

Abstract
Presynaptic differentiation of axons plays a fundamental role in the establishment of neuronal connectivity. However, the mechanisms that govern presynaptic differentiation in the brain remain largely to be elucidated. We report that knockdown of the transcription factor MEF2A in primary neurons and importantly in the rat cerebellar cortex in vivo robustly increases the density of orphan presynaptic sites. Remarkably, the sumoylated transcriptional repressor form of MEF2A drives the suppression of orphan presynaptic sites. We also identify the gene encoding synaptotagmin 1 (Syt1), which acts locally at presynaptic sites, as a direct repressed target gene of sumoylated MEF2A in neurons, and demonstrate that repression of Syt1 mediates MEF2A-dependent elimination of orphan presynaptic sites. Finally, we uncover a role for the MEF2A-induced elimination of orphan presynaptic sites in the accumulation of presynaptic material at large maturing presynaptic boutons. Collectively, these findings define sumoylated MEF2A and Syt1 as components of a novel cell-intrinsic mechanism that orchestrates presynaptic differentiation in the mammalian brain. Our study has important implications for understanding neuronal connectivity in brain development and disease.
Introduction
Synapse formation is essential for the establishment of neural circuitry in the brain. Abnormalities of synapse morphogenesis may contribute to the pathogenesis of diverse disorders from the autism spectrum disorders to neurodegenerative diseases (Selkoe, 2002; Zoghbi, 2003; Abrahams and Geschwind, 2008; Kelleher and Bear, 2008; Südhof, 2008). Therefore, elucidation of the mechanisms that govern synapse morphogenesis will advance our understanding of brain development and diseases.
Presynaptic differentiation of axons represents a fundamental step in synaptogenesis (Scheiffele et al., 2000; Graf et al., 2004; Christopherson et al., 2005; Colón-Ramos et al., 2007). An early event in the process of presynaptic differentiation is the formation of immature release sites, which assemble independently of contact with postsynaptic dendritic structures and are thus termed orphan presynaptic sites (Krueger et al., 2003; Ziv and Garner, 2004; Sabo et al., 2006). Synaptic vesicle and active zone proteins accumulate at orphan presynaptic sites, enabling these sites to support synaptic vesicle exocytosis and endocytosis (Krueger et al., 2003; Sabo et al., 2006). As the process of presynaptic differentiation proceeds, orphan presynaptic sites are eliminated and synaptic vesicle and active zone proteins preferentially localize at presynaptic sites that contact postsynaptic elements (Polo-Parada et al., 2001; Nakamura et al., 2006). However, the mechanisms that orchestrate the suppression of orphan presynaptic sites as neurons mature have remained poorly understood.
The transcription factor myocyte enhancer factor 2A (MEF2A) is highly expressed in differentiated neurons in the brain throughout the period of synaptogenesis (Lyons et al., 1995; Flavell et al., 2006; Shalizi et al., 2006). MEF2A has been implicated in the control of postsynaptic dendritic differentiation (Flavell et al., 2006; Shalizi et al., 2006; Pulipparacharuvil et al., 2008; Pfeiffer et al., 2010). Notably, the transcriptional repressor form of MEF2A that is covalently modified at Lysine 403 by the protein small ubiquitin-related modifier (SUMO) drives postsynaptic dendritic differentiation in the mammalian brain (Shalizi et al., 2006). However, whether and how MEF2A might regulate presynaptic development has remained largely to be elucidated.
In this study, we have discovered that MEF2A plays a critical role in presynaptic development in the mammalian brain. Knockdown of MEF2A robustly increases the density of orphan presynaptic sites in primary neurons and in the rat cerebellar cortex in vivo. Strikingly, the sumoylated transcriptional repressor form of MEF2A mediates the suppression of orphan presynaptic sites. We have also identified synaptotagmin 1 (Syt1) as a direct repressed target gene of sumoylated MEF2A in neurons, and we have found that Syt1 repression operates downstream of MEF2A in the elimination of orphan sites. We have also found that MEF2A-induced elimination of orphan presynaptic sites triggers the maturation of large presynaptic boutons. Together, these findings define a novel transcriptional pathway that orchestrates the coordinate suppression of orphan presynaptic sites and maturation of presynaptic boutons.
Materials and Methods
Plasmids.
The GFP-synapsin and GFP-Munc13 plasmids are described previously (Chi et al., 2001; Andrews-Zwilling et al., 2006). RNAi plasmids were designed as described previously (Gaudilliere et al., 2002). The U6/syt1 plasmid was cloned using the primer: 5′-GTA AGA GGC TGA AGA AGA AA-3′. The RNAi-resistant MEF2A wild-type and K403R mutant plasmids and MEF2A-SUMO plasmid are described previously (Shalizi et al., 2006). Syt1 was cloned from Sprague Dawley rat cDNA and inserted into the pcDNA3 vector (Invitrogen). The pCAG-GFP-synapsin vector was constructed by inserting synapsin into the pCAG-GFP vector (Matsuda and Cepko, 2004) at the BsrGI and BglII sites.
Antibodies.
Antibodies to synapsin (Millipore), PSD95 (Neuromab), GFP (Invitrogen), Flag (Sigma), ERK1/2 (Cell Signaling Technology), Syt1 (Synaptic Systems), Munc13 (Abcam), Bassoon (Assay Designs), synaptophysin (Sigma), NR1 (Millipore), MEF2A (H300, Santa Cruz Biotechnology), and rabbit IgG (Millipore) were purchased. The polyclonal MEF2A antibody for chromatin immunoprecipitation (ChIP) analyses was a gift from Dr. Ron Prywes (Columbia University, New York, NY).
Cerebellar granule neurons.
Granule neurons were prepared from cerebella of P6 Long–Evans rat pups of either sex as described previously (Yang et al., 2009). Neurons were placed in full basal Eagle's medium (BME) supplemented with 5% calf serum at 1 d in vitro (DIV1) and transfected at DIV3 or DIV7 with a modified calcium phosphate method as described previously (Konishi et al., 2002). Granule neurons were fixed 3 d after transfection and immunostained with the GFP and PSD95 antibodies. The density of synapsin clusters was counted along randomized 100–200 μm segments of the distal axon and represented as the density of synapsin clusters per 100 μm. The images were taken in a blinded manner using a NIKON eclipse TE2000 epifluorescence microscope using a digital CCD camera (DIAGNOSTIC Instruments) and imported into the SPOT imaging software.
High-efficiency transfection of granule neurons (maximum efficiency 70%) for biochemical analyses was achieved using a nucleofection method with the Amaxa electroporation device as described previously (Yuan et al., 2008; de la Torre-Ubieta et al., 2010).
Live and functional imaging.
Live confocal imaging analyses were performed as described previously (Puram et al., 2011). Granule neurons were plated on glass-bottom multiwell plates (MatTek) and transfected as indicated. Live imaging of synapsin clusters and presynaptic varicosities was performed using a Yokogawa spinning-disk confocal system. An environment-controlled chamber maintained the neurons at 37°C, 5% CO2. Images were acquired and analyzed using Volocity software (PerkinElmer Life and Analytical Sciences). A 40× objective was used in combination with an automated stage to capture images over a 2 h period. Neurons were chosen randomly and followed by saving their coordinates on the motorized stage.
FM4-64 assays were performed as described previously (Chi et al., 2001; Yang et al., 2009). For FM4-64 imaging, granule neurons transfected with the GFP-synapsin expression plasmid were incubated with 10 μm FM4-64 for 60 s in loading buffer containing 90 mm KCl, 64 mm NaCl, 2 mm MgCl2, 10 mm glucose, 10 mm HEPES, pH 7.4, and 2 mm CaCl2. Cultures were then washed for 10 min with hyperpolarizing buffer, 4 mm KCl, 150 mm NaCl, 2 mm MgCl2, 10 mm glucose, 10 mm HEPES (pH 7.4), and 2 mm CaCl2 to prevent further endocytosis/exocytosis. Neurons were fixed and processed for immunocytochemistry without permeabilization using an antibody targeting an extracellular loop of NR1.
Electron microscopy.
For ultrastructural analysis, lentiviral infected granule neurons were processed for EM as described previously (Litterman et al., 2011; Puram et al., 2011). A Tecnai G2 Spirit BioTWIN electron microscope was used for collection of high-magnification EM images.
Real time RT-PCR.
RNA was extracted from 6 × 106 granule neurons using Trizol (Invitrogen) according to the manufacturer's instructions. Real-time PCR was performed using Lightcycler 480 SYBR Green I Master (Roche) with the following pairs of primers: Syt1 (forward): 5′-TCA ACC CCT ACT ACA ACG AGT CC-3′; Syt1 (reverse): 5′-TTG ACA GCC AGC ATG GCA TCA AC-3′; GAPDH (forward): 5′-TGC TGG TGC TGA GTA TGT CG-3′; GAPDH (reverse): 5′-GCA TGT CAG ATC CAC AAC GG-3′.
Immunoblotting.
Immunoblotting was performed as described previously (Shalizi et al., 2006). Briefly, 293T cells or granule neurons were lysed in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 100 mm NaF, 1 mm DTT, 1 mm sodium vanadate, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 20 mm NEM. Rat cerebella were homogenized with the lysis buffer 25 times and incubated on ice for 20 min.
Microarray analyses.
Total RNA was extracted from granule neurons expressing MEF2A-SUMO and control granule neurons. Three replicates were run per sample category, for a total of 6 arrays. RNA quantity was assessed with Nanodrop (Nanodrop Technologies) and the Agilent Bioanalyzer (Agilent Technologies). Total RNA (200 ng) was amplified, biotinylated, and hybridized on Illumina RatRef-12 Expression BeadChip arrays (Illumina), querying the expression of ∼22,000 RefSeq-curated rat transcripts, as per manufacturer's protocol. Slides were scanned using Illumina BeadStation and signal extracted using Illumina BeadStudio software (Illumina). Raw data were analyzed using Bioconductor packages (www.bioconductor.org) as previously described (Konopka et al., 2009). Briefly, quality assessment was performed by examining the interarray Pearson correlation, and clustering based on top variant genes was used to assess overall data coherence. Contrast analysis of differential expression was performed using the LIMMA package (Smyth, 2005). After linear model fitting, a Bayesian estimate of differential expression was calculated. The threshold for statistical significance was set at p < 0.005.
Electrophoretic mobility shift assay.
Double-stranded oligonucleotide probes containing a MEF2 consensus sequence (Gossett et al., 1989) or a sequence in the Syt1 promoter encompassing a putative MEF2 binding element were generated, the latter with the following pair of primers: (forward): 5′-CCT GAG AGG ATC CAC CTA TTA TTA GTG CTC T-3′; (reverse): 5′-CAG AGA GCA CTA ATA ATA GGT GGA TCC TCT C-3′. After primer annealment, the double-stranded oligonucleotides were labeled with [α-32P]-dGTP (PerkinElmer) using Klenow. In each reaction, 5 μg of lysates of 293T cells transfected with the MEF2A or MEF2A-SUMO expression plasmid or with the control plasmid and 0.4 pmol of probe were mixed in 10 μl of binding buffer (20 mm HEPES, 70 mm NaCl, 10% glycerol, 0.1% Triton-X, 0.5 mm DTT, 1 mm EDTA) and 1 μg of poly dI-dC for 20 min at 25°C. After the reaction, 200 ng of MEF2A antibody (H-300) or rabbit IgG was added and incubated for 20 min at 25°C. The reaction mixture was separated by native 4% polyacrylamide gels in 0.5×Tris borate-EDTA buffer at 4°C and analyzed by Typhoon PhosphorImager.
Chromatin immunoprecipitation.
ChIP assays were performed with rat cerebella fixed with 4% PFA solution and stored at −80°C before use. Brains were chemically cross-linked with 1.1% formaldehyde solution for 15 min at room temperature and homogenized with lysis buffer (10 mm Tris-HCl, 0.25% Triton X-100, 10 mm EDTA and 0.5 mm EGTA). After spin down, the supernatant was removed and the pellet was resuspended using sonication buffer (10 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA and 0.5 mm EGTA) and sonicated to shear crosslinked DNA. Immunoprecipitation followed by qPCR were performed as described previously (Yuan et al., 2008). Primers used for amplifications were as follows: Rat Syt1 promoter (forward): 5′-GTT GCC TGA GAG GAT CCA CCT AT-3′; Rat Syt1 promoter (reverse): 5′-CTC AAG TTG CCC ACT GCT AAC TG-3′; Rat GAPDH promoter (forward): 5′-GTG CTG TGT CAC TAC CGA AGA AC-3′; Rat GAPDH promoter (reverse): 5′-CCA GGA TAG GAC TCA GGG AAT AC-3′.
In vivo electroporation.
In vivo electroporation of postnatal rat pups was performed as described previously (Konishi et al., 2004; Shalizi et al., 2006; Yang et al., 2009). The indicated plasmids were injected into the cerebellum of P4 Sprague Dawley rat pups, and were then subjected to five electric pulses of 160 mV with 950 ms intervals. Electroporated pups were returned to moms and examined 5 d later with immunohistochemistry analyses using the relevant antibodies. The density of orphan or PSD95-apposed synapsin clusters was analyzed in randomized 100–200 μm segments along the parallel fibers. Images of transfected neurons were taken in a blinded manner on an Olympus Fluoview FV1000 confocal microscope and analyzed using the FV10-ASW and SPOT imaging softwares.
Statistics.
Statistical analyses were done using Statview 5.0.1 software. Bar graphs are presented as the mean ±SEM. For experiments in which only two groups were analyzed, the t test was used. Pairwise comparisons within multiple groups were done by ANOVA followed by the Fischer's PLSD post hoc test.
Results
MEF2A suppresses orphan presynaptic sites in granule neurons
To investigate the mechanisms that govern presynaptic development, we used granule neurons of the rat cerebellar cortex. Granule neurons offer a robust system for elucidation of mechanisms of neuronal morphogenesis and connectivity, including synapse differentiation (Palay and Chan-Palay, 1974; Altman and Bayer, 1997; Konishi et al., 2004; Shalizi et al., 2006; Kim et al., 2009; Yang et al., 2009). To visualize presynaptic sites, we expressed the synaptic vesicle protein synapsin fused to green fluorescent protein (GFP-synapsin) in primary granule neurons (Fig. 1A). GFP-synapsin clustered preferentially along the distal portion of granule neuron axons and colocalized with the endogenous synaptic vesicle proteins VAMP2 and synaptophysin and the active zone proteins Bassoon and Munc13 in primary neurons (Fig. 1A–C) (Yang et al., 2009).
Figure 1.

The assembly of orphan presynaptic sites is suppressed in neurons with maturation. A, Rat cerebellar granule neurons were transfected with the GFP-synapsin expression plasmid and subjected to immunocytochemistry using the GFP antibody. A representative image is shown. Arrowheads indicate synapsin clusters. B–C, Granule neurons transfected with the GFP-synapsin expression plasmid were subjected to immunocytochemistry using the GFP antibody and VAMP2 or synaptophysin antibody (B), Bassoon or Munc13 antibody (C). Arrowheads denote synapsin coclusters with synaptic vesicle proteins (VAMP2 or synaptophysin) or active zone proteins (Bassoon or Munc13). D, Granule neurons were transfected with GFP-synapsin and subjected to immunocytochemistry using the GFP, PSD95, and MAP2 antibodies. PSD95 puncta colocalized with the MAP2-positive dendrites. Arrowheads denote synapsin clusters that are unapposed to PSD95. Double arrowheads denote synapsin clusters that colocalize with PSD95. E, Granule neurons cultured for 2, 4, 6, 8, and 12 d in vitro were transfected with the GFP-synapsin expression plasmid and subjected to immunocytochemistry using the GFP and PSD95 antibodies. Quantification of the densities of PSD95-apposed and PSD95-unapposed synapsin clusters is shown. While the density of PSD95-apposed synapsin clusters continued to increase with neuron maturation, the density of orphan PSD95-unapposed clusters failed to increase after DIV4. Scale bars: 10 μm.
To characterize orphan presynaptic sites, we determined the proportion of synapsin clusters that were unapposed with postsynaptic dendritic sites. We found that 50% of synapsin clusters in granule neurons at DIV8 were apposed to the postsynaptic density protein PSD95, and remarkably the remaining 50% of synapsin clusters were unapposed to PSD95 (Fig. 1D,E). The PSD95-apposed and orphan PSD95-unapposed synapsin clusters appeared morphologically similar. However, whereas the PSD95-apposed synapsin clusters were found in axons at sites in close contact with dendrites, the orphan PSD95-unapposed synapsin clusters were found at sites away from dendrites (Fig. 1D).
Interestingly, orphan PSD95-unapposed synapsin clusters appeared within the first four days in primary granule neurons (Fig. 1E). However, in contrast to PSD95-apposed synapsin clusters, which increased in number with neuron maturation, the density of orphan synapsin clusters failed to increase after DIV4 (Fig. 1E), suggesting that a mechanism that suppresses orphan sites is triggered with neuron maturation.
The characterization of orphan PSD95-unapposed synapsin clusters led us to the question of how these sites are regulated. We interrogated the role of the neuron-enriched transcription factor MEF2A in the control of presynaptic differentiation. We used a plasmid-based method of RNA interference (RNAi) to induce the knockdown of MEF2A (Gaudilliere et al., 2002). Expression of MEF2A short hairpin RNAs (shRNAs) using the U6/mef2a RNAi plasmid induced the robust knockdown of endogenous MEF2A in granule neurons (Fig. 2A). We next transfected granule neurons with the U6/mef2a or control U6 RNAi plasmid together with the GFP-synapsin expression plasmid. Strikingly, MEF2A knockdown robustly increased the density of PSD95-unapposed synapsin clusters in granule neurons (Fig. 2B). In contrast, MEF2A knockdown failed to increase the density of synapsin/PSD95 coclusters in neurons (Fig. 2B). These results suggest that MEF2A selectively suppresses the assembly of orphan synapsin clusters.
Figure 2.
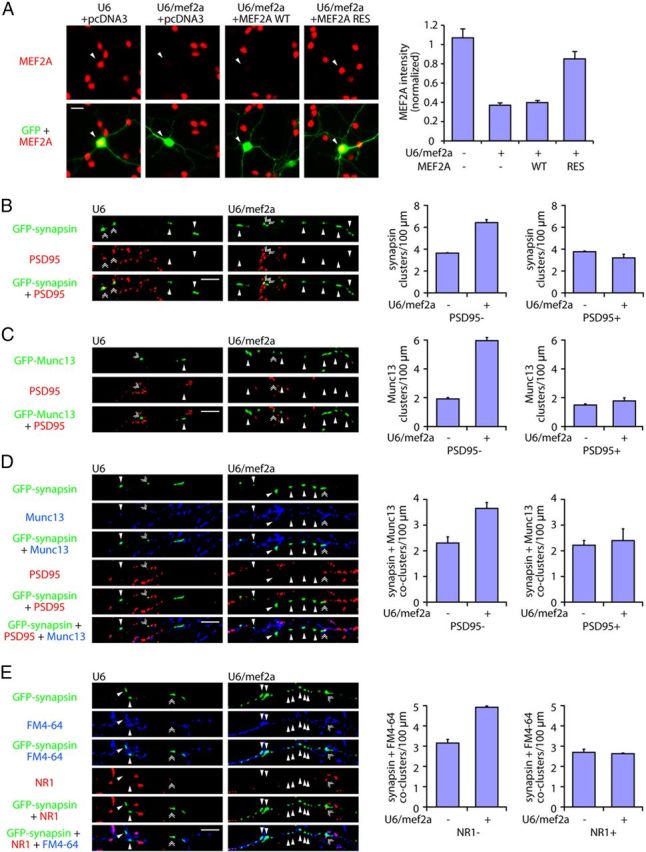
MEF2A suppresses orphan presynaptic sites in granule neurons. A, Left, Granule neurons were transfected with the GFP expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and an expression plasmid encoding MEF2A wild-type cDNA (MEF2A-WT), MEF2A encoded by an RNAi-resistant cDNA (MEF2A-RES), or the control vector and subjected to immunocytochemistry using the GFP and MEF2A antibodies. Arrowheads denote transfected cells. Right, MEFA immunoreactivity was reduced by knockdown of MEF2A (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3). Expression of MEF2A-RES, but not MEF2A-WT, restored MEF2A immunoreactivity in the background of MEF2A RNAi (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3). B, Left, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and subjected to immunocytochemistry using the GFP and PSD95 antibodies. Arrowheads denote orphan PSD95-unapposed synapsin clusters. Double arrowheads indicate synapsin/PSD95 coclusters. Right, Knockdown of MEF2A (U6/mef2a) increased the density of orphan PSD95-unapposed synapsin clusters in granule neurons (p < 0.001, t test, n = 3), but did not increase the density of synapsin/PSD95 coclusters. C, Left, Granule neurons were transfected with the GFP-Munc13 expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and analyzed as in B. Arrowheads denote orphan PSD95-unapposed Munc13 clusters. Double arrowheads indicate Munc13/PSD95 coclusters. Right, MEF2A knockdown increased the density of orphan PSD95-unapposed Munc13 clusters in granule neurons (p < 0.001, t test, n = 3), but failed to increase the density of Munc13/PSD95 coclusters. D, Left, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and subjected to immunocytochemistry using the GFP, Munc13, and PSD95 antibodies. Arrowheads denote orphan PSD95-unapposed synapsin/Munc13 coclusters. Double arrowheads indicate synapsin/Munc13/PSD95 coclusters. Right, MEF2A depletion increased the density of orphan PSD95-unapposed synapsin/Munc13 coclusters in granule neurons (p < 0.05, t test, n = 3), but did not increase the density of synapsin/Munc13/PSD95 coclusters. E, Left, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/mef2a or control U6 RNAi plasmid, depolarized to allow for uptake of the dye FM4-64, and subjected to immunocytochemistry using the NR1 antibody. Arrowheads denote orphan NR1-unapposed synapsin/FM4-64 coclusters. Double arrowheads indicate synapsin/FM4-64/NR1 coclusters. Right, Knockdown of MEF2A increased the density of orphan NR1-unapposed synapsin/FM4-64 coclusters (p < 0.001, t test, n = 3), but had little or no effect on the density of synapsin/FM4-64/NR1 coclusters. Scale bars: 10 μm.
We next asked whether MEF2A regulates other aspects of orphan presynaptic sites. In addition to the clustering of synaptic vesicle proteins, active zones proteins assemble at orphan presynaptic sites (Krueger et al., 2003). To assess the role of MEF2A in the differentiation of active zone specializations, we transfected granule neurons with the U6/mef2a or control U6 RNAi plasmid together with an expression plasmid encoding the GFP-Munc13 fusion protein, the latter to visualize clustering of the active zone protein Munc13. Remarkably, MEF2A knockdown dramatically increased the density of Munc13 clusters at sites unapposed to PSD95, but failed to increase the density of PSD95-apposed Munc13 clusters in neurons (Fig. 2C). In other experiments, MEF2A knockdown increased the density of orphan PSD95-unapposed synapsin clusters that colocalized with clusters of endogenous Munc13 in granule neurons (Fig. 2D). Together, these results suggest that MEF2A suppresses the clustering of synaptic vesicle and active zone proteins at sites unapposed to postsynaptic structures, and thereby inhibits the assembly of orphan presynaptic sites in neurons.
To determine whether MEF2A knockdown induces the formation of orphan presynaptic sites that are functionally active, we measured the ability of granule neuron axons to undergo synaptic vesicle recycling at sites of orphan synapsin clusters. We used an assay of synaptic vesicle recycling, whereby we monitored the incorporation of the dye FM4-64 in granule neurons in response to membrane depolarization. To distinguish between synaptic sites and orphan synapsin sites, granule neurons were analyzed using an antibody to an extracellular loop of the NMDA receptor subunit NR1, a marker of postsynaptic sites. We found that orphan synapsin clusters unapposed to NR1 colocalized with sites of FM4-64 uptake (Fig. 2E). Importantly, MEF2A knockdown increased the density of orphan NR1-unapposed synapsin/FM4-64 coclusters, but had little or no effect on the density of NR1-apposed synapsin/FM4-64 coclusters in neurons (Fig. 2E). Collectively, our results suggest that MEF2A specifically suppresses the assembly of functional orphan presynaptic sites in granule neurons.
Sumoylation of MEF2A suppresses orphan presynaptic sites
We next asked how MEF2A suppresses the assembly of orphan presynaptic sites. Characterization of the temporal profile of MEF2A expression in the rat cerebellum in vivo revealed that sumoylation of MEF2A increased with maturation (Fig. 3A) (Shalizi et al., 2006), suggesting that the sumoylated form of MEF2A might play a role in the suppression of orphan presynaptic sites.
Figure 3.

Sumoylation of MEF2A drives the suppression of orphan presynaptic sites. A, Lysates of cerebella from rat pups at postnatal days 2, 6, 10, 14, 18, and 22 were immunoblotted with the MEF2A or ERK1/2 antibody. MEF2A runs as a doublet at 55 kDa, and the sumoylated form of MEF2A runs as an ∼100 kDa band (Shalizi et al., 2006). The levels of the sumoylated MEF2A increased during the second and third postnatal weeks in the rat cerebellum. B, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and an expression plasmid encoding MEF2A-WT, MEF2A-RES, MEF2A-RES K403R, or a control vector and analyzed as in Figure 2B. MEF2A knockdown increased the density of orphan PSD95-unapposed synapsin clusters (p < 0.01, ANOVA followed by Fisher's PLSD post hoc test, n = 3). Expression of MEF2A-RES, but neither MEF2A-WT nor MEF2A-RES K403R, decreased orphan synapsin cluster density in the background of MEF2A RNAi (p < 0.01, ANOVA followed by Fisher's PLSD post hoc test, n = 3). C, Granule neurons were transfected with the GFP-Munc13 expression plasmid together with the U6/mef2a or control U6 RNAi plasmid and an expression plasmid encoding MEF2A-WT, MEF2A-RES, MEF2A-RES K403R, or a control vector and analyzed as in Figure 2B. MEF2A knockdown increased the density of orphan Munc13 clusters (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3). Expression of MEF2A-RES, but neither MEF2A-WT nor MEF2A-RES K403R, reduced orphan Munc13 cluster density in the background of MEF2A RNAi (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3). D, Granule neurons were transfected with the GFP-synapsin expression plasmid together with an expression plasmid encoding MEF2A-SUMO or the control vector and analyzed as in Figure 2B. Arrowheads denote orphan PSD95-unapposed synapsin clusters. Double arrowheads indicate synapsin/PSD95 coclusters. Expression of MEF2A-SUMO in neurons decreased the density of orphan PSD95-unapposed synapsin clusters (p < 0.05, t test, n = 3), but had little or no effect on the density of synapsin/PSD95 coclusters. Scale bar, 10 μm. E, Granule neurons were transfected with the GFP-Munc13 expression plasmid together with an expression plasmid encoding MEF2A-SUMO or a control vector and analyzed as in Figure 2B. Expression of MEF2A-SUMO decreased the density of orphan PSD95-unapposed Munc13 clusters (p < 0.05, t test, n = 3), but failed to significantly reduce the density of Munc13/PSD95 coclusters. F, Granule neurons transfected with the GFP-synapsin expression plasmid together with the MEF2A-SUMO expression plasmid or the control vector were analyzed as in Figure 2D. Expression of MEF2A-SUMO decreased the density of orphan PSD95-unapposed synapsin/Munc13 coclusters in granule neurons (p < 0.05, t test, n = 3), but had little or no effect on the density of synapsin/Munc13/PSD95 coclusters. G, Granule neurons transfected with the GFP-synapsin expression plasmid together with an expression plasmid encoding MEF2A-SUMO or the control vector were analyzed for synaptic vesicle recycling as in Figure 2E. Expression of MEF2A-SUMO decreased the density of orphan NR1-unapposed synapsin/FM4-64 coclusters (p < 0.05, t test, n = 3), but failed to significantly reduce the density of synapsin/FM4-64/NR1 coclusters. H, Granule neurons were transfected with the GFP-synapsin expression plasmid together with an expression plasmid encoding MEF2A-WT, MEF2A-K403R, or the control vector and analyzed as in Figure 2B. Expression of MEF2A-WT or MEF2A-K403R in neurons had little or no effect on the density of orphan PSD95-unapposed and PSD95-apposed synapsin clusters.
To determine the role of sumoylation in MEF2A-suppression of orphan presynaptic sites, we performed structure—function analyses of MEF2A in the background of MEF2A knockdown in primary neurons. In control analyses, we found that transfection of granule neurons with an expression plasmid encoding a rescue form of MEF2A (MEF2A-RES) using an RNAi-resistant cDNA together with the U6/mef2a RNAi plasmid restored MEF2A expression to similar levels of endogenous MEF2A in control-transfected neurons (Fig. 2A). In contrast, transfection of an expression plasmid encoding MEF2A using wild-type cDNA failed to restore MEF2A levels in neurons transfected with the U6/mef2a RNAi plasmid (Fig. 2A). We next assessed the ability of MEF2A-RES to inhibit the assembly of orphan presynaptic sites in MEF2A knockdown neurons. We found that MEF2A-RES, but not MEF2A-WT, completely reversed the MEF2A RNAi-induced increase in orphan PSD95-unapposed clusters of synapsin and Munc13 (Fig. 3B,C). These results indicate that MEF2A RNAi-induced assembly of orphan presynaptic sites is the result of specific knockdown of MEF2A in neurons. Importantly, the MEF2A rescue experiment allowed the assessment of the role of sumoylation in MEF2A-suppression of orphan presynaptic sites in structure—function analyses. MEF2A contains a conserved SUMO acceptor motif at Lysine 403, whose replacement with arginine prevents the sumoylation of MEF2A (Hietakangas et al., 2006; Shalizi et al., 2006). We found that in contrast to MEF2A-RES, the sumoylation-deficient K403R mutant of MEF2A-RES (MEF2A-RES K403R) failed to reduce the density of orphan PSD95-unapposed clusters of synapsin and Munc13 in granule neurons in the background of MEF2A knockdown (Fig. 3B,C). These data suggest that sumoylation of MEF2A at Lysine 403 is required for the suppression of orphan presynaptic sites.
We also assessed whether sumoylation of MEF2A is sufficient to drive the suppression of orphan presynaptic sites. The fusion of SUMO to a transcription factor mimics the effect of SUMO that is conjugated to the transcription factor on the native lysine (Ross et al., 2002; Shalizi et al., 2006). Expression of a MEF2A-SUMO fusion protein in granule neurons robustly reduced the density of orphan clusters of synapsin or Munc13, orphan synapsin/Munc13 coclusters, and orphan synapsin/FM4-64 coclusters (Fig. 3D–G). In contrast, MEF2A-SUMO expression failed to effectively reduce the density of postsynaptically apposed presynaptic sites in neurons (Fig. 3D–G). Expression of wild-type MEF2A or MEF2A-K403R had little or no effect on the density of orphan synapsin clusters (Fig. 3H).Together, these results support the conclusion that sumoylation of MEF2A plays a key role in the suppression of orphan presynaptic sites in neurons.
Identification of Syt1 as a direct repressed target gene of sumoylated MEF2A in the suppression of orphan presynaptic sites
Having identified a novel function for sumoylated MEF2A in the elimination of orphan presynaptic sites, we next determined the mechanism by which sumoylated MEF2A suppresses orphan presynaptic sites. Because sumoylated MEF2A represses transcription, we performed gene profiling analyses in granule neurons to identify repressed target genes of sumoylated MEF2A. We isolated total RNA from granule neurons expressing MEF2A-SUMO and control granule neurons and analyzed gene expression changes using the Illumina microarray platform. In control immunoblotting analyses, we confirmed that MEF2A-SUMO was expressed in granule neurons (Fig. 4A). We identified 24 genes that were reproducibly downregulated upon MEF2A-SUMO expression (Fig. 4B). Among the MEF2A-SUMO-downregulated genes, several genes encoded proteins enriched at presynaptic sites and along axons including Syt1, Kif1b, and Robo2 (Simpson et al., 2000; Zhao et al., 2001; Tang et al., 2006).
Figure 4.
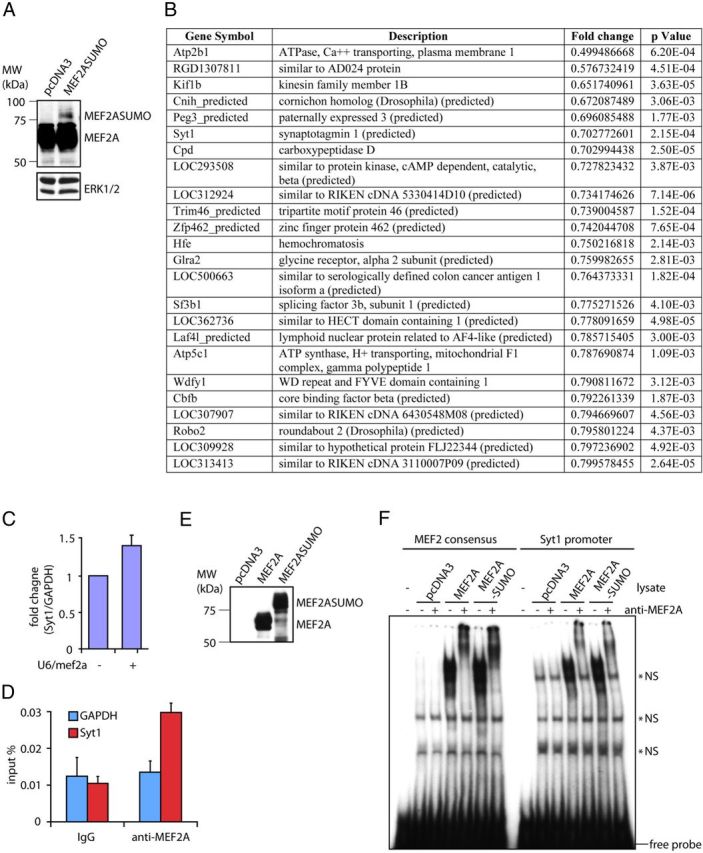
Identification of Syt1 as a direct repressed target gene of MEF2A. A, Granule neurons transfected with an expression plasmid encoding MEF2A-SUMO or the control vector were immunoblotted with the MEF2A or ERK1/2 antibody. B, Microarray analysis using granule neurons transfected with the MEF2A-SUMO expression plasmid or the control vector. Top downregulated genes (p < 0.005) in the MEF2A-SUMO versus control conditions are shown. C, Granule neurons transfected with the U6/mef2a or control U6 RNAi plasmid were subjected to qRT-PCR analyses using primers specific to Syt1 or GAPDH. The levels of Syt1 mRNA normalized to GAPDH are shown relative to the control condition. Knockdown of MEF2A increased Syt1 expression in granule neurons (p < 0.05, t test, n = 3). D, Lysates of rat cerebella were subjected to chromatin immunoprecipitation with the MEF2A or control IgG antibody followed by quantitative PCR analyses using primers specific to the Syt1 or GAPDH promoter. MEF2A was enriched on the Syt1 gene within a promoter region that encompasses a conserved MEF2A binding sequence located 0.3 kbp upstream of transcription start site (p < 0.01, ANOVA followed by Fisher's PLSD post hoc test, n = 4). E, Lysates of 293T cells transfected with an expression plasmid encoding MEF2A, MEF2A-SUMO, or the control vector were immunoblotted with the MEF2A antibody. F, Electrophoretic mobility shift assay (EMSAs) were performed using lysates of 293T cells transfected as in E and incubated with oligonucleotide probes containing the consensus MEF2 binding sequence or a Syt1 promoter sequence encompassing a conserved MEF2A binding element. MEF2A-SUMO and MEF2A bound equally effectively the consensus MEF2 binding sequence and the MEF2A binding element in the Syt1 promoter. Nonspecific signals are denoted (*NS).
Among the candidate MEF2A-SUMO-downregulated genes, the expression of Syt1 was increased upon knockdown of MEF2A (Fig. 4C), suggesting that Syt1 is repressed by endogenous sumoylated MEF2A. We asked whether sumoylated MEF2A directly binds to the promoter of the Syt1 gene. A conserved MEF2-binding sequence was found 0.3 kbp upstream of the transcription start site of the Syt1 gene. Importantly, ChIP analyses using cerebellar lysates revealed that endogenous MEF2A specifically occupied a region of the endogenous Syt1 promoter (Fig. 4D). In electrophoretic mobility shift assays, both MEF2A-SUMO and MEF2A bound to double-stranded oligonucleotide probes containing a canonical MEF2-binding sequence or a conserved MEF2A binding sequence in the Syt1 promoter (Fig. 4E,F), suggesting that sumoylation does not affect MEF2A binding to the Syt1 promoter. Collectively, these results suggest that sumoylated MEF2A directly represses Syt1 transcription in neurons.
We next assessed the role of Syt1 in the control of orphan presynaptic sites. Knockdown of endogenous Syt1 robustly decreased the density of functional orphan presynaptic sites in granule neurons, but had little or no effect on the density of postsynaptically apposed presynaptic sites (Fig. 5A–G). Expression of Syt1 encoded by an RNAi-resistant cDNA, but not by wild-type cDNA, reversed the Syt1 RNAi-induced loss of orphan presynaptic sites (Fig. 5H,I), indicating that the Syt1 RNAi-induced loss of orphan presynaptic sites is the result of specific knockdown of Syt1. In other analyses, we found that expression of Syt1 in granule neurons increased the density of orphan presynaptic sites (Fig. 5J), phenocopying the effect of MEF2A knockdown on presynaptic differentiation. Finally, knockdown of Syt1 suppressed the ability of MEF2A knockdown to increase the density of orphan presynaptic sites in primary granule neurons (Fig. 5K). Together, these findings suggest that sumoylated MEF2A suppresses orphan presynaptic sites via direct repression of the presynaptic gene Syt1.
Figure 5.

Identification of Syt1 as a direct repressed target gene of MEF2A in the suppression of orphan presynaptic sites. A, Granule neurons transfected with the U6/syt1 or control U6 RNAi plasmid were subjected to quantitative RT-PCR analysis using primers specific to Syt1 or GAPDH. The levels of Syt1 mRNA normalized to GAPDH are shown relative to the control condition. Syt1 RNAi induced the knockdown of endogenous Syt1 expression in neurons (p < 0.001, t test, n = 3). B, Granule neurons transfected with the U6/syt1 or control U6 RNAi plasmid were subjected to immunoblotting with the Syt1 or ERK antibody, the latter to serve as a loading control. C, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/syt1 or control U6 RNAi plasmid and analyzed as in Figure 2B. Arrowheads denote orphan PSD95-unapposed synapsin clusters. Double arrowheads indicate synapsin/PSD95 coclusters. Scale bar, 10 μm. D, Knockdown of Syt1 decreased the density of orphan synapsin clusters in granule neurons (p < 0.001, t test, n = 3), but had little or no effect on the density of synapsin/PSD95 coclusters. E, Granule neurons were transfected with the GFP-Munc13 expression plasmid together with the U6/syt1 or control U6 RNAi plasmid and analyzed as in Figure 2B. Knockdown of Syt1 decreased the density of orphan PSD95-unapposed Munc13 clusters in granule neurons (p < 0.05, t test, n = 3), but failed to reduce the density of Munc13/PSD95 coclusters. F, Granule neurons transfected with the GFP-synapsin expression plasmid together with the U6/syt1 or control U6 RNAi plasmid were analyzed as in Figure 2D. Arrowheads denote orphan PSD95-unapposed synapsin/Munc13 coclusters. Double arrowheads indicate synapsin/Munc13/PSD95 coclusters. Knockdown of Syt1 decreased the density of orphan PSD95-unapposed synapsin/Munc13 coclusters in granule neurons (p < 0.05, t test, n = 3), but had little or no effect on the density of synapsin/Munc13/PSD95 coclusters. G, Granule neurons transfected with the GFP-synapsin expression plasmid together with the U6/syt1 or control U6 RNAi were analyzed for synaptic vesicle recycling as in Figure 2E. Arrowheads denote orphan NR1-unapposed synapsin/FM4-64 coclusters. Double arrowheads indicate synapsin/FM4-64/NR1 coclusters. Knockdown of Syt1 decreased the density of orphan NR1-unapposed synapsin/FM4-64 coclusters (p < 0.05, t test, n = 3), but failed to reduce the density of synapsin/FM4-64/NR1 coclusters. H, Lysates of 293T cells transfected with an expression plasmid encoding Syt1 (WT) or an RNAi-resistant Syt1 containing silent mutations (RES) together with the U6/syt1 or control U6 RNAi plasmid were immunoblotted with the Syt1 or ERK antibody, the latter to serve as a loading control. I, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/syt1 or control U6 RNAi plasmid and an expression plasmid encoding Syt1-WT, Syt1-RES, or the control vector and analyzed as in Figure 2B. Syt1 knockdown decreased the density of orphan PSD95-unapposed synapsin clusters (p < 0.05, ANOVA followed by Fisher's PLSD post hoc test, n = 3), while expression of Syt1-RES but not Syt1-WT increased orphan synapsin cluster density in the background of Syt1 RNAi (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3). J, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the Syt1 or the control vector and analyzed as in Figure 2B. Expression of Syt1 increased the density of orphan PSD95-unapposed synapsin clusters (p < 0.05, ANOVA followed by Fisher's PLSD post hoc test, n = 3). K, Granule neurons were transfected with the GFP-synapsin expression plasmid together with the U6/mef2a, U6/syt1, or control U6 RNAi plasmid and analyzed as in Figure 2B. Knockdown of MEF2A increased the density of orphan synapsin clusters (p < 0.001, ANOVA followed by Fisher's PLSD post hoc test, n = 3) but not in the presence of Syt1 knockdown.
The MEF2A/Syt1 pathway suppresses orphan presynaptic sites in the cerebellar cortex in vivo
The identification of a function for the sumoylated MEF2A/Syt1 link in the control of orphan presynaptic sites in primary neurons raised the fundamental question of the role of this pathway in the developing organism in vivo. To address this question, we first characterized orphan presynaptic sites in the rat cerebellar cortex in vivo. Using an electroporation method, we delivered the GFP-synapsin expression plasmid into the cerebellum of P4 rat pups (Fig. 6A) (Konishi et al., 2004; Shalizi et al., 2006; Yang et al., 2009). Pups were returned to moms, and after 5 d animals were killed at P9. Coronal sections of the cerebellar cortex were subjected to immunohistochemistry using the GFP antibody to visualize GFP-synapsin and the calbindin antibody to visualize Purkinje neurons (Fig. 6B). Purkinje neuron somas spanned the Purkinje cell layer (PCL), and their dendrites extended into the molecular layer (ML) (Fig. 6B). Clusters of synapsin were found along granule neuron parallel fiber axons traversing the molecular layer (Fig. 6B).
Figure 6.
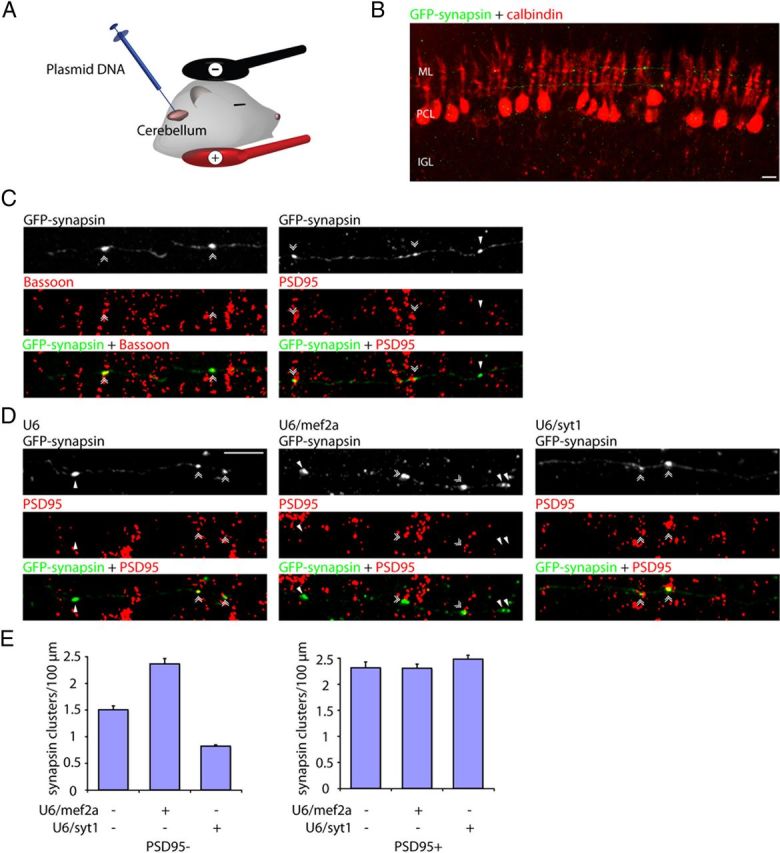
The MEF2A/Syt1 pathway regulates the assembly of orphan presynaptic sites in the rodent brain in vivo. A, B, P4 rat pups were electroporated with the pCAG-GFP-synapsin expression plasmid. Pups were killed 5 d later at P9, and cerebellar sections were subjected to immunohistochemistry using the GFP antibody and the calbindin antibody, the later to visualize Purkinje neurons. Purkinje neuron somas reside in the Purkinje cell layer (PCL) and their dendrites extend into the molecular layer (ML). GFP-synapsin is enriched in clusters along granule neuron parallel fiber axons traversing the molecular layer. C, Cerebellar sections prepared as in A were immunolabeled with the GFP antibody together with the Bassoon or PSD95 antibody. Synapsin clusters colocalized with the active zone protein Bassoon in granule neuron axons (double arrowheads, left), indicating that synapsin clusters represent sites of presynaptic axonal differentiation. Synapsin clusters were found at sites apposed to PSD95 (double arrowheads, right) and at sites unapposed to PSD95 (arrowheads, right), representing respectively synaptic sites and orphan presynaptic sites. D, P4 rat pups were electroporated with the U6/mef2a or control U6 RNAi plasmid together with the pCAG-GFP-synapsin expression plasmid and cerebellar sections prepared as in A were subjected to immunohistochemistry using the GFP antibody and the PSD95 antibody. Arrowheads denote orphan PSD95-unapposed synapsin clusters. Double arrowheads indicate synapsin/PSD95 coclusters. E, Knockdown of MEF2A increased the density of orphan PSD95-unapposed synapsin clusters (p < 0.005 for U6/mef2a, ANOVA followed by Fisher's PLSD post hoc test, n = 3), but did not increase the density of synapsin/PSD95 coclusters. Knockdown of Syt1 decreased the density of orphan PSD95-unapposed synapsin clusters (p < 0.005, ANOVA followed by Fisher's PLSD post hoc test, n = 3), but had little or no effect on the density of synapsin/PSD95 coclusters. Scale bars: 10 μm.
To characterize the parallel fiber synapsin clusters observed in vivo, the cerebellar cortex was subjected to additional immunohistochemical analyses using the GFP antibody together with the Bassoon or PSD95 antibody. PSD95 is expressed postsynaptically in cerebellar granule neuron parallel fiber target neurons, including stellate, basket, and Purkinje cells (Fukaya and Watanabe, 2000; Yamasaki et al., 2011). We found that parallel fiber synapsin clusters colocalized with the active zone protein Bassoon (Fig. 6C), suggesting that synapsin clusters in the cerebellar cortex represent presynaptic sites composed of both synaptic vesicles and active zone proteins. Notably, we observed synapsin clusters both at sites apposed to the postsynaptic protein PSD95 as well as at sites that were unapposed by PSD95 (Fig. 6C). These findings reveal that just as in primary neurons, presynaptic sites in vivo consist of at least two distinct populations, a population that is apposed by postsynaptic elements and a second population that represents orphan presynaptic sites.
We next asked whether the MEF2A/Syt1 pathway regulates orphan presynaptic sites in the cerebellar cortex in vivo. Using the in vivo electroporation approach, we injected the U6/mef2a, U6/syt1, or control U6 RNAi plasmid together with the GFP-synapsin expression plasmid into the cerebellum of P4 rat pups (Fig. 6D,E). Analyses of pups at P9 revealed that knockdown of MEF2A increased the density of orphan presynaptic sites (Fig. 6D,E). In contrast, knockdown of the MEF2A-repressed target gene Syt1 decreased the density of orphan presynaptic sites in vivo (Fig. 6D,E). Knockdown of MEF2A or Syt1 had little or no effect on the density of postsynaptically apposed presynaptic sites (Fig. 6D,E). Together, these results suggest that the MEF2A/Syt1 pathway specifically suppresses the assembly of orphan presynaptic sites in the developing brain in vivo.
MEF2A coordinates the elimination of orphan synapsin clusters with the maturation of presynaptic boutons
The identification of a function for the MEF2A/Syt1 pathway in the suppression of orphan presynaptic sites in primary neurons and in vivo raised the major question of the biological consequences of orphan presynaptic site elimination. In most neurons in the CNS including granule neurons, axonal presynaptic sites mature by forming varicose, bouton-shaped morphological structures that house synaptic vesicles, active zone proteins, and mitochondria (Ziv and Garner, 2004; McAllister, 2007). We asked whether the elimination of orphan presynaptic sites influences the differentiation of mature presynaptic sites. First, to visualize mature presynaptic boutons, we expressed GFP in granule neurons and subjected neurons to immunocytochemistry using the GFP antibody at a time when they have developed further (DIV12–13). Granule neuron axons displayed varicosities distributed along the axon (Fig. 7A), which harbored the synaptic vesicle proteins synapsin and synaptophysin, the active zone protein Munc13, and mitochondria (Fig. 7B–D). In addition, the varicosities colocalized with sites of FM4-64 uptake and were apposed to the postsynaptic protein PSD95 (Fig. 7E,F). Together, these results suggest that granule neuron axon varicosities represent mature functional presynaptic boutons.
Figure 7.

Characterization of maturing presynaptic boutons in primary neurons. A, Granule neurons were transfected with the GFP expression plasmid and subjected to immunocytochemistry using the GFP antibody at DIV12–13. A representative image is shown. Arrowheads indicate presynaptic varicosities along the granule neuron distal axon. B, C, Granule neurons transfected with the GFP plasmid were subjected to immunocytochemistry using the GFP antibody and the synapsin, synaptophysin, or Munc13 antibody. Arrowheads denote presynaptic varicosities colocalized with the endogenous synaptic vesicle protein synapsin or synaptophysin (B), or the active zone protein Munc13 (C). D, Granule neurons transfected with the mCherry expression plasmid were incubated with 100 nm of the Mitotracker Green dye for 30 min before imaging. Arrowheads denote varicosities colocalized with mitochondria as indicated by Mitotracker-positive puncta. E, Granule neurons transfected with the GFP expression plasmid were analyzed for synaptic vesicle recycling as in Figure 2E. Arrowheads denote coclusters of presynaptic varicosities with sites of FM4-64 dye uptake. F, Granule neurons transfected with the GFP plasmid were subjected to immunocytochemistry using the GFP antibody and the PSD95 antibody. Double arrowheads indicate presynaptic varicosities apposed to PSD95. Scale bars: 10 μm.
We next determined the relationship between orphan presynaptic sites and mature presynaptic boutons using environment-controlled live confocal imaging analyses. We expressed in granule neurons GFP-synapsin together with the fluorescent protein mCherry, the latter to visualize presynaptic varicosities. Neurons were continuously imaged for 2 h and subjected to post hoc immunocytochemical analyses using the GFP, mCherry, and PSD95 antibodies. We found that orphan synapsin clusters were eliminated at a high rate in the vicinity of presynaptic varicosities (Fig. 8A). Quantification of live-imaging events revealed that the loss of orphan synapsin clusters near presynaptic varicosities was substantially higher than that in segments randomly selected along the axon (Fig. 8B). In contrast, the loss of PSD95-apposed synapsin clusters was low near presynaptic varicosities and not different from that in randomly selected axon segments (Fig. 8B).
Figure 8.
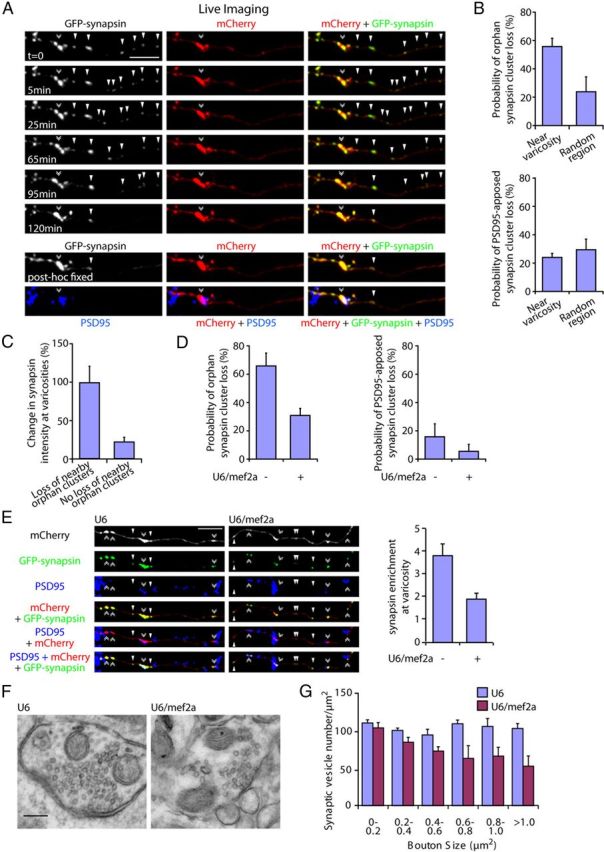
MEF2A coordinately regulates the elimination of orphan synapsin clusters and enrichment of presynaptic material at maturing presynaptic boutons. A, Granule neurons were transfected with the GFP-synapsin and mCherry expression plasmids. Live neurons were imaged using a spinning disk confocal microscope for 2 h and subjected to immunocytochemistry using the GFP, mCherry, and PSD95 antibodies. A representative image sequence is shown. Orphan synapsin clusters unapposed to PSD95 (arrowheads) are lost and coordinately synapsin enrichment is increased at the presynaptic varicosity (double arrowhead). B, Quantification of neurons transfected as in A revealed that the loss of orphan PSD95-unapposed synapsin cluster is significantly higher within a 25 μm region near presynaptic varicosities compared with that in random axon regions (top, p < 0.05, t test, n = 3). The loss of PSD95-apposed synapsin clusters near presynaptic varicosities is not significantly different compared with that in random axon regions (bottom). C, Quantification of the change in synapsin intensity at presynaptic varicosities during the imaging interval as in A. The loss of orphan synapsin clusters within a 25 μm region near presynaptic varicosities was associated with a significant increase in synapsin enrichment at presynaptic varicosities compared with that in presynaptic varicosities with no orphan synapsin cluster loss in their vicinity (p < 0.005, t test, n = 3). D, Granule neurons were transfected with the GFP-synapsin and mCherry expression plasmids together with the U6/mef2a, or control U6 RNAi plasmid and analyzed as in B. Knockdown of MEF2A reduced the loss of orphan PSD95-unapposed synapsin clusters (left, p < 0.01, ANOVA followed by Fisher's PLSD post hoc test, n = 3), but not synaptic PSD95-apposed synapsin clusters, near presynaptic varicosities (right). E, Granule neurons were transfected with the GFP-synapsin and mCherry expression plasmids together with the U6/mef2a or control U6 RNAi plasmid and subjected to immunocytochemistry using the GFP, mCherry, and PSD95 antibodies. Left, Representative images of knockdown and control neurons are shown. Right, Quantification of the intensity of synapsin at presynaptic varicosities normalized to the intensity of synapsin at orphan PSD95-unapposed presynaptic sites. Depletion of MEF2A reduced the enrichment of synapsin at presynaptic varicosities (p < 0.05, ANOVA followed by Fisher's PLSD post hoc test, n = 3). Scale bar, 10 μm. F, Representative presynaptic boutons from control or MEF2A knockdown granule neurons subjected to electron microscopy analyses. Presynaptic boutons contain synaptic vesicles and mitochondria, and are apposed to postsynaptic densities. G, Quantification of synaptic vesicle number in presynaptic boutons normalized to bouton size from control or MEF2A knockdown neurons as in F. Depletion of MEF2A significantly decreased synaptic vesicle number in larger boutons with an area of 0.6–0.8 μm2 (p < 0.01, ANOVA followed by Fisher's PLSD post hoc test, n = 3), 0.8–1.0 μm2 (p < 0.05, ANOVA followed by Fisher's PLSD post hoc test, n = 3), and >1.0 μm2 (p < 0.005, ANOVA followed by Fisher's PLSD post hoc test, n = 3), but not in boutons smaller than 0.6 μm2. Scale bar, 200 nm.
The elimination of orphan synapsin clusters in the vicinity of presynaptic varicosities led us to ask whether orphan clusters might be recruited to the large maturing presynaptic boutons in granule neurons. Therefore, we examined the change in synapsin intensity at the presynaptic varicosities during the 2 h of live imaging. Remarkably, the loss of neighboring orphan synapsin clusters was associated with a dramatic 99% increase in the intensity of synapsin at presynaptic varicosities during the imaging interval (Fig. 8C). In contrast, the absence of orphan synapsin cluster loss led to only a modest 22% increase in synapsin intensity at presynaptic varicosities (Fig. 8C). These results suggest that the elimination of orphan synapsin clusters may serve to enlarge the pool of presynaptic material at large presynaptic boutons.
Since MEF2A suppresses orphan presynaptic sites, we determined whether MEF2A coordinately controls the elimination of orphan synapsin clusters and associated synapsin enrichment at nearby presynaptic varicosities. Strikingly, knockdown of MEF2A reduced orphan synapsin cluster loss in the vicinity of presynaptic varicosities, and had little or no effect on the loss of PSD95-apposed synapsin clusters (Fig. 8D). Importantly, we also found that knockdown of MEF2A reduced the enrichment of synapsin at presynaptic varicosities, as determined by the intensity of synapsin at varicosities relative to orphan sites (Fig. 8E). These results suggest that MEF2A promotes the elimination of orphan synapsin clusters and triggers the concomitant accumulation of presynaptic material at large maturing presynaptic boutons.
To corroborate our live-imaging findings on the function of MEF2A in the enrichment of presynaptic material at presynaptic boutons, we performed electron microscopy analyses of control and MEF2A knockdown granule neurons. Control granule neuron axons displayed large boutons containing synaptic vesicles and mitochondria, which were apposed to postsynaptic densities (Fig. 8F). Importantly, knockdown of MEF2A in granule neurons decreased the number of synaptic vesicles in large boutons (Fig. 8F,G). These data demonstrate that MEF2A plays a critical role in the maturation of presynaptic boutons.
Collectively, our findings define a novel MEF2A-dependent transcriptional mechanism that coordinates the elimination of orphan presynaptic sites and maturation of presynaptic boutons.
Discussion
In this study, we have discovered a MEF2A-dependent transcriptional mechanism that plays an essential role in the regulation of presynaptic differentiation in the mammalian brain. Knockdown of MEF2A increases the density of orphan presynaptic sites in primary granule neurons and importantly in the cerebellar cortex in rat pups in vivo. Remarkably, sumoylation of MEF2A drives the suppression of orphan presynaptic sites. We have also identified the gene encoding Syt1 as a novel direct repressed target gene of MEF2A in neurons, and we have demonstrated that Syt1 repression mediates the ability of sumoylated MEF2A to eliminate orphan presynaptic sites. Finally, we have found that MEF2A coordinates the elimination of orphan synapsin clusters and enrichment of presynaptic material at large mature presynaptic boutons. Collectively, these findings define sumoylated MEF2A and Syt1 as components of a novel transcriptional pathway that orchestrates presynaptic differentiation in the mammalian brain (Fig. 9).
Figure 9.

Schematic of the MEF2A/Syt1 signaling pathway in the regulation of presynaptic development. Sumoylated MEF2A represses transcription of the target gene Syt1 and thereby coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons.
MEF2A has been implicated in the control of postsynaptic dendritic differentiation (Flavell et al., 2006; Shalizi et al., 2006; Pulipparacharuvil et al., 2008; Pfeiffer et al., 2010). Sumoylated MEF2A promotes the morphogenesis of granule neuron dendritic claws that form synapses with mossy fiber terminals and Golgi neuron axons in the cerebellar cortex (Shalizi et al., 2006). Elucidation of a novel function for sumoylated MEF2A in the control of presynaptic differentiation suggests that sumoylation of MEF2A may play a pivotal role in the control of both afferent and efferent connectivity of neurons.
We have uncovered a function for MEF2A in the elimination of orphan presynaptic sites in vivo using RNAi. The in vivo RNAi approach leads to gene knockdown in a small percentage of neurons in the brain, thus facilitating characterization of the cell-autonomous nature of gene function in neuronal morphology in the brain (Stegmuller and Bonni, 2007). It will be important to extend our in vivo studies of MEF2A function in the future to determine MEF2A function in the cerebellar cortex at the level of the organism, e.g., in the control of motor coordination. Mice harboring a mutation in the MEF2A gene have been generated, which do not appear to have impaired coordination (Akhtar et al., 2012). However, the strategy used to generate the MEF2A mutation in these mice allows for the expression of a large truncated MEF2A protein (data not shown). The truncated MEF2A protein retains the transcriptional regulatory C-terminal domain (Black and Olson, 1998; Shalizi and Bonni, 2005). In the future, it will also be crucial to determine MEF2A function in the cerebellar cortex in knock-out mice that are null for MEF2A.
The MEF2A-related transcription factors MEF2C and MEF2D, which are abundantly expressed in the brain, also undergo-sumoylation (Grégoire and Yang, 2005), raising the question of whether these proteins also control presynaptic differentiation. Interestingly, knockdown of MEF2C or MEF2D failed to increase the density of orphan presynaptic sites (data not shown). These data suggest that MEF2A may have specific roles in the elimination of orphan presynaptic sites.
We have identified Syt1, which acts locally at synapses, as a novel repressed target gene of sumoylated MEF2A in neurons. Syt1 has an established role in the regulation of calcium-dependent synaptic vesicle fusion in neurons (Poskanzer et al., 2003; Tang et al., 2006; Xu et al., 2007), raising the intriguing possibility that sumoylated MEF2A may also exert long-range control over neurotransmitter release in mature neurons and thus contribute to adaptive responses in the brain. Although we focused on Syt1 as a target of sumoylated MEF2A, our microarray analyses revealed that expression of sumoylated MEF2A led to the downregulation of other presynaptic genes, including Kif1b. Kif1b plays an important role in axonal transport of synaptic vesicle precursors (Zhao et al., 2001). Thus, our results suggest that sumoylated MEF2A may control the expression of a program of presynaptic genes to orchestrate the process of presynaptic differentiation.
Orphan presynaptic sites have been characterized in primary neurons (Krueger et al., 2003) and in vivo (Pichitpornchai et al., 1994; Shepherd and Harris, 1998; Nakamura et al., 2006). However, the role of orphan presynaptic sites in presynaptic development has remained largely unknown. Our live imaging analyses reveal that orphan presynaptic sites are eliminated at a high rate near presynaptic varicosities and appear to contribute to the accumulation of presynaptic material at large maturing presynaptic boutons. Strikingly, the coordinate elimination of orphan presynaptic sites and maturation of presynaptic boutons is tightly controlled by a cell intrinsic mechanism under the purview of sumoylated MEF2A. It will be interesting in future studies to determine how targets of MEF2A locally coordinate the elimination of orphan presynaptic sites and the maturation of presynaptic boutons.
Transcriptional mechanisms have been linked to the pathogenesis of diverse brain diseases. Interestingly, MEF2 is thought to regulate the expression of a number of autism-linked genes in neurons (Flavell et al., 2008; Morrow et al., 2008). It will be interesting to determine in future studies whether abnormalities in presynaptic development governed by the MEF2A signaling pathway in the brain contribute to neurodevelopmental disorders of cognition.
Footnotes
This work was supported by NIH Grant NS041021 (A.B.), the Japan Society for the Promotion of Science (T.Y.), a National Science Foundation fellowship, a Lefler fellowship, and the Albert J. Ryan foundation (Y.Y.), and the Adelson foundation (D.H.G. and G.C.). We thank Fuying Gao for statistical analyses and members of the Bonni laboratory for helpful discussions and critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, Monteggia LM. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One. 2012;7:e34863. doi: 10.1371/journal.pone.0034863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the cerebellar system: in relation to its evolution, structure, and functions. Boca Raton, FL: CRC; 1997. [Google Scholar]
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Colón-Ramos DA, Margeta MA, Shen K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science. 2007;318:103–106. doi: 10.1126/science.1143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Gaudillière B, Yang Y, Ikeuchi Y, Yamada T, DiBacco S, Stegmüller J, Schüller, Salih DA, Rowitch D, Brunet A, Bonni A. A FOXO-Pak1 transcriptional pathway controls neuronal polarity. Genes Dev. 2010;24:799–813. doi: 10.1101/gad.1880510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya M, Watanabe M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J Comp Neurol. 2000;426:572–586. [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol Cell Biol. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci U S A. 2006;103:45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, Bonni A. A centrosomal Cdc20-APC pathway controls dendritic morphogenesis in postmitoic neurons. Cell. 2009;136:322–336. doi: 10.1016/j.cell.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM. The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron. 2003;40:945–957. doi: 10.1016/s0896-6273(03)00729-3. [DOI] [PubMed] [Google Scholar]
- Litterman N, Ikeuchi Y, Gallardo G, O'Connell BC, Sowa ME, Gygi SP, Harper JW, Bonni A. An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011;9:e1001060. doi: 10.1371/journal.pbio.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Schwarz J, Martin JF, Olson EN. Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J Neurosci. 1995;15:5727–5738. doi: 10.1523/JNEUROSCI.15-08-05727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Tamura H, Horinouchi K, Shiosaka S. Role of neuropsin in formation and maturation of Schaffer-collateral L1cam-immunoreactive synaptic boutons. J Cell Sci. 2006;119:1341–1349. doi: 10.1242/jcs.02862. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. New York: Springer; 1974. [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichitpornchai C, Rawson JA, Rees S. Morphology of parallel fibres in the cerebellar cortex of the rat: an experimental light and electron microscopic study with biocytin. J Comp Neurol. 1994;342:206–220. doi: 10.1002/cne.903420205. [DOI] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Marek KW, Sweeney ST, Davis GW. Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature. 2003;426:559–563. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59:621–633. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Kim AH, Ikeuchi Y, Wilson-Grady JT, Merdes A, Gygi SP, Bonni A. A CaMKIIbeta signaling pathway at the centrosome regulates dendrite patterning in the brain. Nat Neurosci. 2011;14:973–983. doi: 10.1038/nn.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S, Best JL, Zon LI, Gill G. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol Cell. 2002;10:831–842. doi: 10.1016/s1097-2765(02)00682-2. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Gomes RA, McAllister AK. Formation of presynaptic terminals at predefined sites along axons. J Neurosci. 2006;26:10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shalizi AK, Bonni A. Brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3–>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JH, Bland KS, Fetter RD, Goodman CS. Short-range and long-range guidance by Slit and its Robo receptors: a combinatorial code of Robo receptors controls lateral position. Cell. 2000;103:1019–1032. doi: 10.1016/s0092-8674(00)00206-3. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Stegmuller J, Bonni A. Using RNAi in studies of cerebellar development. In: Davidson B, editor. Inhibitory RNAs in neuroscience. San Diego: Society for Neuroscience; 2007. [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M. Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci. 2011;31:3362–3374. doi: 10.1523/JNEUROSCI.5601-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y, de la Iglesia N, Shen J, Bonni A. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science. 2009;326:575–578. doi: 10.1126/science.1177087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–1668. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]