

Abstract
In this review, we outline examples that illustrate the design criteria for achieving macromolecular assemblies that incorporate a combination of two or more chemical, physical or biological stimuli-responsive components. Progress in both fundamental investigation into the phase transformations of these polymers in response to multiple stimuli and their utilization in a variety of pratical applications have been highlighted. Using these examples, we aim to explain the origin of employed mechanisms of stimuli responsiveness which may serve as a guideline to inspire future design of multi-stimuli responsive materials.
Introduction
Responsive polymers, lipids, dendrimers, and other macromolecules that are capable of adapting to changes in the local environment have produced tremendous interest in recent literature, driven by the exponentially increasing demand for “intelligent” materials. Owing to advances in high fidelity transformations such as “click” chemistries, and controlled polymerization techniques such as reversible addition-framentation chain transfer polymerization (RAFT), atom transfer radical polymerization (ATRP), nitroxide mediated radical polymerization (NMP), and ring-opening metathesis polymerization (ROMP), numerous well-defined materials with stimuli-responsive characteristics have been synthesized for explorations in both fundamental and applied science.1-5 The range of potential applications for these materials include catalysis, smart interfaces, tissue engineering, biosensor, diagnostics and drug delivery among others. Several review articles have highlighted investigations of stimuli-responsive polymers and their response mechanism in single stimulus systems.6 Multi-stimuli responsive polymers and macromolecules that are sensitive to two or more stimuli are synthetically challenging, but are materials of emerging interest. Moreover, multifaceted responsiveness could greatly enhance the versatility of these materials in a variety of applications. For example, multi-stimuli responsive polymers could be an ideal platform to mimic biological processes, which are often a result of adaption to multiple environmental changes rather than a single change. Additionally, the complexity of multi-stimuli responsiveness enriches the molecular toolbox, which one can use to manipulate the physical properties of a material, originating from phase transformations due to the co-operative interaction of multiple stimuli within the custom-designed molecule. The impact of such a molecular toolbox will depend on the reliability of “design rules” that one could develop for these materials. For example, achievement of this capability in drug delivery could allow us to precisely tune the guest release kinetics to fit the therapeutic window of an active compound.7 In this review, we attempt to highlight the recent advances in the development of multi-responsive macromolecules and their applications.
This area is diverse, not only in the types of materials used, but also in the nature of the stimulus. Considering this diversity, we have classified the stimuli into three broadly-defined categories: physical, chemical, and biological, and this review has been organized using this classification. Throughout this review we will breifly discuss the material synthesis, but focus on their design, function, and the responsiveness.
Physical-physical dual stimuli-responsive macromolecules
In this section, selected literature reports that investigate the effects of two different physical stimuli will be highlighted. Prominent examples of physical stimuli include temperature and light. Temperature responsive materials have been among the most studied stimuli-responsive materials for several decades due to their ability to change features such as solubility, conformation and hydrophobic/hydrophilic balance, which have a potential for a variety of applications.8 Also, the use of temperature as a stimuli is potentially cheap and convenient. Polymers that undergo hydrophilic-hydrophobic phase transitions are of great interest for researchers who have taken advantage of these transitions to promote the formation, transformation or deformation of aggregates in response to variations in temperature. Poly(N-isopropyl acrylamide) (PNIPAM) has been quite popular among these “smart” materials, since the temperature at which it undergoes phase transition, the so-called lower critical solution temperature (LCST), falls close to that of the human body.9 Below its LCST, PNIPAM exists as an expanded coil state forming hydrogen bonds with water molecules, however upon heating above its LCST this polymer becomes more hydrophobic collapsing to a globular state (Scheme 1).10 In 2006, a three-layer nanostructure containing a unimolecular dendritic core covalently conjugated to dual thermoresponsive coronas composed of PNIPAM and poly (dimethylamino)ethyl methacrylate (PDMA) was developed (Figure 1).11 PDMA undergoes a hydrophilic-to-hydrophobic phase transition at its LCST which is between 40-50 °C.12 These dendritic unimolecular micelles were synthesized by two successive RAFT polymerizations of NIPAM and DMA using a fractionated hyperbranched polyester (Boltorn H40) based macro-RAFT agent. Sizes of these particles ranged from 80-140 nm, based on atomic force microscopy (AFM). The particle core is claimed to be composed of the Boltorn H40 with PNIPAM as the inner layer and PDMA as the corona. Upon heating above the LCST of PNIPAM, the inner shell collapsed; further increase in temperature past the LCST of PDMA caused the corona to collapse as well. During the cooling cycle, a two-stage reswelling process was also observed. Although the double phase transition of these unimolecular dendritic nanostructures was demonstrated, a recent publication also mentions that the second collapse behavior was more complicated since the outer corona was not completely collapsed, presumably because of the strong interaction with the inner PNIPAM layer.13 Based on these results, the authors prepared a new set of dendritic nanostructures composed of PDMA and poly(N,N-diethylacrylamide) (PDEA) instead of PNIPAM, since the latter also possess a LCST of 32 °C and is less toxic than PNIPAM. The ratio of the radius of gyration to the hydrodynamic radius (<Rg>/<Rh>), which reflects the chain density, was found to be ~0.774 at 50 °C suggesting a dense sphere for these nanostructures. A significant decrease in the <Rg>/<Rh> ratio from 1.34 to 1.20 was observed between 29 °C and 33 °C for these nanostructures, indicating the collapse of the inner PDEA chain. A second abrupt decrease of <Rg>/<Rh> from 1.20 to 0.78 was observed at ~44 °C, suggesting the collapse of the outer layer PDMA. Transmittance (using absorbance spectrum), excimer fluorescence and micro-differential scanning calorimetry (micro-DSC) were also used to further confirm this dual transition behavior.
Scheme 1.

LCST behavior of PNIPAM
Figure 1.

Structure of polymers sensitive to dual physical stimuli
Besides LCST, the upper critical solution temperature (UCST) is another property which relates to phase separation that has been considered in material designs. The UCST occurs upon cooling when the unfavorable enthalipic effect becomes dominant, and above which the components of a solution are miscible. Polysulfobetaines and polyoxazolines are among the polymers that possess UCST behavior. Interestingly, polymers composed of units exhibiting LCST and UCST have been used to tune the thermoresponsive phase behavior and reversible self-assembly of polymers.14,15
A recent report illustrates an interesting example of a two-stage thermal transition of polymer chains, leading to the formation of a stable core-shell micellar structure.16 In this work, the authors take advantage of the relationship between oligoethylene glycol chain length and temperature sensitivity. Here, 2-(2-methoxyethoxy) ethyl methacrylate (MEO2MA) and poly(ethylene glycol) methyl ether methacrylate (PEGMA) copolymers were synthesized by ATRP. It was found that the short side chains of this copolymer collapsed at 27 °C, and upon further heating formed stable colloidal dispersions in solution due to the longer ethylene glycol segments which have a higher LCST (Scheme 2). Polymers with the capability to undergo temperature-dependent phase separation have been one of the largest classes of responsive materials studied for decades. A variety of applications have been envisioned including drug delivery and tissue engineering, depending on whether they exhibit LCST and/or UCST behavior.
Scheme 2.
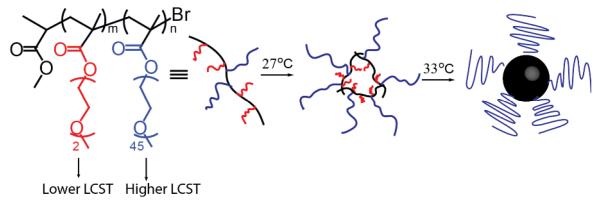
Representation of the two-stage thermally induced aggregation process
Light, another physical stimulus, has been studied in combination with temperature to synergistically manipulate materials’ properties for nearly two decades.17-20 Chromophores that undergo structural isomerization, cyclization or ring-opening upon irradiation at a certain wavelength, are responsible for the light responsiveness of such polymers. Some of these photoinduced transformations can be reverted, upon exposure to light at a different wavelength. Azobenzene is a prominent example of a chromophore that undergoes a rapid trans-to-cis photoisomerization upon irradiation, which results in an increase in the dipole moment (0 D to 3 D) and consequently an increase in the LCST of the polymer chain (Figure 2a).21, 22 A series of polyacrylamide based copolymers incorporating different amounts of azobenzene moieties have been studied (Figure 1). The LCST of these polymers was found to be dependent on the amount of azobenzene introduced into the polymer. After irradiation, the LCST of these polymers increased as the content of azobenzene increased, presumably due to the polarity change of the chromophore. The same pattern was observed when different amounts of the chromophore salicylideneaniline, also known to isomerize from the enol into the keto form upon exposure to UV light, were conjugated directly to the polymer backbone (see Figure 2b).24 The highest ΔLCST obtained for these polymers was ~13 °C, compared to ~7 °C observed for the azobenzene containing polymers.
Figure 2.

Isomerization of azobenzene and salicylideneaniline
Another example that shows the versatility of these smart materials was reported in 2002.25 In this study, the authors switched “on” and “off” the activity of an enzyme (endoglucanase 12A) by utilizing temperature and light responsive polymers, shown in Figure 1, to regulate substrate access. The DMA based polymer containing azobenzene moieties was conjugated to the enzyme by reacting a vinyl sulfone group with a sulfhydryl group. They observed that enzymatic activity was turned “on” when the conjugates were irradiated with visible light at 45 °C, as the polymer was found in its expanded, hydrated state. When irradiated with the UV light, the enzymatic activity decreased significantly due to the collapsed state of the polymer preventing the substrate from reaching the target site.
Other popular photoresponsive moieties include coumarin, o-nitrobenzyl groups, fulgimides and spiropyran.26,27 Coumarin is a benzopyrone molecule that can be crosslinked and uncrosslinked upon irradiation at different wavelengths. Ortho-nitrobenzyl groups are cleaved upon irradiation, while fulgimides and spiropyran are photochromic molecules that undergo photoisomerization. For further information regarding these moieties and temperature/light responsive polymeric materials, we refer the readers to a recently published review.28
Temperature and light have been widely employed as physical stimuli in the design and preparation of responsive materials. The motivation behind these materials design is that external application of stimulus can be controlled and localized to a specific location. The particular focus has been to apply these materials for biomedical applications. The biggest challenge in this field however is engineering the polymers such that the photoresponsive units can undergo transformations at near-IR wavelengths. Note that the tissue penetration depth varies directly with the irradiation wavelength; the penetration depth of the light used for typical photochemical transformations is very poor. Similarly, it is also essential the temperature responsiveness is consistently at physiologically relevant temperatures.
Chemical-chemical dual stimuli-responsive macromolecules
Among current chemically responsive drug delivery systems, pH and redox potential changes are two popular stimuli employed to trigger sharp changes in the properties of a material. The difference in local pH values among various tissues and cellular compartments has frequently been the inspiration for designing delivery vehicles with pH-responsiveness for extracelluar or intracellular drug delivery. For example, the tumor extracellular environment is more acidic (pH 6.5) than in healthy tissues (pH 7.4) due to the Warburg effect, which explains the rapid generation of lactic acid due to the inefficient consumption of glucose in tumor tissues.29,30 Similarly, after delivery to the tumor, vehicles are internalized by the cell through endocytosis. The pH values of the sub-cellular compartments, where delivery vehicles will reside, change dramatically from early endosome to late endosome, and eventually to lysosomes, where the pH can reach 5.0 or even lower.31,32
Redox-sensitive nanoparticles are also interesting because of the presence of higher levels of glutathione (GSH) in cancer cells compared to healthy cells.33 Also in general, GSH is found in micromolar concentrations in the blood plasma, whereas its conccentation is about 10 mM in the cytosol.34 The GSH concentrations of some of the cancer cells have been shown to be even higher.35 Drug delivery vehicles containing disulfide functionalities, which can take advantage of this concentration difference, have been explored.36-39 Drug delivery vehicles based on disulfide functionalities can potentially facilitate extracellular stability and intracellular release of the encapsulated drug molecules.
While polymeric drug delivery vehicles that are designed to respond to single pH or redox stimulus have been extensively investigated, carriers that can respond to both pH and redox stimuli are more scarce.40,41 However, a dual responsive system is more intriguing, and promising in efficacy and selectivity, because of the expected multi-responsiveness in the tumor extracellular environment and intracellular compartments. A recent work reports on a polymer based micelle that exhibits dual sensitivity to pH and redox changes, as shown in Scheme 3.42 At pH > 7, copolymers based on reducible poly(β-amino ester)s (RPAE), which contain disulfide bonds in the polymer backbone and acid sensitive tertiary amines, and poly(ethylene glycol) (PEG), can self-assemble into stable micelles. In these micelles, the hydrophobic RPAE are buried in the core, while the hydrophilic PEG are solvated at the outer shell. The protonation of teriary amines located in RPAE at pH below 6.5 convert the polymer backbones from hydrophobic to hydrophilic, disrupting the hydrophilic-lipophilic balance in the micelles and leading to disassembly. In addition, the triggered dissociation of RPAE-PEG micelles was demonstrated as a result of the degradation of disulfide bonds present in the RPAE backbone. The encapsulated anticancer drug doxorubicin (DOX) can be released from the micelles in either an acidic or a concentrated dithiothreitol (DTT) solution. More importantly, DOX release from the micelles was much faster under highly reductive and acidic conditions.
Scheme 3.

Schematic illustration of RPAE-PEG micelle dissociation and DOX release triggered by changing pH and DTT.
To make the release with high levels of specificity, another example of a dual pH- and redox-sensitive polymer architecture is a core cross-linked micelle. This was described as an “AND” logic gate,43 since drug can be only released when both of pH and redox stimuli are present. In this study, a drug molecule, adriamycin (ADR) was first attached to a hydrazine functionalized poly(ethylene oxide)-b-poly(methacrylic acid) (PEO-b-PMAA) block copolymer (Figure 3) through the formation of hydrazone. Then the ADR-conjugated block copolymer self-assembled into a stable micelle, which was further crosslinked by reaction of the residual hydrazine moieties with the addition of dithiodiethanoic acid. The effective release of double locked ADR could only be triggered by simultaneous treatment with low pH and reducing reagents. No significant ADR release was observed in reducing conditions under neutral pH, due to the intact hydrazone linking, even though the micelles were uncrosslinked due to the reduction of disulfides. Additionally, ADR was not released upon subjecting the crosslinked micelle to an acidic environment, despite the cleavage of ADR from the polymer backbone, due to the trapping of ADR within of the crosslinked micelle.
Figure 3.

pH- and redox- responsive polymers
Similarly, a recent study showed that the release of non-covalently encapsulated hydrophobic molecules can also be triggered by simultaneous application of both low pH and a reducing agent.44 In this paper, polymeric nanoparticles were prepared by cross-linking aldehyde/disulfide-functionalized copolymers and amine/disulfide-functionalized copolymers (Figure 3) through the formation of benzoic imine and disulfide bonds. It is reported that the benzoic imine bond is stable at pH 7.4 but hydrolyzes in an acidic environment. No disassembly of nanoparticles, or release of encapsulated hydrophobic guest molecules, can be observed when low pH or reducing conditions were individually applied, because the nanoparticles were still crosslinked via either imine bonds or disulfide bonds. In order to increase the practicality of these nanoparticles as drug delivery vehicles, the possiblity of surface functionalization of nanoparticles was demonstrated by immobilizing PEG onto the particles.
Shell cross-linked (SCL) micelles has also attracted great interest as an alternative scaffold, where uncrosslinking the shell can trigger molecular release.45-47 The use of dual stimuli-sensitive SCL micelles, to release non-covalently loaded molecules in response to either acidic pH or a reductive microenviroment, has been recently demonstrated.48 In aqueous media, the diblock copolymer PCL-b-P(OEGMA-co-MAEBA) self-assembles into micelles consisting of hydrophobic PCL cores and a hydrophilic P(OEGMA-co-MAEBA) corona (Figure 3). Dithiol-bis(propanoic dihydrazide) (DTP) was used as the shell cross-linker, which contains both a disulfide linkage and hydrazide termini. DTP was conjugated with the micelle corona via aldehydes to form dynamic acylhydrazone bonds. Stimuli-induced drug release was demonstrated in vitro using camptothecin loaded SCL micelles by both acylhydrazone cleavage in pH 5.0 and disulfide cleavage in 10 mM DTT. Drug leakage was minimized under physiological pH 7.4, however release profiles at the pH of extracellular tumor tissues (~6.5) was not examined.
Though the majority of studies on dual chemical responsive systems have focused on pH and redox variations, others have been investigated that are sensitive to dual pH, dual redox, or to a combination of pH and salt. An intriguing example of such materials is a nanoparticle that can respond to both pHe (pH of the tumor extracellular matrix) and endo/lysosomal pH for enhanced drug delivery.49 As shown in Scheme 4, the polymer-doxorubicin conjugate (PPC-Hyd-DOX-DA) was pH-induced and charge-conversional. A polymer-doxorubicin conjugate was synthesized starting with the diblock copolymer monomethoxyl poly(ethylene glycol)-b-poly-(allyl ethylene phosphate) (mPEG-b-PAEP). This polymer was modified using a UV-induced thiolene “click” chemistry to synthesize cysteamine-modified mPEG-b-PAEP (mPEG-b-PAEP-Cya, PPC). Then a portion of amine groups were converted to sulfhydryl groups through reaction with 2-iminothiolane, which was used to conjugate DOX to the polymer by a hydrazone bond. The remaining amino groups were reacted with DMMA to obtain the polymer-doxorubicin conjugate. The amide bond formed between the amine group and 2,3-dimethylmaleic anhydride (DMMA) was cleavable under slightly acidic conditions, pH~6.8, which is comparable the pHe. The positive charge generated serves to enhance cellular uptake in the ECM specifically, where the pH is lower. On the other hand, DOX release occurs when the pH is decreased to endo/lysosomal pH due to the acid-labile hydrazone bond between DOX and the polymer. The DOX release at a pH relevant to endocytosed carriers promotes drug diffusion into the nuclei. This design proposes the employment of tumor site accumulation via the EPR effect, facilitated cell internalization by charge-conversion due to pHe, and intracellular drug release due to endosomal pH.
Scheme 4.

Chemical structure of the dual pH-responsive polymer-doxorubicin conjugate and pH triggered cellular internalization and intracellular drug release
Dual redox responsive assemblies have also been exploited for controlled drug delivery system upon specific biological events such as inflammation or physiological oxidative stress. An amiphiphilic triblock compolymer, shown in Figure 4, was synthesized by end-capping the diselenide containing polyurethane (PUSeSe) block with two poly(ethylene glycol) (PEG) chains.50 The polymer was found to form stable micelles that stably encapsulate the guest moelcules. The loaded guest molecules can be released from the micelles upon addition of an oxidant (i.e. H2O2) or a reductant (i.e. glutathione) due to the degradation of micelles caused by the cleavage of Se-Se bond either in oxidation or reduction conditions. It opens a new type of assembly for the programmed release of cargos in different environments. Variations in salt concentrations can also be used for disassembly in guest release, which provides biomedical significance to these assemblies. High intracellular salt concentrations (up to 451±6 mmol/kg dry weight) are associated with oncogenesis.51,52 Rapid drug release in tumor sites can be achieved if drug delivery systems respond not only to acidic pH, but also to the high salt concentration. For this purpose, dual-responsive micelles sensitive to variations in salt concentration and pH were prepared by using an amphiphilic brush copolymers, poly(2-hydroxyethylmethacrylate)-g-(poly(e-caprolactone)-adenine:uracil-poly(ethylene glycol)) (PHEMA-g-(PCL-A:U-PEG)) into which DOX was encapsulated (Figure 4).53 They showed that the hydrogen bonding interaction between PCL-A and U-PEG could be easily reduced by either changing the pH or increasing the salt concentration, leading to the disassembly of the micelles. Their results showed that 90% of the DOX was released in 25 hours at low pH and high salt concentration. This new type of smart micelle might have potential as anticancer delivery vehicles.
Figure 4.

Structure of dual redox responsive polymer and pH/salt dual responsive polymer
The examples above clearly illustrate the significant progress made in the field of dual chemical stimuli responsive carriers in terms of the design and synthesis of complex materials with dual chemical stimuli responsive properties. However, most researchers still focus on the design of carriers that respond to pH and redox, despite the fact that it is impossible to find one position in the tumor environment, where the pH is low and the GSH concentration is high concurrently.
For that reason, the dual pH and redox responsive polyplex carriers for enhanced intracellular delivery of plasmid DNA (pDNA), where these responses are sequentially tailored into a system (Scheme 5). In this study, ternary corsslinked polyplexes were prepared by coating a pH-sensitive membrane-active polyanion on positively charged binary crosslinked polyplexes, which were formed via electrostatic interaction between cationic poly (L-lysine) containing thiol groups (PLys(PDP)) and anionic pDNA, and then crosslinked by disulfide formation.54 The polyanion can be charge-conversional due to the degradation of cis-aconitic amides in late endosomal/lysosomal pH, helping the endosomal escape of polyplexes without associated cytotoxicity. At last, pDNA was released following disulfide reduction in the cytoplasm, where is reductive.
Scheme 5.
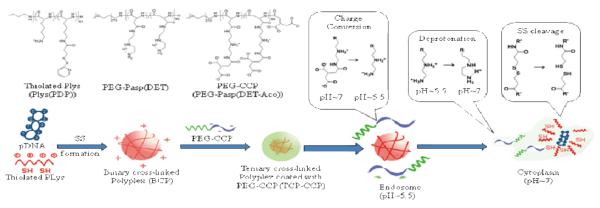
a) Chemical structure of polymers, b) Schematic illustration for preparation and intracellular trafficking of ternary cross-linked polyplex
Physical-chemical dual responsive macromolecules
Combining physical and chemical stimuli in designing responsive polymer systems is often aimed to take advantage of the key features in both internal and external stimuli. The combination of the two stimuli greatly improves the flexibility to trigger the stimuli-responsive polymers temporally and spatially, and therefore have the capability to achieve the desired properties on demand. One of the most investigated combinations of physical and chemical stimuli for dual responsive polymers is temperature and pH. Thermo and pH dual responsive polymers typically contain PNIPAM or poly(ethyleneglycol) (PEG) unit as the thermoresponsive component and a polyamine or polycarboxylic acid unit as the pH sensitive component. Hydrophilicity of both components undergoes drastic change in response to the stimuli and shifts the hydrophilic lipophilic balance (HLB) of the polymer to act as a driving force to mediate the assembly and disassembly process. By modifying the surface with dual responsive polymers, “intelligent” soft interfacial surface layers that exhibit the desired features can be achieved.55 Surface properties, such as wettability, have been tuned by altering the polymer chain conformation, by treating the surface at various pHs and temperatures.56 As is ubiquitous with stimuli-sensitive polymeric materials, these dual responsive polymers are also thought to be useful as drug delivery vehicles. Co-formulating lipids with PNIPAM-co-PAA (Figure 5) was found to efficiently reduce the thermo dose that is required to achieve appreciable cargo release from thermo-sensitive liposomes.57 The influence of polymer co-formulation on the threshold temperature of thermo-sensitive liposomes can be enhanced by lowering the pH of the solution. Unlike previous examples, where both responsive units are merged in the same copolymer, a micellar complex was achieved by co-assembly of a copolymer composed of ethylated 4-vinylpyridine, NIPAM with deoxycholic acid (DA) under basic conditions through Coulombic interactions, shown in Scheme 6.58 The assembled complex is disrupted under acidic conditions, where DA is protonated leading to a decrease in electrostatic interactions the driving force for the assembly. Appreciable release of guest molecules was observed at pH 5.2 at 37 °C; elevating the temperature enhanced the release percentage at the same pH. When heated above the LCST at pH 7.4, NIPAM presumably became less hydrophilic causing inter-particle aggregation, which likely caused the guest molecules to be squeezed out.
Figure 5.

Chemical and physical dual stimuli responsive polymers
Scheme 6.

Temperature and pH mediated disassembly of electrostatically assembled nanoparticles.
Temperature and pH sensitive dual responsive systems have drawn attention, because of the abnormal pH and elevated temperatures observed in many diseased tissues. These systems are often studied using a combination of a temperature-responsive moiety and a pH-sensitive dye. Optical properties of the dye should be such that it is highly sensitive to the local environment, so that minor variations in pH or temperature would be conveniently read using spectroscopic changes. An elegant example to illustrate this concept is the design of a pH/temperature dual sensor, based on a random copolymer composed of a pH-sensitive solvatochromic moiety, poly(disperse red 1 methacrylate) (PDR1MA), and a thermoresponsive component, poly(oligoethylene glycol methacrylate) (POEGMA) (Figure 5).59 The change in the optical properties of disperse red 1 in response to pH is not surprising. However, it is noteworthy that in acidic conditions, both a red shift of the polymer absorbance and color intensity enhancement was observed, when the polymer passed the LCST. In contrast, neither the color nor color intensity of disperse red 1 monomer changed with the solution temperature. This indicated that the temperature-induced solubility transition of the polymer has a significant effect on changing the polarity of co-polymerized dye, which was attributed to the enhanced protonation of the dye, caused by better solubility of the dye in the more hydrophobic polymer above the cloud point.
Using a similar concept, specific ion recognizable fluorogenic moieties have also been incorporated into the POEGMA chain to afford PEG-b-P(MEO2MA-co-OEGMA-co-ZQMA) shown in Figure 5, generating an ion and temperature sensitive dual sensor.60 Zn2+ coordinates with quinoline moiety in ZQMA generating the fluorescent complex to suggest the presence of zinc ions. The fluorescent complex is also sensitive to the polarity of the local environment, which is associated with the temperature-dependent solubility of POEGMA; therefore, the temperature change can also be monitored by the fluorescence change.
Another example of a dual ratiometric probe design that employs a FRET “turn-on” strategy was given by the same group, as shown in Scheme 7.61 In this study, a poly(oligoethyleneglycol methacrylate) containing double hydrophilic block copolymer was engineered to bear the 7-nitro-2,1,3-benzoxadiazole (NBD) moiety in the middle of the polymer chain, and rhodamine B (RhB) at both chain termini. Here, NBD serves as a fluorescent donor, while RhBs are pH switchable FRET acceptors. RhB exists as a lactam in basic conditions, which is non-fluorescent. However, RhB converts to an acyclic fluorescent form in acidic conditions and serves as a FRET acceptor for NBD, if the NBD and RhB units are in close proximity. It is important to note that the polymer stays in solution in a single chain state. Upon heating, the polymer chain collapses bringing NBD and RhB close to each other, which results in enhanced FRET. In comparison, only the fluorescence of NBD can be observed in basic conditions, across the temperature range, regardless of whether the polymer is in its extended or collapsed form, because RhB is non-fluorescent under these conditions.
Scheme 7.

Schematic representation of FRET processes modulated by pH and temperature
Understanding the effect of polymer’s architecture on their assembly behavior is also fundamentally interesting. An elegant candidate to investigate this releationship is pH/temperature dual sensitive dendronized polymers. Dendronized polymers, which merge both dendrimeric and polymeric architectures, were hypothesized to exhibit certain unique stimuli-responsive behavior. Poly(amidoamine) dendronized poly(2-hydroxyethyl) methacrylate (PHEMA) has been synthesized through a so-called “macromonomer” approach that quantitatively immobilizes the dendron to the polymer chain (Figure 5).62 The dendronization endows the polymer with a molecular weight dependent LCST behavior, which has not been clearly observed in traditional thermo-responsive polymers like PNIPAM. This dependence is attributed to the difference in hydrophilic lipophilic balance (HLB) between different generations. The G2 dendron has more amide functionalities than G1, rendering G2 more hydrophilic, and therefore having a higher LCST. The polymers also exhibit pH responsiveness due to the presence of tertiary amine moieties in the dendrons. The phase transition pH value of the G2 dendronized polymer is higher than the G1 polymer, which is similarly attributed to the higher hydrophilicity of the G2 dendron.
Interestingly, a recent report has shown that poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) which contains only pH sensitive functional groups havs been shown to demonstrate both pH and temperature sensitivity (see Figure 6).63 These block copolymers have been shown to form a variety of nanostructures with differential morphologies at different temperatures, which is attributed to the change in the packing parameters of the block copolymer. The packing parameter is directly associated with the effective volume fraction of hydrophobic PDPA block which is determined by the degree of ionization of the block. As the ionization of PDPA block was found to be a function of both solution pH and temperature, the morphology of the nanostructure can be modulated by the pH and temperature.
Figure 6.

Chemical and physical dual stimuli responsive polymers
External stimuli has not only been utilized to trigger the disintegration of nanoassemblies, but also to assist the self-assembly process. Capability to use the stimuli to manipulate the assembly process can also provide a robust avenue to control the physical and chemical characteristics of the obtained materials. Taking advantage of the LCST behavior of polyethylene glycol, our group has successfully obtained a variety of redox sensitive nanogels by varying the temperature at which we cross-link the nanoassemblies, based on poly(polyethylene glycol methacrylate)-co-poly(pyridyl disulfide ethylmethacrylate) poly(PEGMA-co-PDSEMA) (Figure 6).64 The size of the assembly of poly(PEGMA-co-PDSEMA) in solution is dependent on temperature, due to the LCST behavior of the PEG side chains. The assemblies can be cross-linked via disulfide reduction of PDS, to lock the assembled structure, thus achieving nanogel size variations. It is worth noting that nanogels prepared at higher temperatures are much more rigid and dense when compared with those synthesized at lower temperature. This is likely due to the compact packing of PEG chains in the assembly resulting from the increasing hydrophobicity as a result of temperature increase. The release of the cargo from these nanogels can be triggered under reducing conditions, similar to those found in tumor cells.
A material with similar responsiveness was synthesized by grafting PNIPAM from pullulan by RAFT polymerization, which provides this modified polysaccharide with LCST behavior that is utilized to drive the polysaccharide self-assembly in solution above the LCST of PNIPAM (Figure 6).65 Aminolysis of RAFT end groups, followed by oxidation of the generated thiol, affords the cross-linked nanogel as a product of disulfide formation. Dual responsiveness was indicated by the observation of temperature-triggered swelling, shrinking and disintegration induced by the reducing agent.
Control over the response kinetics of stimuli sensitive polymers is an important requirement for the designed material. Tunable guest release kinetics were achieved from micelles assembled from an amphiphilic block copolymer bearing both light and redox responsive moieties, by taking advantage of the different rate of responsiveness toward different stimuli.66 The middle hydrophobic block of PEO-b-poly(disulfide-alt-nitrobenzene)-b- PEO is susceptible to both reducing agents and photolysis (Scheme 8). Reduction of the disulfide backbone degrades the polymer and drives disassembly of micelles and guest release. Similarly, fast photolysis of o-nitrobenzyl methyl ester by exposure to UV light can also induce polymer degradation. In addition, the generated photolysis products of the o-nitrobenzyl block are alcohol and acid, both of which dramatically enhanced the hydrophilicity of the degraded polymer. The combined effect of these two driving forces was demonstrated to further accelerate the disintegration of micelles and guest release, when compared with exposure to a reducing agent. Manipulation of the guest release kinetics was achieved by combining fast-slow profiles using the two stimuli. Through a similar strategy, tunable guest release kinetics from micelles built up from PEG-S-S-PLA, shown in Figure 6, have also been achieved by treating micelles with ultrasound and a reducing agent.67 Disulfide linkage between hydrophilic and hydrophobic blocks is not only chemically reducible, but also can serve as a mechanophore. Cleavage of disulfide linkages, upon subjecting micelles to a reducing agent or ultrasound, breaks the amphiphilicity of the polymer inducing the disassembly of micelles, but with quite a different rate. Combining the two stimuli, the guest release kinetics can be finely tuned. One can expect such systems will have great implications in drug delivery for which release kinetics of a drug is required to be coordinated with the therapeutic window of the drug.
Figure 8.

Responsive molecules sensitive to more than two stimuli
Bio-related dual stimuli responsive macromolecules
Macromolecules that respond to biochemical stimuli can find use in many applications including drug and gene delivery, imaging, sensing, and bio-mimicry as artificial membranes, among others. Biochemical stimuli can induce physical and chemical responses in macromolecules such as sol-gel and gel-sol transitions, triggered assembly or disassembly, structural reorganization, and surface modification.68 The advantage of incorporating biochemical stimuli-induced responses to a molecular design is that they have the potential to be specific for a biomedical function, particularly when combined with a second cooperative biochemical stimulus or a complementary physical or chemical stimuli. The works discussed here will include examples utilizing enzymes and sugars as stimuli in dual-responsive materials. For more expansive information on enzyme responsive materials refer to the tutorial review “enzyme-responsive polymeric assemblies, nanoparticles and hydrogels”.68
Matrix Metalloproteinases (MMPs) are endopeptidases that are capable of degrading extracellular matrix proteins. The expression of MMPs are increased in the tumor environment as well as in rheumatoid arthritis, and are thus interesting enzymes to be used for drug delivery, imaging, and sensing functions. Multi-stimuli sensitive systems have been demonstrated using MMPs, including a dual MMP-induced surface-modified iron-oxide nanoparticle self-assembly, as well as gelatinase-and pH-driven disintegration of doxorubicin-DNA-polymer aggregates.69,70 Similarly, an MMP2 or MMP9 degradable and thermo responsive triblock copolymer pOEGMA-b-peptide-b-pNIPAM, shown in Figure 7 was recently synthesized by ATRP and studied for potential drug delivery applications.71 The triblock copolymer, comprised of a hydrophobic pNIPAM core and hydrophilic pOEGMA corona, can self-assemble into a micelle above the LSCT of thermosensitive pNIPAM. An azide functionality was incorporated at the termini of the pOEGMA block for click coupling to an alkyne containing florescent probe Alexa Fluor 555. In the presence of MMP, the protease-specific peptide linkage between the hydrophilic block and the hydrophobic block is cleaved, removing the poly(oligo(ethylene)) unit and Alexa Fluor probe. The polymer cleavage was demonstrated using GPC with fluorescence detection. After incubation with MMP, the peak at the retention time of pOEGMA-labeled pOEGMA-b-peptide-b-pNIPAM peak is greatly reduced, and the cleavage product pOEGMA is observed at a later retention time, comparable to the retention time of peptide-pOEGMA. Micelles were imaged using the Nanosight LM10-HS microscopy system using both light scattering and fluorescence. Following enzymatic degradation, the particles could still be visualized using the light scattering mode. However in the fluorescent mode, the shedding of the pOEGMA corona was apparent by the disappearance of fluorescence into the background noise. The authors propose that this pOEGMA corona “shedding” and has potential for enzyme triggered drug delivery applications.
Figure 7.

Bio-related dual stimuli-responsive polymers
Micellar nanoparticles that are morphologically responsive to both protease and phosphatase have recently been reported and are schematically represented in Scheme 9.72 Amphiphilic peptide hybrid copolymers that are comprised of a hydrophobic backbone and hydrophilic head groups containing consecutive substrates for cancer-associated enzymes protein kinase A (PKA), protein phosphatase-1 (PP1), and MMP-2 and -9, were synthesized. Two amphiphiles were made: amphiphile 1 containing polymer-conjugated distal PKA-substrate and terminal MMP-substrate, and amphiphile 2 containing the reversed sequence of polymer-conjugated distal MMP-substrate and terminal PKA-substrate. The peptide hybrid copolymers form stable spherical micelles due to their amphiphilic nature. For amphiphile 1, the addition of MMP and subsequent cleavage of the substrate functionalities, resulted in a retained spherical nanostructure. For amphiphile 2 however, the MMP-induced cleavage reduced the peptide hydrophilicity, and the micelles formed network aggregates. Morphological switching of the micelles was observed by a phosphorylation-dephosphorylation cycle. Phosphorylation of PKA-substrate formed larger amorphous aggregates, which is reversible in the presence of phosphatase.
Scheme 9.

Enzymatic modulation of polymer morphologies
An amphiphilic alternating polyester that is responsive to both pH and esterase, was synthesized by living ring-opening polymerization.73 The polyester, p(PGA-co-ME2MO), self-assembles into micelles and can encapsulate hydrophobic molecules due to the amphiphilicity from the hydrophilic oligo(ethylene glycol) side chains and the hydrophobic N-phthaloyl groups (Figure 7). These assemblies were loaded with rifampin and assessed for percent release over time at pH 7.4 (physiological pH) and 5.5 (endosomal pH), and in the presence of proteinase K. Results indicate that the assembly is stable at pH 7.4, but drug release accelerates at pH 5.5 due to hydrolysis of the polyester backbone. Drug release also accelerates in the presence of the esterase at pH 7.4 due to the degradation of the backbone. The experiments were repeated with the micelles loaded with DOX resulting in similar pH and enzyme dependent release profiles. DOX-loaded polyester micelles were observed to have a higher toxicity than free Dox in HeLa cells in vitro, which was hypothesized to be due to effective endocytosis of the cells and quick DOX release within the endosome.
The use of boronic acids in macromolecules has attracted increasing interest due to their ability to bind diols reversibly to form boronate esters in aqueous environments. Boronate esters are sensitive to pH and responsive to competing diols, such as carbohydrates, providing reason to explore their use in a variety of contexts.74-76 Incorporation of boronic acids into polymersomes, which are both pH and sugar responsive, were demonstrated to behave as nanoreactors with controllable membrane permeability (Scheme 10).77 A poly(boronic acid) containing block copolymer, poly(ethylene glycol)-block-poly(styrene boronic acid) (PEG-b-PSBA), was developed with a PEG-Br precursor by ATRP polymerization of a pinacol ester protected boronic acid styrene block, followed by pinacol deprotection. This amphiphilic block copolymer was co-assembled with poly(ethylene glycol)-b-polystyrene (PEG-b-PS) to form polymersomes. At high pH (12.6, 0.5 M aqueous NaOH), the boronic acid is ionized to boronate and the PEG-b-PSBA polymer becomes water-soluble. Similarly, the complexation of glucose to boronic acid imparts water solubility to the polymer and reduces the pH at which ionization to boronate occurs. Stimuli-induced permeability of these nanoreactors can be tuned by changing the weight percentage of the stimuli responsive polymer PEG-b-PSBA polymer to the co-assembled PEG-b-PS polymer. A model application of these nanoreactors was demonstrated by catalytic activity assessment of polymersome inclusion of Candida Antarctica Lipase B (CALB) and hydrolysis of substrates DiFMU octanoate and p-nitrophenyl acetate.
Scheme 10.

Sugar and OH− responsive permeable nanoreactor
A recent study has investigated the use of dual responsive boronate cross-linked telodendrimer-based micelles for drug delivery.78 The telodendrimers, comprised of linear polyethylene glycol and dendritic cholic acid block copolymers, were functionalized with boronic acid or catechols for crosslinking at the core-shell interface (Figure 7). The highly crosslinked boronate esters (BCM4) were stable at physiological pH and in the presence of blood proteins and sodium dodecyl sulfate (SDS). In the presence of SDS at pH 5.0, endosomal pH, the particle size decreased rapidly due to boronate ester hydrolysis. The particle size persisted in the presence of SDS and glucose (containing one cis-diol), however, in the presence of SDS and 100 mM mannitol (containing three cis-diol pairs) the particle size decreased. Using in vivo studies, it has been shown that DiD and PTX co-loaded BCM4 were able to preferentially accumulate in a SKOV-3 ovarian tumor as opposed to healthy organs, mainly due to the EPR effect. It will be interesting to see if the efficacy of drug delivery for these systems can be improved by mannitol co-administration in vivo.
A variety of applications, particularly those pertinent to biomedical functions, can benefit from well-defined and well-controlled biochemically-responsive materials. In the context of drug delivery, for instance, effective macromolecule design requires that chemical or physical response occurs selectively and efficiently at the desired location. When the biochemical response is paired with a second chemical or physical stimulus the duality should be clear in function (cooperative or complimentary) and location of response (same or different). Materials that are bio-responsive are exciting due to their supposed increase in specificity. Also, it has been noted that biochemical stimuli are caused by the primary imbalances of diseased states, where the commonly targeted chemical and physical stimuli are secondary responses biological imbalances.79,80to Obtaining fine control over these materials, to exploit inherent biological features, will be vital to gaining this specificity.
Multiple stimuli-responsive macromolecules
Multiple stimuli responsive materials, those that are responsive to three or more stimuli, are fundamentally interesting and are less explored. A boronic acid block copolymer, synthesized by RAFT polymerization of boronic acid acrylamido monomer (APBA) with NIPAM, was demonstrated to self-assemble in response to 81 changes in pH, sugar concentration, and temperature (Figure 8). Temperatures above the LCST of the PNIPAM block (32 °C) caused the PAPBA-b-PNIPAM block copolymers to dehydrate and aggregate into micelles in solution; below the LCST of the PNIPAM block, the copolymer is dissolved as unimers. The PAPBA block is neutralized and hydrophobic under acidic conditions and polyanionic and hydrophilic under basic conditions. The formation of boronate esters, in the presence of diols, also increases the hydrophilicity of the polymer. The multi responsive nature of PAPBA-b-PNIPAM was demonstrated by reversible particle aggregation and disintegration according to temperature, pH and glucose concentration. The dehydration of each block was also examined by NMR spectroscopy with change in pH and temperature.
Multi-responsive metallo-supramolecular polyelectrolyte gel-like materials that exhibit thermo-, chemo-and mechano-responses, have been reported using a combination of metal ions with a bis-ligand monomer.82 Pentaethylene glycol linked tridentate ligand bis(2,6-bis(1′-methyl-benzimidazoyl)-4-hydroxypyridine) (BIP-PEG-BIP), shown in Figure 8, was mixed with either Co(II) or Zn(II) ions as linear chain binding units or with La(III) or Eu(III) ions as cross-linking units. Four gel-like metallo-supramolecular materials were prepared with the bis-ligand monomer (1): 1:Co/La, 1:Zn/La, 1:Co/Eu, and 1:Zn/Eu, and tested for thermoresponsive behavior. A reversible gel-sol transition was observed for heating of 1:Co/La to 100 °C. The materials exhibited a thixotropic (shear-thinning) behavior, supporting that they are also mechano-responsive. The lanthanide-containing systems were also demonstrated as chemo-responsive. The addition of formic acid to 1:Zn/Eu broke down the gel-like material due to the binding of lanthanides to carboxylic acids; the process could be reversed by drying and re-swelling the material.
A viral capsid protein (CP) and elastin-like polypeptide (ELP) fusion protein that assembles in response to pH, temperature, and salt has been reported.83 Virus-like capsids of cowpea chlorotic mottle virus (CCWV) exist as soluble dimmers at pH 7.5, and can self-assemble into nanocapsules at pH 5.0 without its viral RNA. The nanocapsules are only stable at low pH, however, so an ELP was conjugated to the CP to incorporate its stimuli-responsive properties to the nanocapsule formation. The ELP is composed of repeating peptide V-P-G-Xaa-G for which Xaa is a natural amino acid other than proline and noted ELP[XiYjZk-n], where X, Y, or Z represent the amino acids Xaa, i, j, and k are the ratio of those amino acids at Xaa, and n is the number of pentapeptide repeats. ELPs have LCST behavior and a transition temperature (Tt) that is dependent on the particular peptide, protein concentration, and salt concentration. Using TEM, the conjugated CP-ELP[V4L4G1-9] was shown to form 28 nm assemblies at pH 5.0 due to the pH responsiveness of CP. The size exclusion chromatography (SEC) and multi-angle laser light scattering (MALLS) of the nanocapsules indicated that the molecular weight was consistent with 180 CP units, like those RNA containing CCMW with T=3 icosahedral architecture. The fusion protein also formed aggregates in response to increase in temperature from 20 °C to 35 °C due to the LCST behavior of ELP. The Tt of ELP was decreased to below room temperature for the fusion protein by the increase of NaCl concentration, so that nanocapsules are able to form at pH 7.5 without the application of heat. These “ELP-induced” capsules were found to have a diameter of 18 nm and T=1 icosahedral architecture. The CP-ELP fusion protein was thus able to form defined and different nanocapsules based on the applied stimuli.
A block copolymer that is triply responsive to changes in temperature, pH, and redox potential, has micellar properties, and can encapsulate guest molecules has been reported by our group.84 The multiple stimuli responsiveness of the block copolymer is schematically shown in Scheme 11. The design consists of a PNIPAM block attached to a THP-protected HEMA block with a disulfide linker. The THP-protected HEMA block changes from hydrophobic to hydrophilic following THP deprotection in acidic conditions. Release of encapsulated Nile red was found to increase over time as the acetal cleaves due to the decrease in pH. The PNIPAM block changes from hydrophilic to hydrophobic above its lower critical solution temperature (LCST). Turbidity measurements were conducted to obtain an LCST of about 35 °C for the block copolymer (BCP). Additionally, reduction of the disulfide linker can cleave the BCP into its respective homopolymers under mild reducing conditions. GPC analysis confirmed dissociation into homopolymers. Turbidity of the solution was also found to increase after DTT addition, as a result of the precipitation of the p-HEMA and an LCST that resembles only PNIPAM. Nile red containing BCP micelles were treated with glutathione to monitor percent dye retention, and it was found that dye precipitation increased with time. To assess the ability to fine-tune the release kinetics, the responsiveness to a combination of stimuli was investigated. An acidic environment was demonstrated to affect the thermal behavior of the polymer. The combination of acidic and redox environments resulted in a more rapid micelle collapse and release of encapsulated Nile red, as compared to either stimuli alone. Lastly, Nile red containing BCPs were treated with acid and DTT and incubated at room temperature, then the temperature of the solution was increased. A curious result was that the solution did not become cloudy as expected due to PNIPM dehydration, so it was hypothesized that HEMA blocks may be engaged in hydrogen bonding in PNIPAM, affecting the LCST behavior.
Scheme 11.

Schematic representation of block copolymer combined pH, thermo and reduction sensitivity
A series of amphiphilic oligomers were synthesized by our group that exhibit sensitivity to temperature, enzymatic reaction, and pH (Figure 8).85 The response to stimuli for well-defined oligomers was compared to the corresponding monomers, in an attempt to identify the reasons that polymer sensitivity is greater than amphiphilic small molecules. The amphiphiles consist of a penta(ethylene glycol) and alkyl moiety attached to the meta-positions of a benzoyl building block, which were then attached to oligoamine scaffolds to yield monomeric to hexameric oligomers. Temperature sensitivity of oligomers was assessed by turbidity measurements using the high-tension voltage response of the photomultiplier on a CD spectrometer, while keeping the concentration of OEG units constant. As oligomeric length increased, both the transition temperatures (Tt) and the full-width at half max (FWHM) decreased. The latter factor is related to the rate of coil-to-globule transition. The difference in transition rate was attributed to the difference in cooperativity in the dehydration of OEG units with temperature variation. Additionally, because the alkyl moiety is terminated with an ester, the oligomers are also esterase sensitive. The product of esterase cleavage is a pH sensitive carboxylic acid, which can affect the HLB and LCST of the oligomer. The pentamer incubated with porcine liver esterase, resulted in a temperature dependent turbidity generation about 10 °C higher than the same pentamer solution without enzyme. Consistent with predictions for effect on the HLB, protonation of PLE treated pentamer in acidic conditions decreased the Tt and FWHM, while the Tt increased under basic conditions.
Finally, our group reported the formation of micellar aggregates from the complexation of a polyanionic polymer and a non-protonable quaternary ammonium cation.86 The polyelectrolyte composed of a homopolymer containing an ionic carboxylate unit as shown in Scheme 12 was connected to the polymer backbone by a disulfide bond. After complexation of the polyelectrolyte with the surfactant, dodecyl(trimethyl)azanium bromide (DTAB), the critical aggregate concentration (CAC) was lower when compared to that of DTAB alone, suggesting that these micelles can encapsulate guest molecules at a low concentration. It was also demonstrated that the addition of a reducing agent such as GSH disrupted the polyvalent interactions, resulting in disassembly of the aggregate and release of the encapsulated guest, Nile red. Interestingly, it was reported that these aggregates were also disassembled by low pH and high salt concentration by weakening the electrostatic interactions of the surfactant and the polymer.
Scheme 12.

Multiple stimuli responsive polyelectrolyte/surfactant complex micelles.
Primary investigations of multiple responsive materials have provided considerable insights into the factors that need to be tuned to incorporate three or more responsive elements in one design. The molecular design requirements for a system that needs to respond to three more stimuli becomes certainly more complex. However, these systems do hold the promise of being even more specific in its response to three or more stimuli concurrently. Enhanced understanding of the design principles should lead to promising materials both for biological and abiological applications.
Conclusions and outlook
We have attempted to outline the design principles that underlie polymer-based stimuli responsive materials with multi-stimuli responsive capabilities. The rather simplified “design rule” is that one can simply incorporate two or more functional groups that respond to different stimuli into the polymeric system. A main requirement is that these moieties, which are responsive to physical, chemical, or biological stimuli, be mutually compatible in the polymer during the syntheses, assembly, and use. The response to these stimuli could be structural or functional. The structural responses have often involved phase transformations, assembly formations or disassembly. The functional responses have involved molecular release, solubility variations, or color/fluorescence changes. The investigations that led to our current understanding, have provided valuable insights into these “design rules”. Guided by these, custom-designed sitmuli responsive polymers have the potential to profoudly influence a broad spectrum of fields including surface science, sensing, tissue engineering, and drug delivery.
Though exciting progress has been made in the area of multi-stimuli resopnsive polymers, these materials are yet to reach the practical realm. From a fundamental perspective, while significant information has been obtained about the initial and final states of the assemblies in response to a stimulus or combination of stimuli, relatively little known about the pathways through which these stimuli-induced changes occur. Such an understanding will be instrumental in greatly expanding repertoire of stimuli-responsive assemblies. From an applications perspective, relatively little attention has been paid to translate the fundamental understanding in molecular design criteria to develop polymers that would be biocompatible and biodegradable. Nonetheless, considering our current understanding of the structure-property relationships of these materials and the ever-improving analytical capabilities to visualize time-dependent evolution of nanostructures, the custom-designed multi-stimuli responsive materials are well-positioned to make a significant impact in a variety of areas. Prominent among these areas include drug delivery, sensing, self-healing materials, tissue engineering, and adaptable surfaces.
Scheme 8.
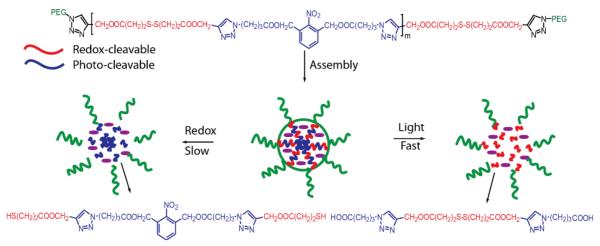
Fast and slow disassembly of micelles induced respectively by light and redox stimuli.
Acknowledgment
Support from the NIGMS of the National Insitutes of Health (GM-065255) and the U.S. Army Research Office (57858-CH) are acknowledged. Support from the National Science Foundation through the Materials Research Science and Education Center (MRSEC) and the Center for Hierarchical Manufacturing Nanoscale Science and Engineering Center (NSEC) are also acknowledged.
Biographies

Jiaming Zhuang
Jiaming Zhuang received his B.Eng. (2005) and M.Sc. (2008) degree from Xiamen University, Fujian, PR China. He has been pursuing his Ph. D. degree under the guidance of Professor Thayumanavan in Department of Chemistry, UMass Amherst since 2009. He is currently working on design, synthesis and self-assembly of stimuli responsive amphiphilic copolymers and their biological applications.

Mallory R. Gordon
Mallory R. Gordon received her B.A. degree from Franklin and Marshall College in 2011 with a major in Chemistry. She is currently a Ph.D. student in the Chemistry Department at the University of Massachusetts under the direction of Professor S. Thayumanavan. Her current research is focused on enzyme-responsive amphiphilic polymeric nanogels.

Judy Ventura
Judy Ventura was born in Mayagüez, Puerto Rico in 1983. In 2008 she received her B. S. degree in science majoring Industrial Biotechnology at the University of Puerto Rico, Mayagüez Campus. She then joined the Chemistry Department from the University of Massachusetts, Amherst where she is currently pursuing her PhD degree under the supervision of Professor Thayumanavan. Her research interests focuses on designing and developing polymeric vehicles for drug and protein delivery applications.

Longyu Li
Longyu Li was born in Jiangxi, China in 1988. He received his B.Sc degree in chemistry from Nankai University (2009), Tianjin, China. Then he spent almost one year as a research assistant in Fudan Univeristy, Shanghai, China. He joined the University of Massachusetts Amherst, as a Ph.D. student in the fall of 2010 and is currently working under the guidance of Professor S. Thayumanavan. His current research is focused on the design and synthesis of polymeric nanogel for drug delivery and also the preparation of monodisperse nanoparticles based on boronate esters.

S. “Thai” Thayumanavan
S. “Thai” Thayumanavan is a Professor in the Department of Chemistry at the University of Massachusetts Amherst. He received his B.Sc. and M.Sc. degrees from The American College in Madurai, India. He received his Ph.D. from the University of Illinois at Urbana-Champaign in 1996 under the direction of Professor Peter Beak. Following a postdoctoral stint with Professor Seth R. Marder at Caltech, he started his independent career at Tulane University in 1999 and moved to UMass Amherst in 2003. His research work involves the design and syntheses of new macromolecules, including dendrimers, to obtain novel supramolecular assemblies that are of interest in stimuli-sensitive materials, delivery of hydrophobics and biologics, sensing, and renewable energy applications.
Notes and references
- 1.Binder WH, Sachsenhofer R. Macromol. Rapid Commun. 2007;28:15–54. [Google Scholar]
- 2.Moad G, Rizzardo E, Thang SH. Aust. J. Chem. 2009;62:1402–1472. [Google Scholar]
- 3.Matyjaszewski K. Macromolecules. 2012;45:4015–4039. [Google Scholar]
- 4.Hawker CJ, Bosman AW, Harth E. Chem. Rev. 2001;101:3661–3688. doi: 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- 5.Hilf S, Kilbinger AFM. Nat. Chem. 2009;1:537–546. doi: 10.1038/nchem.347. [DOI] [PubMed] [Google Scholar]
- 6.Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk V.v., Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nat. Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 7.Chacko RT, Ventura J, Zhuang J, Thayumanavan S. Adv. Drug Delivery Rev. 2012;64:836–851. doi: 10.1016/j.addr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riess G. Prog. Polym. Sci. 2003;28:1107–1170. [Google Scholar]
- 9.Schild G. Prog. Polym. Sci. 1992;17:163–249. [Google Scholar]
- 10.Zhang Y, Cremer PS. Curr. Opin. Chem. Biol. 2006;10:658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Luo S, Shi W, Liu S. Langmuir. 2006;22:989–997. doi: 10.1021/la0522707. [DOI] [PubMed] [Google Scholar]
- 12.Lee AS, Gast AP, Butun V, Armes SP. Macromolecules. 1999;32:4302–4310. [Google Scholar]
- 13.Luo S, Ling C, Hu X, Liu X, Chen S, Han M, Xia J. J. Colloid Interface Sci. 2011;353:76–82. doi: 10.1016/j.jcis.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 14.Chang Y, Chen WY, Yandi W, Shih YJ, Chu WL, Liu YL, Chu CW, Ruaan RC, Higuchi A. Biomacromolecules. 2009;10:2092–2100. doi: 10.1021/bm900208u. [DOI] [PubMed] [Google Scholar]
- 15.Roth PJ, Davis TP, Lowe AB. Macromolecules. 2012;45:3221–3230. [Google Scholar]
- 16.Peng B, Grishkewich N, Yao Z, Han X, Liu H, Tam KC. ACS Macro Lett. 2012;1:632–635. doi: 10.1021/mz300135x. [DOI] [PubMed] [Google Scholar]
- 17.Kungwatchakun D, Irie M. Makromol. Chem. Rapid Commun. 1988;9:243–246. [Google Scholar]
- 18.Salonen A, Langevin D, Perrin P. Soft Matter. 2010;6:5308–5311. [Google Scholar]
- 19.Feng Z, Lin L, Yan Z, Yu Y. Macromol. Rapid Commun. 2010;31:640–644. doi: 10.1002/marc.200900777. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama H, Tamaoki N. J. Polym. Sci., Part A: Polym. Chem. 2004;42:5200–5214. [Google Scholar]
- 21.Hartley GS, Le Févre JW. J. Chem. Soc. 1939:531–535. [Google Scholar]
- 22.Kumar GS, Neckers DC. Chem. Rev. 1989;89:1915–1925. [Google Scholar]
- 23.Jochum FD, Theato P. Polymer. 2009;50:3079–3085. [Google Scholar]
- 24.Jochum FD, Theato P. Macromolecules. 2009;42:5941–5945. [Google Scholar]
- 25.Shimoboji T, Larenas E, Fowler T, Kulkarni S, Hoffman AS, Stayton P. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16592–16596. doi: 10.1073/pnas.262427799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He J, Yan B, Tremblay L, Zhao Y. Langmuir. 2011;27:436–444. doi: 10.1021/la1040322. [DOI] [PubMed] [Google Scholar]
- 27.Woodcock JW, Wright RAE, Jiang X, O’Lenick TG, Zhao B. Soft Matter. 2010;6:3325–3336. [Google Scholar]
- 28.Jochum FD, Theato P. Chem. Soc. Rev. 2013 doi: 10.1039/c2cs35191a. Advanced article. [DOI] [PubMed] [Google Scholar]
- 29.Heiden MGV, Cantley LC, Thompson CB. Science. 324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HJ, Bae Y. Biomacromolecules. 2011;12:2686–2696. doi: 10.1021/bm200483t. [DOI] [PubMed] [Google Scholar]
- 31.Zhou ZX, Shen YQ, Tang JB, Fan MH, Van Kirk EA, Murdoch WJ, Radosz M. Adv. Funct. Mater. 2009;19:3580–3589. [Google Scholar]
- 32.Liu T, Li XJ, Qian YF, Hu XL, Liu SY. Biomaterials. 2012;33:2521–2531. doi: 10.1016/j.biomaterials.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 33.Ou M, Xu RZ, Kim SH, Bull DA, Kim SW. Biomaterials. 2009;30:5804–5814. doi: 10.1016/j.biomaterials.2009.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakravarthi S, Jessop CE, Bulleid NJ. EMBO Rep. 2006;7:271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito G, Swanson JA, Lee KD. Adv. Drug Deliv. Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 36.Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH, Lee SC. Chem. Commun. 2008;48:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Liu W, Lin L, Chen D, Stenzel MH. Biomacromolecules. 2008;9:3321–3331. doi: 10.1021/bm800867n. [DOI] [PubMed] [Google Scholar]
- 38.Ryu, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J. Am. Chem. Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 39.Ryu J-H, Bickerton S, Zhuang J, Thayumanavan S. Biomacromolecules. 2012;13:1515–1522. doi: 10.1021/bm300201x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai S, Ravi P, Tam KC. Soft Matter. 2008;4:435–449. doi: 10.1039/b714741d. [DOI] [PubMed] [Google Scholar]
- 41.McCarle RL. Annu Rev Anal Chem. 2012;5:391–411. doi: 10.1146/annurev-anchem-062011-143157. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Qiu X, Ouyang J, Kong J, Zhong W, Xing MM. Biomacromolecule. 2011;12:3601–3611. doi: 10.1021/bm200804j. [DOI] [PubMed] [Google Scholar]
- 43.Wei C, Guo J, Wang C. Macromol Rapid Commun. 2011;32:451–455. doi: 10.1002/marc.201000708. [DOI] [PubMed] [Google Scholar]
- 44.Jackson AW, Fulton DA. Macromolecules. 2012;45:2699–2708. [Google Scholar]
- 45.Thurmond KB, Remsen EE, Kowalewski T, Wooley KL. Nuc. Acids Res. 1999;27:2966–2971. doi: 10.1093/nar/27.14.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Remsen EE, Wooley KL. J. Am. Chem. Soc. 2000;122:3642–3651. [Google Scholar]
- 47.Wooley KL. J. Polym. Sci., Part A: Polym. Chem. 2000;38:1397–1407. [Google Scholar]
- 48.Hu X, Li H, Luo S, Liu T, Jiang Y, Liu S. Polym. Chem. 2013;4:695–706. [Google Scholar]
- 49.Du JZ, Du XJ, Mao CQ, Wang J. J. Am. Chem. Soc. 2011;133:17560–17563. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 50.Ma N, Li Y, Xu H, Wang Z, Zhang X. J. Am. Chem. Soc. 2010;132:442–443. doi: 10.1021/ja908124g. [DOI] [PubMed] [Google Scholar]
- 51.Cameron I, Smith N, Pool T, Sparks R. Cancer Res. 1980;40:1493–1500. [PubMed] [Google Scholar]
- 52.Luciani A. Eur. J. Cell Biol. 2001;80:187–195. doi: 10.1078/0171-9335-00102. [DOI] [PubMed] [Google Scholar]
- 53.Wang D, Huan X, Zhu L, Liu J, Qiu F, Yan D, Zhu X. RSC Adv. 2012;2:11953–11962. [Google Scholar]
- 54.Sanjoh M, Miyata K, Christie RJ, Ishii T, Maeda Y, Pittella F, Hiki S, Nishiyama N, Kataoka K. Biomacromolecules. 2012;13:3641–3649. doi: 10.1021/bm301095a. [DOI] [PubMed] [Google Scholar]
- 55.Rahane SB, Floyd, Metters AT, Kilbey SM. Adv. Funct. Mater. 2008;18:1232–1240. [Google Scholar]
- 56.Xia F, Feng L, Wang S, Sun T, Song W, Jiang W, Jiang L. Adv. Mater. 2006;18:432–436. [Google Scholar]
- 57.Ta T, Convertine AJ, Reyes CR, Stayton PS, Porter TM. Biomacromolecules. 2010;11:1915–1920. doi: 10.1021/bm1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui W, Lu X, Cui K, Niu L, Wei Y, Lu Q. Langmuir. 2012;28:9413–9420. doi: 10.1021/la3016436. [DOI] [PubMed] [Google Scholar]
- 59.Pietsch C, Hoogenboom R, Schubert US. Angew. Chem. Int. Ed. 2009;48:5653–5656. doi: 10.1002/anie.200901071. [DOI] [PubMed] [Google Scholar]
- 60.Liu T, Liu S. Anal. Chem. 2011;83:2775–2785. doi: 10.1021/ac200095f. [DOI] [PubMed] [Google Scholar]
- 61.Wan X, Liu S. J. Mater. Chen. 2011;21:10321–10329. [Google Scholar]
- 62.Gao M, Jia X, Li Y, Liang D, Wei Y. Macromolecules. 2010;43:4314–4323. [Google Scholar]
- 63.Pearson RT, Warren NJ, Lewis AL, Armes SP, Battaglia G. Macromolecules. 2013;46:1400–1407. [Google Scholar]
- 64.Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J. Am. Chem. Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 65.Morimoto N, Qiu X, Winnik FM, Akiyoshi K. Macromolecules. 2008;41:5985. [Google Scholar]
- 66.Han D, Tong X, Zhao Y. Langmuir. 2012;28:2327–2331. doi: 10.1021/la204930n. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Tong R, Xia H, Zhang H, Xuan J. Chem. Commun. 2010;46:7739–7741. doi: 10.1039/c0cc02628j. [DOI] [PubMed] [Google Scholar]
- 68.Hu J, Zhang G, Liu S. Chem. Soc. Rev. 2012;41:5933–5949. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 69.von Maltzahn G, Harris TJ, Park J-H, Min D-H, Schmidt AJ, Sailor MJ, Bhatia SN. J. Am. Chem. Soc. 2007;129:6064–6065. doi: 10.1021/ja070461l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong L, Xia S, Huang Z, Chen H, Chen J, Zhang J. Biomaterials. 2010;31:6309–6316. doi: 10.1016/j.biomaterials.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 71.de Graaf AJ, Mastrobatttista E, Vermonden T, van Nostrum CF, Rijkers DTS, Liskamp RM, Hennink WH. Macromolecules. 2012;45:842–851. [Google Scholar]
- 72.Ku T-H, Chien M-P, Thompson MP, Sinkovits RS, Olson NH, Baker TS, Gianneschi NC. J. Am. Chem. Soc. 2011;133:8392–8395. doi: 10.1021/ja2004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang W, Ding J, Xiao C, Tang Z, Li D, Chen J, Zhuang X, Chen X. Biomacromolecules. 2011;12:2466–2474. doi: 10.1021/bm200668n. [DOI] [PubMed] [Google Scholar]
- 74.Pasparakis G, Cockayne A, Alexander C. J. Am. Chem. Soc. 2007;129:11014–11015. doi: 10.1021/ja074349z. [DOI] [PubMed] [Google Scholar]
- 75.Pasparakis G, Vamvakaki M, Krasnogor N, Alexander C. Soft Matter. 2009;5:3839–3841. [Google Scholar]
- 76.Wu W, Mitra N, Yan ECY, Zhou S. ACS Nano. 2010;4:4831–4839. doi: 10.1021/nn1008319. [DOI] [PubMed] [Google Scholar]
- 77.Kim KT, Cornelissen JJLM, Nolte RJM, van Hest JCM. Adv. Mater. 2009;21:2787–2791. [Google Scholar]
- 78.Li Y, Xiao W, Xiao K, Berti L, Luo J, Tseng HP, Fung G, Lam KS. Angew. Chem. Int. Ed. 2012;51:2864–2869. doi: 10.1002/anie.201107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azagarsamy M, Sokkalingam P, Thayumanavan S. J. Am. Chem. Soc. 2009;131:14184–14185. doi: 10.1021/ja906162u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azagarsamy M, Yesilyurt V, Thayumanavan S. J. Am. Chem. Soc. 2010;132:4550–4551. doi: 10.1021/ja100746d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roy D, Cambre JN, Sumerlin BS. Chem. Commun. 2009:2106–2108. doi: 10.1039/b900374f. [DOI] [PubMed] [Google Scholar]
- 82.Beck JB, Rowan SJ. J. Am. Chem. Soc. 2003;125:13922–13923. doi: 10.1021/ja038521k. [DOI] [PubMed] [Google Scholar]
- 83.Van Eldijk MB, Wand JC-Y, Minten IJ, Li C, Zlotnick A, Nolte RJM, Comelissen JLM, van Hest JCM. J. Am. Chem. Soc. 2012;134:18506–18509. doi: 10.1021/ja308132z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klaikherd A, Nagamani C, Thayumanavan S. J. Am. Chem. Soc. 2009;131:4830–4838. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang F, Klaikherd A, Thayumanavan S. J. Am. Chem. Soc. 2011;133:13496–13503. doi: 10.1021/ja204121a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gosh S, Yesilyurt V, Savariar EN, Irvin K, Thayumanavan S. J. Polym. Sci., Part A: Polym. Chem. 2009;47:1052–1060. doi: 10.1002/pola.23204. [DOI] [PMC free article] [PubMed] [Google Scholar]