

Abstract
HIV-1 Tat protein plays a crucial role in perturbations of the dopamine (DA) system. Our previous studies have demonstrated that Tat decreases DA uptake, and allosterically modulates DA transporter (DAT) function. In the present study, we have found that Tat interacts directly with DAT, leading to inhibition of DAT function. Through computational modeling and simulations, a potential recognition binding site of human DAT (hDAT) for Tat was predicted. Mutation of tyrosine470 (Y470H) attenuated Tat-induced inhibition of DA transport, implicating the functional relevance of this residue for Tat binding to hDAT. Y470H reduced the maximal velocity of [3H]DA uptake without changes in the Km and IC50 values for DA inhibition of DA uptake but increased DA uptake potency for cocaine and GBR12909, suggesting that this residue does not overlap with the binding sites in hDAT for substrate but is critical for these inhibitors. Furthermore, Y470H also led to transporter conformational transitions by affecting zinc modulation of DA uptake and WIN35,428 binding as well as enhancing basal DA efflux. Collectively, these findings demonstrate Tyr470 as a functional recognition residue in hDAT for Tat-induced inhibition of DA transport and transporter conformational transitions. The consequence of mutation at this residue is to block the functional binding of Tat to hDAT without affecting physiological DA transport.
Keywords: Dopamine transporter, HIV-1 Tat, mutation, uptake, computational modeling, allosteric modulation
Introduction
The estimated prevalence of HIV-1-associated neurocognitive disorders (HAND) is about 70% of HIV-1 positive individuals with antiretroviral therapy (Robertson et al., 2007; Tozzi et al., 2007; Ernst et al., 2009). Cocaine has been shown to increase the incidence and exacerbate the severity of HAND by enhancing viral replication (Nath et al., 2001; Ferris et al., 2008). Antiretroviral agents cannot prevent the production of HIV-1 viral proteins, such as Tat protein, in HIV-1 infected brains in the early stage of HIV-1 infection (McArthur et al., 2010; Nath and Clements, 2011). Tat has been detected in the brains (Del Valle et al., 2000; Hudson et al., 2000; Lamers et al., 2010) and the sera (Westendorp et al., 1995; Xiao et al., 2000) of HIV-1 infected patients. Furthermore, Tat interacting with cocaine exacerbates the progression of neurocognitive impairment (Buch et al., 2011; Gannon et al., 2011).
Accumulating clinical evidence supported by imaging (Chang et al., 2008; Meade et al., 2011a), neurocognitive (Kumar et al., 2011; Meade et al., 2011b), and postmortem examinations (Kumar et al., 2009; Gelman et al., 2012) reveals that abnormal neurocognitive function observed in HAND is associated with dysfunctions in dopamine (DA) neurotransmission (Berger and Arendt, 2000; Purohit et al., 2011). The DA transporter (DAT) terminates DA signaling and thus is central to control synaptic dopaminergic tone (Torres and Amara, 2007). DAT activity is strikingly reduced in HIV-1-infected patients with a history of cocaine use (Wang et al., 2004; Chang et al., 2008). We have demonstrated that Tat allosterically modulates DAT function and reduces DAT cell surface expression in rat striatal synaptosomes (Zhu et al., 2009; Zhu et al., 2011; Midde et al., 2012).
Viral replication within HIV-1 infected brain regions results in Tat release, which elevates DA levels via inhibiting DAT function (Gaskill et al., 2009). Exposure of HIV-1 infected patients to cocaine further impairs DAT function and increases synaptic DA levels (Ferris et al., 2010). Importantly, the elevated DA induced by Tat and cocaine stimulates viral replication and Tat release (Gaskill et al., 2009), which has been implicated in the neuropathogenesis of HAND (Li et al., 2009). Considering oxidative stress-induced damage to dopaminergic neurons, long lasting exposure to viral proteins and elevated DA eventually lead to a DAT deficit that potentiates HAND severity and accelerates its progression (Purohit et al., 2011). To the best of our knowledge, the mechanism(s) of Tat and cocaine interaction with hDAT have been virtually unexplored. In order to explore the molecular mechanism(s) underlying the interplay of Tat with cocaine in disrupting DAT-mediated DA neurotransmission, we performed computational modeling and simulations to predict potential recognition binding sites of human DAT (hDAT) for Tat. Identifying the functional recognition residues in hDAT for Tat may provide therapeutic insights into HAND in concurrent cocaine users. Upon prediction and validation of the functional relevance of tyrosine 470 (Tyr470) in hDAT, we determined the mechanisms that underlie mutation of Tyr470 in Tat-induced inhibition of DA transport and transporter conformational transitions.
Materials and Methods
Construction of plasmids
Plasmid pcDNA3.1+/Tat1-72 that encodes Tat1-72 protein was provided by Dr. Avindra Nath (NINDS/NIH). Plasmid GFP-tagged Tat1-86 was a gift from by Dr. Mauro Giacca (Molecular Medicine Laboratory, ICGEB, Italy). Plasmid pcDNA3.1+/Tat1-101 that encodes Tat1-101 protein was provided by NIH AIDS Reagent Program. Mutation in hDAT (tyrosine to histidine, Y470H-hDAT) was generated based on WT hDAT sequence (NCBI, cDNA clone MGC:164608 IMAGE:40146999) by site-directed mutagenesis. Synthetic cDNA encoding hDAT subcloned into pcDNA3.1+ (provided by Dr. Haley E Melikian, University of Massachusetts) was used as a template to generate Y470H-hDAT using a QuikChange™ site-directed mutagenesis Kit (Agilent Tech, Santa Clara CA). The sequence of the mutant construct was confirmed by restriction enzyme mapping and DNA sequencing.
Cell culture and DNA transfection
CHO cells (ATCC #CCL-61) were maintained in F12 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/mL streptomycin) at 37°C in a 5% CO2 incubator. For hDAT transfection, cells were seeded into 24 well plates at a density of 1×105 cells/cm2. After 24 h, cells were transfected with WT or mutant DAT plasmids using Lipofactamine 2000 (Life Tech, Carlsbad, CA). Cells were used for the experiments after 24 h of transfection.
Co-immunoprecipitation (Co-IP) of DAT and Tat
To determine whether Tat directly binds to DAT, Co-IP of Tat and DAT assays were performed in rat synaptosomes after exposure to recombinant Tat1-86 as described previously (Li et al., 2008). In brief, rat anti-DAT antibody (6 μg, MAB369, Millipore, Temecula, CA) was incubated with 20 μl protein A/G agarose beads (SC2003, Santa Cruz Biotechnology Inc., Santa Cruz, CA) for 5–6 h at 4°C with constant rotating and were centrifuged at 8,000 g for 5 min. The agarose-anti DAT antibody complex was washed five times with immunoprecipitation buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, 1 mM EDTA, 1 mM EGTA, 0.2 mM sodium ortho-vanadate, 0.2 mM PMSF, 0.5% NP-40) to remove the unbound antibody. Rat synaptosomes from striatum and cerebellum, and spleen homogenates were prepared as described previously (Zhu et al., 2009) and adjusted to equal protein concentration (1.5 mg/ml) using the Bradford protein assay (Bradford, 1976). Then, aliquots (500 μg) of synaptosomes or homogenates were incubated in Krebs-Ringer-HEPES (KRH) buffer (final concentration in mM: 125 NaCl, 5 KCl, 1.5 MgSO4, 1.25 CaCl2, 1.5 KH2PO4, 10 D-glucose, 25 HEPES, 0.1 EDTA, 0.1 pargyline, and 0.1 L-ascorbic acid; pH 7.4) containing recombinant Tat1-86 (350 nM, final concentration, Clade B, # REP0002a, DIATHEVIA, Fano, Italy) for 1 h at room temperature and were then centrifuged at 8,000 g for 5 min. The resulting pellets were washed 5 times with KRH buffer. The pellets were resuspended and added to the agarose-antibody complex and incubated with agitation at 4°C overnight. These samples (agarose-antibody-protein complex) were centrifuged at 8,000 g for 1 min and the resulting pellets were washed with immunoprecipitation buffer 5 times. These samples were then mixed with 2 × Laemmli sample buffer and boiled 5 min. To detect immunoreactivity of DAT or Tat protein, the samples were then subjected to Western blotting with either goat polyclonal DAT antibody (1:200, Cat # SC-1433, Santa Cruz Biotechnology Inc., Santa Cruz, CA) or mouse anti HIV-1 Tat (1:1000, Cat # ab24778, Abcam, Cambridge, MA) using our published method (Zhu et al., 2009).
GST-pull-down assay
To confirm whether Tat interacts with DAT through a protein-protein interaction, GST-Tat fusion protein was used as bait for pull-down DAT to show their interaction as described previously (Li et al., 2008). In brief, BL21 E. coli expressing pGEX-Tat1-86 (obtained from Dr. Virginie W Gautier, University College Dublin, Ireland) and GST only (as negative control) were grown in liquid culture media and induced GST protein expression by 100 mM IPTG. These GST proteins were added to glutathione sepharose beads (17-0756-01, GE Healthcare) and then incubated with the cell lysates from CHO cells transfected with hDAT. The beads were washed with the immunoprecipitation buffer described above and mixed with protein sample buffer. The eluted proteins were subjected to immunoblotting with anti-DAT antibody (Cat # SC-1433, Santa Cruz Biotechnology Inc., Santa Cruz, CA).
Predicting the site for hDAT binding with Tat
The binding structure of hDAT with HIV-1 clade B type Tat was modeled and simulated based on the nuclear magnetic resonance (NMR) structures of Tat (Peloponese et al., 2000) and the constructed structure of DAT(DA), as reported previously (Huang and Zhan, 2007; Huang et al., 2009). Briefly, Brownian dynamics (BD) simulations were performed to obtain the initial binding structure of the hDAT-Tat complex. Starting from the available 11 NMR structures of Tat, the BD simulations were launched from a spherical surface around the extracellular side of hDAT, and the electrostatic interaction energy was calculated for each BD trajectory by multiplying the electrostatic potential of hDAT with the atomic charges of Tat. The initial complex for hDAT binding with Tat was identified from the BD trajectories, with the lowest interaction energy and the best geometric matching quality. The identified initial hDAT-Tat complex structures were energy-minimized in the same way as described in our previous studies on hDAT binding with DA and cocaine (Huang and Zhan, 2007; Huang et al., 2009). Molecular dynamics (MD) simulations were performed to further relax and equilibrate the energy-minimized structure of the hDAT-Tat binding complex. Finally, the MD-simulated hDAT-Tat binding structure was energy-minimized and analyzed.
Preparation of released Tat from Tat-expressing cells
To generate released Tat from Tat-expressing cells, CHO cells were seeded into 60 mm plates at a density of 1 × 106/cm2. After 24 h, cells were transfected with different amounts (5 or 10 μg) of plasmid DNAs for Tat1-72, GFP-tagged Tat1-86 and Tat1-101 using Lipofactamine 2000. Cells transfected with pcDNA3.1+ were used as a negative control. After transfection, culture media from Tat- transfected cells were collected at 24, 48 and 72 h.
[3H]DA uptake assay
Twenty four hours after transfection, [3H]DA uptake in CHO cells transfected with wild type hDAT (WT hDAT) and Y470H-hDAT was performed in KRH buffer using a modified procedure as reported previously (Zhu et al., 2009). To determine whether mutated hDAT alters the maximal velocity (Vmax) or Michaelis-Menten constant (Km) of [3H]DA uptake, kinetic analyses were conducted in WT hDAT versus Y470H-hDAT in the presence or absence of recombinant Tat1-86. To generate saturation isotherms, [3H]DA uptake was conducted in duplicate wells containing one of six concentrations of unlabeled DA (final DA concentrations, 1.0 nM–5 μM) and a fixed concentration of [3H]DA (500,000 dpm/well, specific activity, 31 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA). In parallel, nonspecific uptake of each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. To determine the effect of Tat on DA uptake, cells transfected with WT or Y470H-hDAT were preincubated with each concentration of [3H]DA in the presence or absence of the concentrations of released Tat or Tat1-86 (350 nM). The reaction was terminated by washing twice with ice cold uptake buffer. Cells were solubilized in 1% SDS and radioactivity was measured using a liquid scintillation counter (model Tri-Carb 2900TR; PerkinElmer Life and Analytical Sciences, Waltham, MA). Kinetic parameters (Vmax and Km) were determined using Prism 5.0 (GraphPad Software Inc., San Diego, CA).
For the competitive inhibition experiment, assays were performed in duplicate in a final volume of 500 μl. Cells in each well were incubated in 450 μl buffer containing 50 μl one of final concentrations of unlabeled DA (1 nM-1 mM), GBR12909 (1 nM-10 μM), cocaine (1 nM-1 mM), or ZnCI2 (10 μM) at 37°C for 10 min and [3H]DA uptake was determined by addition of 50 μl of [3H]DA (0.1 μM, final concentration) for an additional 5 min.
Immunodepletion
Released Tat was prepared as described above. Seventy-two hours after transfection with Tat1-72 plasmid, aliquots (500 μl) of the conditioned media were incubated with mouse anti-Tat antibody (1:200, # ab6539, Abcam, Cambridge, MA) at 4°C for 2 h on a shaking platform, followed by incubation with 20 μl of Protein A/G – PLUS Agarose beads (# SC2003, Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 4°C for 2 h. After incubation, the agarose-antibody-Tat complex was pelleted at 12,000 g for 2 min at 4°C and supernatants were collected. For the immunodepletion assay, CHO cells transfected with hDAT were incubated with either Tat-conditioned media or supernatant at 37°C for 2 h, followed by [3H]DA uptake assay, as described above. Anti-Tat antibody specificity for the Tat protein was determined by using mouse IgG1 kappa monoclonal antibody (1:200, # ab18447, Abcam, Cambridge, MA) as an isotype control.
Cell surface biotinylation
To determine whether decreased DA uptake in Y470H-hDAT is due to a reduction of cell surface DAT, biotinylation assays were performed as described previously (Zhu et al., 2005). CHO cells expressing hDAT and Y470H-hDAT were plated on 6 well plates at a density of 105 cells/well. Cells were incubated with 1 ml of 1.5 mg/ml sulfo-NHS-SS biotin (Pierce, Rockford, IL) in PBS/Ca/Mg buffer (In mM: 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, 0.1 CaCl2, pH 7.3). After incubation, cells were washed 3 times with 1 ml of ice-cold 100 mM glycine in PBS/Ca/Mg buffer and incubated for 30 min at 4°C in 100 mM glycine in PBS/Ca/Mg buffer. Cells were then washed 3 times with 1 ml of ice-cold PBS/Ca/Mg buffer and then lysed by addition of 500 ml of Triton X-100, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μM pepstatin, 250 μM phenylmethylsulfonyl fluoride), followed by incubation and continual shaking for 20 min at 4 °C. Cells were transferred to 1.5 ml tubes and centrifuged at 20,000 g for 20 min. The resulting pellets were discarded, and 100 μl of the supernatants was stored at −20 °C for determination of immunoreactive total DAT. Remaining supernatants were incubated with continuous shaking in the presence of monomeric avidin beads in Triton X-100 buffer (100 μl/tube) for 1 h at room temperature. Samples were centrifuged subsequently at 17,000 g for 4 min at 4°C, and supernatants (containing the nonbiotinylated, intracellular protein fraction) were stored at −20°C. Resulting pellets containing the avidin-absorbed biotinylated proteins (cell-surface fraction) were resuspended in 1 ml of 1.0% Triton X-100 buffer and centrifuged at 17,000 g for 4 min at 4°C, and pellets were resuspended and centrifuged twice. Final pellets consisted of the biotinylated proteins adsorbed to monomeric avidin beads. Biotinylated proteins were eluted by incubating with 50 μl of Laemmli sample buffer for 20 min at room temperature. If further assay was not immediately conducted, samples were stored at −20 °C.
[3H]WIN 35,428 Binding Assay
For the competitive inhibition experiment, cells transfected with hDAT and Y470H-hDAT were incubated in KRH buffer containing 50 μl of [3H]WIN 35,428 (5 nM, final concentration, specific activity, 85 Ci/mmol) and ZnCI2 (10 μM) using our published method (Zhu et al., 2009). The reaction was terminated by washing twice with ice cold KRH buffer. Nonspecific binding at each concentration of [3H]WIN 35,428 was determined in the presence of 30 μM cocaine (final concentration). Cells were solubilized in 1% SDS and radioactivity was measured using a liquid scintillation counter.
DA efflux assay
Basal efflux from CHO cells transfected with hDAT or mutated hDAT was measured, as described previously (Guptaroy et al., 2009). Cells were incubated in 24 well plates at a density of 105 cells/well for 24 h. Before assays, cells were washed 3 times with KRH buffer and preloaded with [3H]DA (0.05 μM, final concentration) for 20 min at room temperature. After loading, cells were washed 3 times with KRH buffer. To obtain an estimate of the total amount of [3H]DA in the cells at the zero time point, cells from a set of wells (four wells/sample) were lysed rapidly in 1% SDS after preloading with [3H]DA. Buffer (500 μl) was added into separate set of cell wells and transferred to scintillation vials after 1 min as fractional efflux at 1 min, and another 500 μl buffer was added to the same wells (where the buffer was just removed for 1 min time point) and collected to vials after 10 min. Additional fractional efflux at 20, 30, 40 and 50 min, respectively, was repeated under the same procedure. After 40 or 50 min, cells were lysed and counted as total amount of [3H]DA remaining in the cells from each well. To determine whether exposure to Tat alters basal DA efflux, CHO cells transfected with hDAT were incubated with Tat-conditioned media from Tat-transfected cells collected at 72 h after transfection at 37°C for 2 h, followed by DA efflux assay.
Data Analysis
Descriptive statistics and graphical analyses were used as appropriate. Results are presented as mean ± S.E.M., and n represents the number of independent experiments for each experiment group. IC50 values for DA, cocaine and GBR12909 inhibiting specific [3H]DA uptake were determined from inhibition curves by nonlinear regression analysis using a one-site model with variable slope. Kinetic parameters (Vmax or Km) of [3H]DA uptake were determined from saturation curves by nonlinear regression analysis using a one-site model with variable slope. For experiments involving comparisons between unpaired samples, unpaired Student's t test was used to assess any difference in the kinetic parameters (IC50, Vmax or Km) between WT and mutant; log-transformed values of IC50 or Km were used for the statistical comparisons. Significant differences between samples were analyzed with separate ANOVAs followed by post-hoc tests, as indicated in the results section of each experiment. All statistical analyses were performed using IBM SPSS Statistics version 20, and differences were considered significant at p < 0.05.
Results
Tat protein directly binds to hDAT
Exposure of rat striatal synaptosomes to Tat protein inhibits DA uptake (Zhu et al., 2009). To determine whether Tat protein directly binds to DAT, we performed Co-IP of hDAT and Tat assays. As depicted in Fig. 1A, recombinant Tat1–86 bound to Tat antibody was able to immunoprecipitate hDAT in rat striatal synaptosomes but not in spleen and cerebellum where the density of DAT was low. To confirm this finding, we also used GST-Tat fusion protein (as bait) to pull down hDAT to show their interaction. Figure 1B shows that GST-Tat1–86 bound to hDAT protein. These data strongly suggest that the influence of Tat on DAT function involves a protein-protein interaction between Tat and DAT, which provides an experimental base for us to perform the following computational modeling analysis of the bindings between Tat and hDAT.
Figure 1.

A direct interaction between Tat and DAT and the energy-minimized hDAT(DA) binding complex following the MD simulation. Co-IP of DAT and Tat was performed by immunoprecipitation (IP) with anti-DAT antibody as bait and immunoblot (IB) with anti-Tat antibody. (A) Co-IP of DAT and Tat. Rat synaptosomes from spleen, cerebellum, striatum were preincubated with (+, lanes 1, 2 and 4, from left) or without (−, lane 3) 350 nM recombinant Tat1-86 (rTat1-86). Top panel: DAT immunoreactivity was detected in striatum but not in spleen and cerebellum. Bottom panel: rTat1-86 bound to agarose beads was able to immunoprecipitate DAT in rat striatum but not in spleen and cerebellum. rTat1-86 (10 ng) was loaded in lane 5 as the positive control for Tat immunoreactivity. (B) GST-Tat1-86 bound to WT hDAT protein. Top panel: The GST-Tat1-86 fusion proteins were bound to glutathione-sepharose beads, and then incubated with cell lysates from CHO cells transfected with WT hDAT at room temperature for 1 h following Western Blot using anti-DAT. GST-Tat fusion protein bound to glutathione-sepharose was able to pull down DAT, but GST alone was not. Bottom panel: DAT immunoreactivity in CHO cells expressing hDAT was shown in all lanes. (C) Side view of the complex structure. Tat is shown as the ribbon in cyan color and hDAT(DA) as the ribbon in gold color. Atoms of residue C22 (Cys22) of Tat are shown as overlapped balls in cyan color. Atoms of substrate dopamine (DA) and the Cl− ion are shown as overlapped balls in green color. 2 Na+ ions are shown as balls in blue color. The vestibule (colored in purple) is represented as the molecular surface calculated by using the program HOLLOW (Ho and Gruswitz, 2008). (D) Local view of the anchoring residues Lys 19 (K19) and Cys22 (C22) of Tat inside the vestibule of hDAT(DA). Residues K19 and C22 of Tat are shown in ball-and-stick style, and colored by the atom types. Residue Tyr470 (Y470) of hDAT(DA) is also shown in ball-and-stick style and colored by the atom types. The hydrogen bonding between the K19 side chain of Tat and the hydroxyl oxygen atom on Y470 side chain of hDAT(DA) is indicated with the dashed line. Non-popar hydrogen atoms are not shown for clarity. The positions of transmembrane domain11 and 12 (TM11 and TM12) of hDAT(DA) are also labeled.
Binding structure of hDAT with HIV-1 Tat
The energy-minimized binding structure of hDAT with Tat following the MD trajectory was shown in Fig. 1C and 1D. Tat protein is located on the gate of the vestibule of hDAT(DA). A loop (formed from residues #19 to #22) of Tat is plunged into the vestibule of hDAT(DA), blocking the central pore of the substrate-entry tunnel of hDAT(DA). Tat and DAT molecules bind with each other through both electrostatic interactions and shape complementarity. Particularly, the side chain of Cys22 (C22) of Tat is located inside the vestibule of hDAT(DA), contacting closely with the side chain of Tyr470 residue of hDAT and Lys19 (K19) side chain of Tat. The positively charged head group of Lys19 side chain of Tat is hydrogen-bonded with the hydroxyl oxygen on Tyr470 side chain of hDAT(DA). The positively charged side chain of Lys19 also interacts with the aromatic side chain of Tyr470 through the cation-π interaction; the modeled distance between the N atom of Lys19 side chain and the center of the aromatic ring of Tyr470 side chain of hDAT(DA) is 4.55 Å. Based on the modeled hDAT-Tat complex structure, we predicted that residue Tyr470 is critical for the hDAT binding for Tat.
Extracellularly Released Tat is more potent than recombinant Tat in inhibiting hDAT function
Most previous studies of Tat-induced inhibition of DAT function have been performed using recombinant Tat. To mimic the nature of Tat released from HIV-1 infected cells, we have established a technique to ensure that clade B type Tat can be released from Tat-expressing cells, and the effects of released Tat on DA uptake were examined. CHO cells were transfected with different amounts of plasmid Tat1–72, GFP-tagged Tat1–86, and Tat1–101 DNA, and subsequently the conditioned media from these transfected cells were collected as a source of released Tat. The estimated amount (~1 ng/ml) of released Tat in culture media was measured by the density of immunoreactive bands and quantitated by comparison to a known amount of recombinant Tat1–86.
To determine the effects of released Tat on DA uptake, we first performed the concentration and time-dependent studies for released Tat. Different amounts of released Tat from conditioned media collected at 24, 48 and 72 h were tested in [3H]DA uptake in CHO expressing hDAT. A maximal effect of released Tat on DA uptake was observed when 100 μl conditioned media collected at 72 h were used (data not shown). As shown in Fig. 2A, released Tat from cells transfected with Tat1–72, Tat1–86 or Tat1–101 produced a similar magnitude of change from control in [3H]DA uptake (F(3, 12) = 29.6; p < 0.01, one-way ANOVA with Dunnett's multiple comparison test), suggesting that Tat1–72, Tat1–86 and full length Tat1–101 exhibit an equal ability in Tat-induced inhibitory effect on DA transport. We next determined whether the inhibitory effect on DA uptake was specific for released Tat by immunodepletion assay (Fig. 2B). Exposure to released Tat (1 ng/ml) produced a significant reduction (31 ± 2.7%) of specific [3H]DA uptake compared to control (media collected from cells transfected with vector alone). The released Tat-induced decrease in DA uptake was diminished by immunodepletion with anti-Tat antibody but not with an isotype control antibody (F(3, 12) = 13.4; p<0.001, one-way ANOVA with Tukey's multiple comparison test). These data also confirmed that the inhibitory effect of incubation with conditioned media on DA uptake was specific for released Tat.
Figure 2.
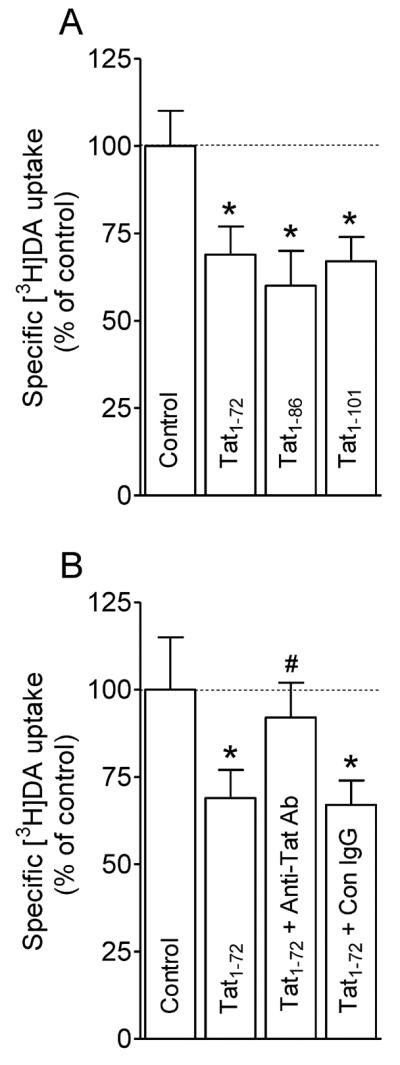
Inhibition of DA uptake by released Tat from Tat-expressing cells. (A) CHO cells transfected with WT hDAT were preincubated in KRH buffer including 100 μl conditioned media collected at 72 h from cells transfected with plasmid Tat1-72, Tat1-86, Tat1−101 DNAs and vector alone (Control) followed by addition of [3H]DA uptake. * p < 0.05 different from control (Dunnett's Multiple comparison test). (B) Specificity of released Tat in inhibition of [3H]DA uptake. Conditioned media collected at 72 h from cells transfected with Tat1-72 were preincubated with anti-Tat antibody or isotype control IgG at 4°C for 3 h, followed by incubation with protein A/G – Agarose beads 4°C for 2 h. Media collected at same time from cells transfected with vector alone was used as control. Cells transfected with WT hDAT were preincubated in KRH buffer containing supernatants from the agarose-antibody-medium-beads complex, followed by [3H]DA uptake. Released Tat1-72 caused significant decrease in [3H]DA uptake, which was attenuated by immunodepletion with anti-Tat antibody but not isotype control antibody (one-way ANOVA followed by Tukey's multiple comparison test). * p < 0.05 different from control. # p < 0.05 different from Tat1-72 and Tat1-72 + Con IgG. (n = 4).
Mutation of Tyr470 alters DA uptake kinetics and potency of substrate and inhibitors
To validate the feasibility of the computational model of the DAT(DA)-Tat complex, we determined whether a specific residue (Tyr470, which was predicted by the computational modeling as one of favorable intermolecular interactions between Tat and hDAT) in hDAT is important for intermolecular interaction between Tat and DAT. A mutation in hDAT (tyrosine to histidine, Y470H-hDAT) was generated by site-directed mutagenesis. We first determined the pharmacological profiles of [3H]DA uptake in CHO cells transfected with equal amounts of plasmid DNA for WT hDAT and mutated hDAT. As shown in Fig. 3A, the Y470H-hDAT displayed a decrease in the Vmax values (2.8 ± 0.8 pmol/min/105 cells) compared with WT hDAT [15.7 ± 0.9 pmol/min/105 cells; t(3) = 15.6, p<0.001, unpaired Student's t test]; no difference in the Km values was observed.
Figure 3.
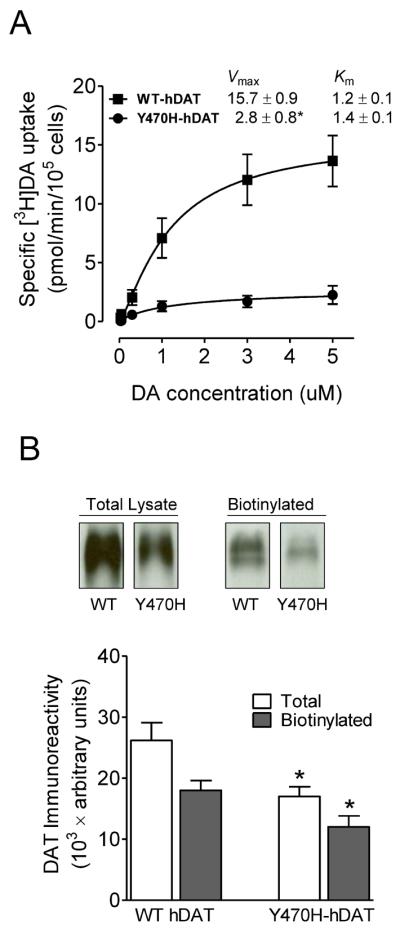
[3H]DA uptake and DAT surface expression in WT hDAT and mutant. (A) Kinetic analysis of [3H]DA uptake in WT hDAT and Y470H-hDAT. CHO cells transfected with WT hDAT or Y470H-hDAT were incubated with one of six mixed concentrations of the [3H]DA as total rate of DA uptake. In parallel, nonspecific uptake of each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. *p < 0.05 compared to control value (unpaired Student's t test) (n = 5). (B) Cell surface of WT hDAT (WT) or Y470H-hDAT (Y470H) was analyzed by biotinylation. Top panel: representative immunoblots in CHO cells expressing WT hDAT or Y470H-hDAT. Bottom panel: DAT immunoreactivity is expressed as mean ± S.E.M. densitometry units from three independent experiments (n = 3). *p < 0.05 compared to WT hDAT (unpaired Student's t test).
We have reported that Tat protein influences selective binding sites on the DAT, with differential impact on binding to GBR12909, WIN35,428 and cocaine (Zhu et al., 2009; Zhu et al., 2011). To explore the potential relationship between the binding sites of Tat in DAT and the binding sites of DAT substrate and inhibitors, we also tested the ability of DA, cocaine and GBR12909 to inhibit [3H]DA uptake in WT hDAT and Y470H-hDAT (Table 1). The apparent affinity (IC50) for DA was not significantly different between the WT hDAT (895 ± 80 nM) and Y470H-hDAT (737 ± 72 nM). However, the potencies of cocaine and GBR12909 for inhibition of [3H]DA uptake were ~3.5-fold greater in Y470H-hDAT as compared with WT hDAT (unpaired Student's t test).
Table 1.
Summary of inhibitory activities in [3H]DA uptake in WT and mutated hDAT in the presence of DA, cocaine or GBR12909
| DA | cocaine | GBR12909 | |
|---|---|---|---|
| IC50 (nM) | |||
| WT hDAT | 895 ± 80 | 370 ± 40 | 160 ± 30 |
| Y470H-hDAT | 737 ± 72 | 100 ± 30* | 50 ± 10* |
Data are calculated as a percentage of DA uptake in the absence of substrate or inhibitor and analyzed by nonlinear regression. Data are presented as mean ± S.E.M. of IC50 values from three to four independent experiments performed in duplicate.
p < 0.05 compared with WT hDAT (unpaired Student's t test).
To assess whether the decreased Vmax in this mutant was caused by decreased surface DAT expression, we determined DAT surface expression in CHO cells transfected with WT or Y470H-hDAT using cell surface biotinylation. As shown in Fig. 3B, despite no difference in the ratio of surface DAT to total DAT between WT and Y470-hDAT (biotinylated/total: WT, 0.70 ± 0.06; and Y470H, 0.68 ± 0.1; p >0.05, one-way ANOVA), the absolute surface DAT in the mutant hDAT was decreased slightly compared to WT hDAT (unpaired Student's t test). Thus, the reduction of available DAT on the cell surface may also partially contribute to the decreased DA uptake actually measured in Y470H-hDAT, relative to WT DAT.
Mutation of Tyr470 attenuates Tat-induced inhibitory effects on DA transport
To determine whether the mutation of Tyr470 alters inhibitory effects of Tat on DA uptake, we examined the specific [3H]DA uptake in WT hDAT and Y470H-hDAT in the presence or absence of released Tat1–72 (1 ng/ml) or recombinant Tat1–86 (350 nM). As shown in Figure 4A, two-way ANOVA on the specific [3H]DA uptake in WT and Y470H-hDAT revealed a significant main effect of mutation (F(1, 24) = 6.5; p < 0.05), Tat treatment (F(1, 24) = 7.5; p < 0.05) and a significant mutation × Tat interaction (F(1, 24) = 8.9; p < 0.05). A subsequent simple effect analysis revealed a dramatic decrease (80%) in [3H]DA uptake in Y470H-hDAT (F(1, 12) = 25; p < 0.001) compared to WT hDAT in the absence of Tat. Exposure to Tat decreased [3H]DA uptake by 50% in hDAT (F(1, 12) = 16.1; p < 0.01; Fig. 4A); however, no effect of Tat was observed in Y470H-hDAT (F(1, 12) = 0.05; p > 0.05), suggesting that mutation of Tyr470 in hDAT attenuates Tat-induced reduction of hDAT function.
Figure 4.

Effects of Tat on kinetic analysis of [3H]DA uptake in WT hDAT and mutant. (A) CHO cells transfected with WT or Y470H-hDAT were preincubated with or without released Tat1-72 (1.0 ng/ml) at room temperature for 20 min followed by the addition of one of six mixed concentrations of the [3H]DA. In parallel, nonspecific uptake at each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. (B) [3H]DA uptake in cells transfected with WT or Y470H-hDAT was determined in the presence or absence of recombinant Tat1-86 (rTat1-86, 350 nM, final concentration). (C) [3H]DA uptake in cells transfected with WT (0.8 μg plasmid cDNA) or Y470H-hDAT (2.4 μg plasmid cDNA) was determined in the presence or absence of rTat1-86 (350 nM). Data are expressed as means from five independent experiments ± S.E.M. *p < 0.05 compared with the respective control values. # p < 0.05 compared to WT hDAT. (n = 5)
With regard to the effect of recombinant Tat1–86 on DA uptake (Fig. 4B), a separate two-way ANOVA analysis revealed a significant main effect of mutation (F(1, 24) = 7.4; p < 0.05) and Tat treatment (F(1, 24) = 9.5; p < 0.05) as well as a significant mutation × Tat interaction (F(1, 24) = 12.9; p < 0.05). [3H]DA uptake in Y470H-hDAT was 19% of that in WT hDAT in cells transfected with equal amount of plasmid DNA for WT and mutated DAT, which is consistent with the low DAT expression observed in Fig. 3B. Exposure to Tat1–86 decreased [3H]DA uptake by 38% in WT hDAT (F(1, 12) = 12.8; p < 0.01); however, no effect of Tat on [3H]DA uptake was observed in Y470H-hDAT. Since DA uptake is linear with DAT expression, in order to rule out whether the lack of effect of Tat on DA uptake in this mutant hDAT is due to a low DAT expression level in Y470H-hDAT relative to WT hDAT, we corrected the Vmax value of Y470H-hDAT to 40% of that in WT hDAT using 3× amount of plasmid Y470H DNA in transfection (Fig. 4C), as reported previously (Chen et al., 2004). A two-way ANOVA revealed a significant main effect of mutation (F(1, 24) = 6.4; p < 0.05) and Tat treatment (F(1, 24) = 5.5; p < 0.05) as well as a significant mutation × Tat interaction (F(1, 24) = 11.2; p < 0.05). Similarly, exposure to recombinant Tat1–86 decreased Vmax by 35% and 6% in WT hDAT and Y470H-hDAT, respectively. Thus, this result supports the inference that Tyr470 in hDAT is critical for HIV-1 Tat-induced inhibition of dopamine transport.
Mutation of Tyr470 affects Zinc regulation of DAT conformational transitions and basal DA efflux
We hypothesize that Tat, via allosteric modulation sites, alters conformational states of DAT, thereby decreasing DA transport. To test this possibility, we determined whether mutation of Tyr470 affects zinc regulation of DAT conformational transitions and basal DA efflux. In general, the conformational changes in DA transport processes involve conversions between outward- and inward-facing conformations (Zhao et al., 2010). Occupancy of the endogenous Zn2+ binding site in WT hDAT (His193, His375, and Glu396) stabilizes the transporter in an outward-facing conformation, which allows DA to bind but inhibits its translocation, thereby increasing [3H]WIN 35,428 binding (Norregaard et al., 1998; Moritz et al., 2013), but decreasing DA uptake (Loland et al., 2003). Addition of Zn2+ is able to partially reverse an inward-facing state to an outward-facing state (Norregaard et al., 1998; Loland et al., 2003). On the basis of this principle, the addition of Zn2+ to WT hDAT would inhibit DA uptake, whereas in a functional mutation in DAT Zn2+ might diminish the preference for the inward-facing conformation and thus enhance DA uptake. To explore this possibility, we examined the effects of Tyr470 mutation on Zn2+ modulation of [3H]DA uptake and [3H]WIN35,428 binding that are thought to reflect stabilization of outwardly facing transporter forms (Richfield, 1993; Norregaard et al., 1998). For these experiments, CHO cells expressing WT and Y470H-hDAT were treated with 10 μM ZnCl2 and assayed for both [3H]DA uptake and [3H]WIN 35,428 (Fig. 5A and 5B). As shown in Fig. 5A, two-way ANOVA on the specific [3H]DA uptake in WT and Y470H-hDAT revealed a significant main effect of mutation (F(1, 24) = 11.5; p < 0.05), zinc (F(1, 24) = 9.1; p < 0.05) and a significant mutation × zinc interaction (F(1, 24) = 9.9; p < 0.05). The addition of Zn2+ decreased [3H]DA uptake in WT and Y470H-hDAT by 89% versus 32%, respectively (Fig. 5A, p< 0.001 relative to control, unpaired Student's t test). A two-way ANOVA on the specific [3H]WIN35,428 binding in WT and Y470H-hDAT revealed a significant main effect of mutation (F(1, 24) = 6.5; p < 0.05), zinc (F(1, 24) = 4.3; p < 0.05) and a significant mutation × zinc interaction (F(1, 24) = 4.2; p < 0.05). Zn2+ caused a 40% increase in [3H]WIN 35,428 binding in WT but had no effect on Y470H-hDAT (Fig. 5B, p< 0.001 relative to control, unpaired Student's t test). The data suggest that Tyr470 mutation disrupts an intermolecular interaction key for maintenance of the outward-facing conformation.
Figure 5.

Mutation of Tyr470 alters transporter conformational transitions. Tyr470 mutation of DAT affects zinc regulation of DA uptake (A) and [3H]WIN 35,428 binding (B). CHO cells transfected with WT or Y470H-hDAT were incubated with KRH buffer alone (control) or with ZnCI2 (10 μM, final concentration) followed by [3H]DA uptake or [3H]WIN 35,428 binding (n = 4). The histogram shows [3H]DA uptake and [3H]WIN 35,428 binding expressed as mean ± S.E.M. of the respective controls set to 100% for the mutant. *p < 0.05 compared to control. #p < 0.05 compared to WT hDAT with ZnCI2. (C) Functional DA efflux properties of WT hDAT and mutant. CHO cells transfected WT or Y470H-hDAT were preincubated with [3H]DA (0.05 μM, final concentration) at room temperature for 20 min. After incubation, cells were washed and incubated with fresh buffer at indicated time points. Subsequently, the buffer was separated from cells, and radioactivity in the buffer and remaining in the cells was counted. Each fractional [3H]DA efflux in WT hDAT and Y470H-hDAT was expressed as percentage of total [3H] in the cells at the start of the experiment. Fractional [3H]DA efflux at 1, 10, 20, 30 and 40 min are expressed as the percentage of total [3H]DA with preloading with 0.05 μM (WT hDAT: 15379 ± 1800 dpm and Y470H-hDAT: 2488 ± 150 dpm) present in the cells at the start of the experiment (n = 4). ×p < 0.05 and ××p < 0.01, compared to WT hDAT (Bonferroni t-test). (D) Functional DA efflux properties of WT hDAT in the presence or absence of Tat1-72. CHO cells transfected with WT hDAT were preincubated with released Tat1-72 (1 ng/mg) followed by DA efflux assay. Fractional [3H]DA efflux at 1, 10, 20, 30, 40 and 50 min are expressed as the percentage of total [3H]DA with preloading with 0.05 μM (control: 14200 ± 1448 dpm and released Tat: 10102 ± 1505 dpm) present in the cells at the start of the experiment (n = 6). ^p < 0.05 and ^^p < 0.01, compared to WT hDAT in the absence of Tat (Bonferroni t-test).
To further determine the role of Y470H-hDAT in the transition between outward-facing and inward-facing states, we also examined basal DA efflux in WT hDAT and this mutant. As shown in Fig. 5C, after preloading with 0.05 μM [3H]DA for 20 min at room temperature, cells were washed and fractional DA efflux samples were collected at the indicated times. A two-way ANOVA revealed significant main effects of mutation (F(1, 14) = 170; p<0.001) and time (F(4, 56) = 145; p<0.001). A significant mutation × time interaction (F(4, 56) = 78; p<0.001) was also found. Post-hoc analysis revealed robust increases in DA efflux at 1, 10, 20, and 30 min compared to WT hDAT (p < 0.05, Bonferroni t-test). To determine whether exposure to Tat represents similar results, basal DA efflux in WT hDAT was determined in the presence or absence of released Tat1–72 (Fig. 5D). The fractional basal DA efflux data in WT hDAT were expressed as a percentage change in the respective controls of total DA content in cells with or without Tat. Two-way ANOVA revealed a significant main effect of treatment (F(1, 10) = 18.7; p < 0.01) and time (F(5, 50) = 291.9; p<0.001) as well as a significant treatment × time interaction (F(5, 50) = 9.9; p<0.001). Although a lower magnitude of DA efflux in response to Tat treatment was found in WT hDAT in comparison to DA efflux in Y470H-hDAT (Fig. 5D), post-hoc analysis revealed that exposure to released Tat1–72 significantly increased basal DA efflux at 1, 10, 20 and 30 min compared to control (p < 0.05, Bonferroni t-test). These data further support the possibility that Tyr470 mutation causes a regional conformational change that affects hDAT associated with Tat.
Discussion
In the current study, we used an integrated approach including computational modeling and simulations, protein mutagenesis and molecular pharmacological function assays to explore a key residue in the intermolecular interactions between HIV-1 Tat and hDAT. Our data provide additional evidence showing a direct interaction between Tat and hDAT as suggested in our previous report (Zhu et al., 2009). Through modeling and simulations, the site for Tat interaction with DAT has predicted that residue Tyr470 in hDAT is crucial for HIV-1 Tat-induced inhibition of DA transport. Tyr470 mutation did not alter the affinity for DA uptake but increased DA uptake potency for cocaine and GBR12909, suggesting that Tyr470 does not overlap with the substrate binding site but disrupts the binding sites on DAT for these inhibitors. Importantly, mutation of Tyr470 alters Zn2+ modulation of DAT and basal DA efflux, compared to WT hDAT, implying a mechanistic context for the transporter conformational transitions by this mutant. Collectively, our results provide a relatively comprehensive molecular insight into this important residue for DA translocation and the underlying allosteric mechanism in DAT for Tat binding.
In response to the fundamental question of how Tat interacts with hDAT through their recognition binding sites to interrupt DAT-mediated DA transmission, our computational model has predicted Tyr470 of hDAT and Cys22 as well as Lys19 in Tat as the favorable intermolecular interactions between hDAT and Tat protein. Data from Co-IP and GST pull-down experiments demonstrate a direct interaction between Tat and DAT, which is consistent with the predictions from computational modeling and simulations. This study, based on the computational prediction, demonstrated that mutation of Tyr470 changes hDAT conformation and attenuates Tat-induced inhibition of DA transport. Despite the importance of Tyr470, our computational modeling does not anticipate that a single residue in hDAT is sufficient to control the interaction of Tat with hDAT. Therefore, once all recognition residues in hDAT are identified, an essential task in our future studies will be to determine the influence of combined recognition residues on Tat-DAT interaction. The computational prediction of the binding mode was based on a series of computational modeling studies including homology modeling, Brownian dynamics simulations (Gabdoulline and Wade, 1998), and molecular dynamics simulations. Considering the fact that the Tat molecule has a large positive electrostatic potential, and hDAT(DA) bears negative charge, the long-range electrostatic attraction can be viewed as the driving force for the association of Tat with hDAT. The binding mode of hDAT with Tat demonstrates that the Tat molecule is associated with DAT through inter-molecular electrostatic attractions and complementary hydrophobic interactions. In support of our proposed model, the current study and our previous report (Zhu et al., 2009) demonstrate that mutation of either Tyr470 in hDAT or Cys22 in Tat leads to attenuation of WT Tat-induced inhibitory effects on DA transport, implicating a structural and functional relationship between Tat and hDAT. Since Lys19 is also predicted as a critical residue for Tat interaction with the Tyr470 residue in DAT, we will also determine whether mutation of Lys19 produces a similar effect to mutation of Cys22 in Tat on DAT function in our future investigation. These data also qualitatively support that our computationally simulated model of the hDAT(DA)-Tat complex is reliable to be used to predict the intermolecular interactions between Tat and DAT.
Although both recombinant Tat and Tat released from cells expressing Tat produced a strong inhibitory effect on DA transport, released Tat was ~4000-fold more potent than recombinant Tat. This finding is consistent with a previous report showing this form of Tat was more neurotoxic than recombinant Tat protein (Li et al., 2008). We also found that Tat1–72, Tat1–86 and full length Tat1–101 exhibited an equivalent inhibitory effect on DAT function, which is consistent with previous studies showing the equal ability of Tat protein in Tat-induced neurotoxicity (Li et al., 2008; Aksenov et al., 2009). The concentration of released Tat (1 ng/ml) used in this study is similar to native HIV-1 Tat actually detected in the serum of patients with HIV infection (Westendorp et al., 1995) and in the conditioned medium of HIV-infected cells (Albini et al., 1998). Thus, our data support that physiologically secreted Tat is more neurotoxic to neuronal targets, such as DAT. This approach is feasible for further determination of the recognition sites of Tat (WT versus mutated) that functionally interact with hDAT.
The present results show that mutation of Tyr470 decreased the Vmax with no changes in Km value and IC50 value for DA inhibiting DA uptake compared to WT hDAT, demonstrating that the Tyr470 residue does not affect substrate transport characteristics. These data are consistent with the aforementioned computational prediction based on the modeled hDAT(DA)-Tat complex structure: Tat binding site in the hDAT(DA) complex does not overlap with the binding site of substrate DA (Fig. 2). In contrast, the DA uptake potency of cocaine and GBR12909 is increased in Y470H-hDAT compared to WT hDAT. While GBR12909 labels the classic DA uptake site in rodent brain, binding to the piperazine acceptor site (Andersen et al., 1987) is affected much less by mutation of hDAT than cocaine (Loland et al., 2002; Guptaroy et al., 2011), consistent with the fact that cocaine preferentially stabilizes the hDAT in the outward-facing conformational state, resulting in a reduction of DA uptake (Reith et al., 2001; Loland et al., 2002). One interpretation of our finding is that Tat allosterically modulates DA transport rather than overlaps DA uptake sites on DAT as previously suggested (Zhu et al., 2011). However, the Tat binding site in hDAT may be close to the binding sites in hDAT for cocaine and GBR12909, increasing their DA uptake potency. Thus, the results may suggest synergistic influences of Tat and cocaine on DAT function: Tat, via allosteric modulation of the DAT, enhances the inhibitory effects of cocaine on DA transport. These findings also provide evidence to support our previous reports that Tat and cocaine synergistically inhibit DAT function in vivo and in vitro (Harrod et al., 2008; Ferris et al., 2010).
Our previous work provides evidence that Tat allosterically modulates DAT function (Zhu et al., 2009; Zhu et al., 2011). The allosteric modulation of DAT is responsible for conformational transitions via substrate- and ligand-binding sites on DAT (Zhao et al., 2010; Shan et al., 2011). In the present study, we found that mutation of Tyr470 diminished Zn2+-induced inhibition of [3H]DA uptake and attenuated increased [3H]WIN 35,428 binding compared to WT hDAT. The endogenous Zn2+ binding sites in hDAT have been widely employed to investigate whether mutations of hDAT alter transporter conformational transitions in DA transport (Norregaard et al., 1998). The Zn2+-mediated inhibition of DA transport and stimulation of WIN35,428 binding to DAT occur by stabilization of outwardly facing transporter conformations (Richfield, 1993; Norregaard et al., 1998; Moritz et al., 2013). Although analysis of Zn2+ regulation of DAT function only reflects one mechanistic aspect of DAT mutant-induced conformational change, our data provide strong evidence for the context of an allosteric mechanism responsible for conformational transitions in DAT. To explore the possible mechanism, we also examined the effects of mutant hDAT on basal DA efflux and observed extremely low accumulation of DA over time in Y470H-hDAT relative to WT hDAT. One possible explanation for the low accumulation of DA is that basal DA efflux can be elevated in this mutant, resulting in reduced accumulation of intracellular DA. One caveat is that DA efflux data may reflect not only DA moving out of the cell through the transporter, but also non-specific diffusion and reuptake. However, there is evidence (Guptaroy et al., 2009; Guptaroy et al., 2011) that this measurement largely reflects basal DA efflux through DAT because such basal DA efflux is consistent with amphetamine- or voltage-stimulated efflux of intracellular DA in cells expressing hDAT and its mutant. Amphetamine, a substrate for DAT, competitively inhibits DA reuptake and elicits outward transport of DA by reversal of the transporter (Sulzer et al., 2005). Thus, it is possible that Y470H mutation shifts the conformation of DAT from physiologically favored substrate influx mode to an efflux one; a conformational switch promoted by Tat protein, and disturbs transition between inward- and outward-facing conformations. Taken together, these findings infer a potential mechanism that mutation of Tyr470 alters the transporter conformational transitions, which is consistent with our previous findings that Tat mediates allosteric modulation of DAT (Zhu et al., 2011). Additionally, the enhanced DA efflux was also observed in WT hDAT in the presence of Tat. Interestingly, the magnitude of DA efflux in WT hDAT was lower in response to Tat compared to that in Y470H-hDAT. One possible explanation for the discrepancy is that in addition to residue Tyr470, other recognition residues of the DAT may be involved in the effects of Tat on DA efflux. Therefore, to fully understand the mechanisms by which Tat inhibits DAT function, future studies based on the combined experimental and computational approaches will be necessary to further analyze the changes in the conformational transition attributed to the identified residues in DAT (i.e. an outward-facing form and an inward-facing form).
In conclusion, we have begun to identify the specific intermolecular interactions between Tat and DAT and the molecular mechanism(s) that underlie how Tat, via the recognition binding sites in DAT, interrupts DA transport. Particularly, our results provide a relatively comprehensive molecular validation on this important residue of DA translocation and the underlying allosteric mechanism in DAT for Tat. We propose that multiple recognition residues in the DAT are involved in the dynamic and complex interactions between Tat and DAT. Results obtained from Tyr470 only reflect the role of this residue in DAT-Tat interaction. The current findings have shed light on further mapping and validating the predicted sites for Tat interaction with DAT towards an ultimate goal to develop compounds that specifically block Tat binding site(s) in DAT without affecting physiological DA transport. Ideally, these compounds would be therapeutic candidates for stabilizing physiological dopaminergic tone.
Acknowledgements
This research was supported by grants from the National Institutes of Health to Jun Zhu (DA024275, DA026721, DA035714), Chang-Guo Zhan (DA032910, DA035552), and Rosemarie Booze (DA013137, HD043680).
Footnotes
Conflicts of Interest The authors declare no conflicts of interest.
References
- Aksenov MY, Aksenova MV, Mactutus CF, Booze RM. Attenuated neurotoxicity of the transactivation-defective HIV-1 Tat protein in hippocampal cell cultures. Exp Neurol. 2009;219:586–590. doi: 10.1016/j.expneurol.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albini A, Ferrini S, Benelli R, Sforzini S, Giunciuglio D, Aluigi MG, Proudfoot AE, Alouani S, Wells TN, Mariani G, Rabin RL, Farber JM, Noonan DM. HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci U S A. 1998;95:13153–13158. doi: 10.1073/pnas.95.22.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PH, Jansen JA, Nielsen EB. [3H]GBR 12935 binding in vivo in mouse brain: labelling of a piperazine acceptor site. Eur J Pharmacol. 1987;144:1–6. doi: 10.1016/0014-2999(87)90002-1. [DOI] [PubMed] [Google Scholar]
- Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. Journal of psychopharmacology. 2000;14:214–221. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buch S, Yao H, Guo M, Mori T, Su TP, Wang J. Cocaine and HIV-1 interplay: molecular mechanisms of action and addiction. J Neuroimmune Pharmacol. 2011;6:503–515. doi: 10.1007/s11481-011-9297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Wang GJ, Volkow ND, Ernst T, Telang F, Logan J, Fowler JS. Decreased brain dopamine transporters are related to cognitive deficits in HIV patients with or without cocaine abuse. Neuroimage. 2008;42:869–878. doi: 10.1016/j.neuroimage.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Rickey J, Berfield JL, Reith ME. Aspartate 345 of the dopamine transporter is critical for conformational changes in substrate translocation and cocaine binding. J Biol Chem. 2004;279:5508–5519. doi: 10.1074/jbc.M306294200. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- Ernst T, Yakupov R, Nakama H, Crocket G, Cole M, Watters M, Ricardo-Dukelow ML, Chang L. Declined neural efficiency in cognitively stable human immunodeficiency virus patients. Ann Neurol. 2009;65:316–325. doi: 10.1002/ana.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Mactutus CF, Booze RM. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev. 2008;32:883–909. doi: 10.1016/j.neubiorev.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM. Hyperdopaminergic tone in HIV-1 protein treated rats and cocaine sensitization. J Neurochem. 2010;115:885–896. doi: 10.1111/j.1471-4159.2010.06968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabdoulline RR, Wade RC. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 1998;14:329–341. doi: 10.1006/meth.1998.0588. [DOI] [PubMed] [Google Scholar]
- Gannon P, Khan MZ, Kolson DL. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Current opinion in neurology. 2011;24:275–283. doi: 10.1097/WCO.0b013e32834695fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskill PJ, Calderon TM, Luers AJ, Eugenin EA, Javitch JA, Berman JW. Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol. 2009;175:1148–1159. doi: 10.2353/ajpath.2009.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB, Lisinicchia JG, Chen T, Johnson KM, Jennings K, Freeman DH, Jr, Soukup VM. Prefrontal Dopaminergic and Enkephalinergic Synaptic Accommodation in HIV-associated Neurocognitive Disorders and Encephalitis. J Neuroimmune Pharmacol. 2012;7:686–700. doi: 10.1007/s11481-012-9345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guptaroy B, Fraser R, Desai A, Zhang M, Gnegy ME. Site-directed mutations near transmembrane domain 1 alter conformation and function of norepinephrine and dopamine transporters. Mol Pharmacol. 2011;79:520–532. doi: 10.1124/mol.110.069039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guptaroy B, Zhang M, Bowton E, Binda F, Shi L, Weinstein H, Galli A, Javitch JA, Neubig RR, Gnegy ME. A juxtamembrane mutation in the N terminus of the dopamine transporter induces preference for an inward-facing conformation. Mol Pharmacol. 2009;75:514–524. doi: 10.1124/mol.108.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod SB, Mactutus CF, Fitting S, Hasselrot U, Booze RM. Intra-accumbal Tat1-72 alters acute and sensitized responses to cocaine. Pharmacol Biochem Behav. 2008;90:723–729. doi: 10.1016/j.pbb.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BK, Gruswitz F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC structural biology. 2008;8:49. doi: 10.1186/1472-6807-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhan CG. How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophysical journal. 2007;93:3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Gu HH, Zhan CG. Mechanism for cocaine blocking the transport of dopamine: insights from molecular modeling and dynamics simulations. J Phys Chem B. 2009;113:15057–15066. doi: 10.1021/jp900963n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Ownby RL, Waldrop-Valverde D, Fernandez B, Kumar M. Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol. 2011;17:26–40. doi: 10.1007/s13365-010-0003-4. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Fernandez JB, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M. Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol. 2009;15:257–274. doi: 10.1080/13550280902973952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers SL, Salemi M, Galligan DC, Morris A, Gray R, Fogel G, Zhao L, McGrath MS. Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J Neurovirol. 2010;16:230–241. doi: 10.3109/13550281003735709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland CJ, Norgaard-Nielsen K, Gether U. Probing dopamine transporter structure and function by Zn2+-site engineering. Eur J Pharmacol. 2003;479:187–197. doi: 10.1016/j.ejphar.2003.08.068. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Norregaard L, Litman T, Gether U. Generation of an activating Zn(2+) switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transport cycle. Proc Natl Acad Sci U S A. 2002;99:1683–1688. doi: 10.1073/pnas.032386299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Ann Neurol. 2010;67:699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]
- Meade CS, Conn NA, Skalski LM, Safren SA. Neurocognitive impairment and medication adherence in HIV patients with and without cocaine dependence. J Behav Med. 2011a;34:128–138. doi: 10.1007/s10865-010-9293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade CS, Lowen SB, MacLean RR, Key MD, Lukas SE. fMRI brain activation during a delay discounting task in HIV-positive adults with and without cocaine dependence. Psychiatry Res. 2011b;192:167–175. doi: 10.1016/j.pscychresns.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midde NM, Gomez AM, Zhu J. HIV-1 Tat Protein Decreases Dopamine Transporter Cell Surface Expression and Vesicular Monoamine Transporter-2 Function in Rat Striatal Synaptosomes. J Neuroimmune Pharmacol. 2012;7:629–639. doi: 10.1007/s11481-012-9369-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz AE, Foster JD, Gorentla BK, Mazei-Robison MS, Yang JW, Sitte HH, Blakely RD, Vaughan RA. Phosphorylation of dopamine transporter serine 7 modulates cocaine analog binding. J Biol Chem. 2013;288:20–32. doi: 10.1074/jbc.M112.407874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Clements JE. Eradication of HIV from the brain: reasons for pause. AIDS. 2011;25:577–580. doi: 10.1097/QAD.0b013e3283437d2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR. Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol. 2001;7:66–71. doi: 10.1080/135502801300069737. [DOI] [PubMed] [Google Scholar]
- Norregaard L, Frederiksen D, Nielsen EO, Gether U. Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 1998;17:4266–4273. doi: 10.1093/emboj/17.15.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peloponese JM, Jr, Gregoire C, Opi S, Esquieu D, Sturgis J, Lebrun E, Meurs E, Collette Y, Olive D, Aubertin AM, Witvrow M, Pannecouque C, De Clercq E, Bailly C, Lebreton J, Loret EP. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C R Acad Sci III. 2000;323:883–894. doi: 10.1016/s0764-4469(00)01228-2. [DOI] [PubMed] [Google Scholar]
- Purohit V, Rapaka R, Shurtleff D. Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Molecular neurobiology. 2011;44:102–110. doi: 10.1007/s12035-011-8195-z. [DOI] [PubMed] [Google Scholar]
- Reith ME, Berfield JL, Wang LC, Ferrer JV, Javitch JA. The uptake inhibitors cocaine and benztropine differentially alter the conformation of the human dopamine transporter. J Biol Chem. 2001;276:29012–29018. doi: 10.1074/jbc.M011785200. [DOI] [PubMed] [Google Scholar]
- Richfield EK. Zinc modulation of drug binding, cocaine affinity states, and dopamine uptake on the dopamine uptake complex. Mol Pharmacol. 1993;43:100–108. [PubMed] [Google Scholar]
- Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS. 2007;21:1915–1921. doi: 10.1097/QAD.0b013e32828e4e27. [DOI] [PubMed] [Google Scholar]
- Shan J, Javitch JA, Shi L, Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS One. 2011;6:e16350. doi: 10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Torres GE, Amara SG. Glutamate and monoamine transporters: new visions of form and function. Curr Opin Neurobiol. 2007;17:304–312. doi: 10.1016/j.conb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, Visco-Comandini U, Vlassi C, Giulianelli M, Galgani S, Antinori A, Narciso P. Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr. 2007;45:174–182. doi: 10.1097/QAI.0b013e318042e1ee. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chang L, Volkow ND, Telang F, Logan J, Ernst T, Fowler JS. Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain. 2004;127:2452–2458. doi: 10.1093/brain/awh269. [DOI] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Apparsundaram S, Bardo MT, Dwoskin LP. Environmental enrichment decreases cell surface expression of the dopamine transporter in rat medial prefrontal cortex. J Neurochem. 2005;93:1434–1443. doi: 10.1111/j.1471-4159.2005.03130.x. [DOI] [PubMed] [Google Scholar]
- Zhu J, Mactutus CF, Wallace DR, Booze RM. HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J Pharmacol Exp Ther. 2009;329:1071–1083. doi: 10.1124/jpet.108.150144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Ananthan S, Mactutus CF, Booze RM. Recombinant human immunodeficiency virus-1 transactivator of transcription1-86 allosterically modulates dopamine transporter activity. Synapse. 2011;65:1251–1254. doi: 10.1002/syn.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]