PubMed] [Google Scholar]
- 40.Garner P, Park JM. J Org Chem. 1987;52:2361–2364. [Google Scholar]
- 41.Dondoni A, Perrone D. Org Synth Coll Vol. 2004;10:320–326. [Google Scholar]
- 42.Sagawa S, Abe H, Hase Y, Inaba T. J Org Chem. 1999;64:4962–4965. doi: 10.1021/jo9900883. [DOI] [PubMed] [Google Scholar]
- 43.Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. J Med Chem. 2003;46:5271–5276. doi: 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- 44.Varlet JM, Fabre G, Sauveur F, Collignon N, Savignac P. Tetrahedron. 1981;37:1377–1384. [Google Scholar]
- 45.Zanella Y, Berte-Verrando S, Diziere R, Savignac P. J Chem Soc, Perkin Trans. 1995;1:2835–2838. [Google Scholar]
- 46.Girgis NS, Cottam HB, Larson SB, Robins RK. J Heterocycl Chem. 1987;24:821–827. [Google Scholar]
- 47.Mark V, Wazer JRV. J Org Chem. 1964;29:1006–1008. [Google Scholar]
- 48.Lindberg J, Svensson SCT, Påhlsson P, Konradsson P. Tetrahedron. 2002;58:5109–5117. [Google Scholar]
- 49.Ward DE, Rhee CK. Tetrahedron Lett. 1991;32:7165–7166. [Google Scholar]
- 50.Morrison JF. Biochim Biophys Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]