

Development of resistance to targeted drugs and immunotherapy limits improvement in response rates and disease-free survival of cancer patients. In advanced melanoma, for example, multiple mechanisms of resistance emerge during BRAF inhibitor drug therapy that affect both tumor cell properties and microenvironment.1 In addition, just as immunoediting during the de novo immune response against cancer selects for immune-resistant tumor variants,2 active and passive immunotherapies can create selective pressures that lead to outgrowth of resistant tumor cells or new metastases (Figure 1). Understanding the mechanisms of resistance is critical to developing therapeutic strategies to counteract them as well as preventive measures to avoid their onset. In this issue of Molecular Therapy, Boisgerault and colleagues3 nicely elucidate these points using a tumor vaccination model to induce a T-cell response that places a selective pressure on the tumor facilitating the emergence or selective survival of resistant cancer stem cells or cells undergoing epithelial–mesenchymal transition (EMT). The study further shows how acquired or intrinsic mechanisms of resistance can reveal molecular vulnerabilities that may allow responses even to chemotherapy drugs that would have been considered ineffectual.
Figure 1.
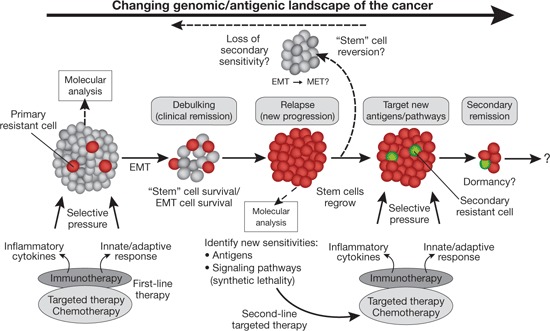
The evolving tumor genomic and antigenic landscape and cancer stem cell resistance and vulnerability during therapy. Immunotherapy and other types of cancer therapy can debulk tumors but also put selective pressure on tumors leading to the survival and outgrowth of resistant cells (relapse) that have cancer stem cell properties or properties of cells undergoing epithelial–mesenchymal transition (EMT). However, these resistant cells (shown in red) overexpress key antigens or metabolic vulnerabilities that can be targeted by newer antigen-specific immunotherapies or drugs. These new targets can be identified by molecular analysis of this changing genomic and antigenic landscape (represented by the solid arrow across the top). Immunotherapies could thus be combined with chemotherapies either simultaneously or in succession sooner to both debulk the original tumor and eradicate any emerging resistant cells. However, although relapsed tumors can be treated to induce a secondary remission, additional resistant cells could emerge (shown as the green cells). The cancer stem cell or EMT phenotype is plastic, and these cells can differentiate through mesenchymal–epithelial transition (MET) or revert to a molecular profile similar to that of the bulk of the original tumor (indicated at the top of the diagram and by the dotted arrow in reverse). Thus, these new stem cell vulnerabilities can be transient, and sensitivity to second-line therapies based on the new molecular or antigenic profile can be rapidly lost. Intermittent dosing of different regimens may be needed to overcome this problem.
Boisgerault et al. made use of their previously described vesicular stomatitis virus (VSV) tumor complementary DNA (cDNA) library approach to vaccinate against prostate cancer and melanoma.4,5 A multiantigen VSV vaccine made from a normal human prostate cDNA library (called ASEL) was used to treat large established TC2 prostate tumors in mice (derived from the TRAMP-C2 spontaneous mouse tumor model) to uncover synthetic lethality in cells resistant to the vaccine. The authors had previously reported that a more aggressive regimen of nine vaccinations with ASEL eradicated all tumors without relapse and was associated with a strong interleukin-17 T helper cell (Th17) response against prostate antigens.4 However, in this new study they found that a more limited course of vaccination with ASEL (six injections), associated with a greater Th1 response and limited involvement of Th17 cells, led to complete macroscopic tumor regressions followed in many mice by aggressive tumor recurrence. Interestingly, these recurrent tumors (TC2R) were enriched with cells that had lost prostate epithelial markers (e.g., E-cadherin) but had gained mesenchymal markers (e.g., vimentin and N-cadherin) associated with tumor stem cells and cells with enhanced invasive and metastatic properties. These resistant cells also exhibited characteristics of cancer stem cells based on colony-forming assays. The workers further found that coincubation of splenocytes from ASEL-vaccinated mice with TC2 tumor cells in vitro led to the appearance of cells with mesenchymal properties. Using mixtures of fluorescence-tagged TC2 and TC2R cells, they also observed preferential outgrowth of TC2R cells in vitro after incubation with ASEL vaccine-sensitized spleen cells.
With the aim of identifying the mechanisms of resistance, the authors then generated a secondary VSV–cDNA library vaccine from the in vivo immune escape variants (called IEEL) that was used to effectively immunize against the recurrent tumors. Subcloning of the VSV–cDNA library identified the antigen specificities driving the secondary IEEL response and two new antigens overexpressed in the resistant cells: CD44 and topoisomerase IIα (TOPO-IIα). The overexpression of TOPO-IIα was both unexpected and interesting because it raised the prospect of treating resistant cells with doxorubicin or etoposide. Indeed, treatment with doxorubicin prevented the growth of TC2R explants in vivo as well as in culture and also prevented the generation of immune escape variants from TC2 tumors emerging after ASEL vaccination in vivo. These results suggest that either intrinsically resistant cancer stem cell–like tumor cells overexpressing TOPO-IIα were present in the primary tumors and survived the strong adaptive immune response or EMT induced by the immune pressure led to the outgrowth of a subpopulation of cells expressing high levels of TOPOIIα.
To determine whether this phenomenon was unique to their prostate cancer system, the authors next evaluated whether the acquired synthetic lethality to TOPO-IIα inhibition extended to nonepithelial B16 melanoma cells expressing herpes simplex virus thymidine kinase, which are susceptible to the drug ganciclovir. They found a similar subset of drug-resistant cells in subcutaneous tumors that overexpressed TOPO-IIα that could also be selectively eradicated using doxorubicin.3 Thus, surprisingly, using two different tumors (one epithelial and the other neuroectodermal), they found a subpopulation of resistant cancer stem cell–like cells overexpressing TOPO-IIα that made both of them susceptible to a drug considered clinically ineffective for either. Importantly, in the prostate cancer system, the resistant mesenchymal stem cell–like cells lost both their TOPO-IIα overexpression and sensitivity to doxorubicin after a week of culture, indicating that the phenotype and therapeutic window were transient.
The findings indicate that immunotherapy can lead to novel and unexpected forms of resistance that should be carefully monitored using biomarkers. The results also underscore the possible danger of a suboptimal immune response that fails to induce “sterilizing immunity” and instead promotes tumor escape. However, monitoring of changes in tumor gene expression and subsequent targeting of a changing antigen profile with a tumor cDNA vaccine approach may overcome these resistance mechanisms. The nature of the changing antigenic profiles during therapy will require further study in different tumor types, as will the role for changes in the mutational landscape of the tumor that can generate missense and nonsense mutations recognized by the immune system as strong nonself (neoepitope) rejection antigens.2
The results further suggest an adaptive cancer vaccination approach in which multiple cDNA vaccines can be made at different times as a personalized therapy for each patient. VSV may be an ideal vehicle in that it has been found safe in nonhuman primates and generates strong cell-mediated immune responses.6,7 An alternative approach could make use of tumor RNA–complementary RNA transduced dendritic cells, which have been developed as an effective clinical-grade cancer vaccine platform.8,9 Although iterative production of personalized vaccines could be cumbersome, the library vaccination approach obviates the need to identify specific antigens. In patients undergoing effective resistant tumor control, one could monitor T-cell responses to identify the specific antigens involved. A potential caveat to this adaptive vaccination approach lies in the molecular heterogeneity of tumor cells in different sites of disease in humans and whether there exist common overexpressed genes or mutations that can be exploited as antigenic targets at different stages of disease progression.
Because the resistant cells are mesenchymal-like and overexpress TOPO-IIα EMT may be a mechanism of immune resistance to T-cell–based immunotherapies. Inflammatory cytokines such as tumor necrosis factor-α and interleukin-1β can induce EMT in breast cancer cells and other epithelial tumors,10 and tumor cells with stem cell properties are relatively resistant to killing by cytotoxic lymphocytes.11 The resistant cells may also be the same as the slow-cycling stemlike cells recently found in melanoma and other forms of cancer resistant to chemotherapy and mitogen-activated protein kinase inhibitors that upregulate mitochondrial oxidative phosphorylation and downmodulate glycolysis.12
It is tempting to speculate that ASEL vaccine-resistant cells in the TC2 model and the ganciclovir-resistant cells in the melanoma model are similar subsets of slow-cycling cancer stem cells that others have found resistant to other types of drugs.12 If so, this would suggest an important intersection of immunotherapy with chemotherapy in terms of inducing the selective survival of slow-cycling stemlike cells or cells that have undergone EMT. New immunotherapy clinical trials should therefore monitor the emergence of such cells so as to identify possible markers of resistance and newly emergent vulnerabilities. A caveat is that enrichment of stemlike or EMT cells in tumors during therapy can be transient and reversible. Theoretically this could lead to tertiary resistance against the second-line stemlike or EMT tumor cell target and suggests that multiple cycles of different intermittent therapies may need to be juxtaposed over a long period of time to contain development of these resistance phenotypes. Strategically spaced drug holidays may allow any resistant cells to lose their stemlike properties and regain sensitivity to the original therapy.
Conversely, Boisgerault and colleagues' demonstration of the synthetic lethality (doxorubicin sensitivity) suggests the power of combining immunotherapy with chemotherapy, and that immunotherapy may uncover pathways that can be effectively targeted by existing drugs that would otherwise not be used as a consequence of the primary resistance of the tumors to these drugs. This contrasts with the more traditional approach of using chemotherapy or radiation to kill tumor cells and release antigen so as to facilitate an immune response using active immunotherapy. Recent studies have shown that some chemotherapy drugs activate an innate immune response in the tumor microenvironment via release of ligands for Toll-like and purinergic receptors that facilitate antitumor T-cell responses.13,14 Earlier studies have used chemotherapy (e.g., cyclophosphamide) before immunotherapy as a way of transiently depleting CD4+ T-regulatory cells.15 Moreover, in recent clinical trials, patients who received chemotherapy after immunotherapy due to disease progression had improved responses and longer overall survival. For example, patients with progressive non–small cell lung cancer who had an immune response to previous vaccination with an adenovirus–p53 formulation saw improvement in their response to oxaloplatin and longer overall survival.16 Immunotherapy may have facilitated the expansion of an oxaloplatin-sensitive subset of tumor cells, or the chemotherapy may have enhanced the sensitivity of the tumor cells to an ongoing memory T-cell response. Similar results in other clinical trials in which salvage chemotherapy used after immunotherapy led to unexpected improvement in clinical responses should be reinterpreted in this light.
In summary, the new study underscores an emerging view of cancer therapy as an interplay of forces regulating EMT and cancer stem cells and identifying their vulnerabilities to overcome therapeutic resistance. These results also emphasize the need to perform strong biomarker-driven studies to identify emerging molecular mechanisms of resistance and vulnerabilities on an ongoing basis and tailor each round of therapy for our patients according to these changing molecular and antigenic characteristics.
Acknowledgments
Research in the author's laboratory is generously supported by grants from the National Cancer Institute, the Melanoma Research Alliance, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
References
- Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–1304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ.et al. (2012Cancer exome analysis reveals a T-cell–dependent mechanism of cancer immunoediting Nature 482400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisgerault N, Kottke T, Pulido J, Thompson J, Diaz RM, Rommelfanger-Konkol D.et al. (2013Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse Mol Ther 211507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P.et al. (2011Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors Nat Med 17854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM.et al. (2012Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma Nat Biotechnol 30337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G, Beenhakker N, Hofman S, Walther-Jallow L, Mäkitalo B, Mooij P.et al. (2012Immunization with apoptotic pseudovirus transduced cells induces both cellular and humoral responses: a proof of concept study in macaques Vaccine 302523–2534. [DOI] [PubMed] [Google Scholar]
- Mire CE, Miller AD, Carville A, Westmoreland SV, Geisbert JB, Mansfield KG.et al. (2012Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates PLoS Negl Trop Dis 6e1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa E, Vieweg J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev. 2004;199:251–263. doi: 10.1111/j.0105-2896.2004.00139.x. [DOI] [PubMed] [Google Scholar]
- Liao X, Li Y, Bonini C, Mair S, Gilboa E, Greenberg PD.et al. (2004Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes Mol Ther 9757–764. [DOI] [PubMed] [Google Scholar]
- Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim F, Dombrowski Y, Ritter C, Buttmann M, Häusler S, Ossadnik M.et al. (2009Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast cancer stem cells Cancer Res 698058–8066. [DOI] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D.et al. (2013Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) Cells Cancer Cell 23811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL.et al. (2007Calreticulin exposure dictates the immunogenicity of cancer cell death Nat Med 1354–61. [DOI] [PubMed] [Google Scholar]
- Rowswell-Turner RB, Harden JL, Nair RE, Gu T, Kilinc MO, Egilmez NK. Chronic chemoimmunotherapy achieves cure of spontaneous murine mammary tumors via persistent blockade of posttherapy counter-regulation. J Immunol. 2011;187:4109–4118. doi: 10.4049/jimmunol.1101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther. 2010;10:983–991. doi: 10.1517/14712598.2010.484801. [DOI] [PMC free article] [PubMed] [Google Scholar]