

Abstract
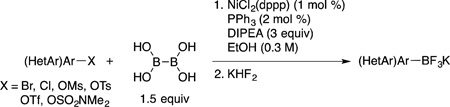
Arylboronic acids are gaining increased importance as reagents and target structures in a variety of useful applications. Recently, the palladium-catalyzed synthesis of arylboronic acids employing the atom economical tetrahydroxydiboron (BBA) reagent has been reported. The high cost associated with palladium, combined with several limitations of both palladium and copper-catalyzed processes, prompted us to develop an alternative method. Thus, the nickel-catalyzed borylation of aryl and heteroaryl halides and pseudo-halides using tetrahydroxydiboron (BBA) has been formulated. The reaction proved to be widely functional group tolerant and applicable to a number of heterocyclic systems. To the best of our knowledge, the examples presented here represent the only effective Ni-catalyzed Miyaura borylations conducted at room temperature.
Introduction
Arylboronic acids are becoming increasingly important reagents and target structures.1 This unique class of compounds is known to undergo a diverse variety of reactions, including the Petasis-Borono Mannich reaction,2 Chan-Lam coupling,3 conjugate addition reactions,4 and most importantly a variety of cross-coupling reactions.5 In addition to their use as reagents, boronic acids are finding an increasing role in biological and medicinal applications.6
Traditional methods for the synthesis of arylboronic acids from the corresponding halides and trialkyl borates rely on a metal-halogen exchange approach and require the use of organolithium or organomagnesium reagents.7 Although widely utilized, this process presents limitations regarding functional group tolerability, being incompatible with molecules containing sensitive functional groups embedded within their structures.8 Transition metal-catalyzed borylation has emerged as a viable alternative to afford boron species containing a high degree of molecular complexity. Rh9 and Ir-10 catalyzed C-H borylations provide access to many aryl and heteroarylboron derivatives. However, the selectivities of these reactions are determined by steric and electronic effects within the aryl system, making them limited to specific substitution patterns. To overcome these limitations, Cu,11 Ni12 and Pd13-catalyzed Miyaura borylations of aryl and heteroaryl halides have been developed. Regardless of the high selectivity and functional group compatibility, these methods often require the use of bis(pinacolato) diboron (B2Pin2), pinacolborane (HBPin), or neopentylglycolborane as a boron source, thus resulting in the initial formation of boronates. To access the boronic acids, an additional deprotection step is required. Representative examples of deprotection conditions to unveil the desired boronic acid include acidic hydrolysis,14 oxidation15 or reduction16 (Scheme 1). In addition to the requisite deprotection, all of the boronate-based reagents release diols on conversion to the desired arylboronic acids, and these diol-byproducts must be removed from the reaction mixture through often laborious procedures.17 This, combined with the inherent lack of atom economy in these processes, greatly diminishes the appeal of these approaches.
Scheme 1.

Commonly Employed Borylating Agents and BBA
In an effort to devise a method to provide direct access to arylboronic acids, we recently developed a Pd-catalyzed borylation of aryl- and heteroaryl halides utilizing tetrahydroxydiboron (BBA).18 Under the conditions that evolved, aryl and heteroaryl chlorides and bromides were efficiently borylated. The reaction required 0.1 – 5 mol % of a palladium-based pre-formed catalyst, 3 equivalents of BBA, and reaction temperatures of 80 °C. Although highly effective in most cases, aryl halides containing ketones and aldehydes afforded the desired product along with undesired byproducts resulting from reduction of these embedded functional groups. Furthermore, the substrate scope for heteroaryl systems was restricted to nitrogen-containing molecules, such as quinolines and indoles, as the borylation of furans and thiophenes derivatives could not be achieved.
Because of the high cost of palladium and the problematic substrates cited above, we were interested in developing a nickel-catalyzed borylation of aryl and heteroaryl halides utilizing BBA. Previously reported nickel-catalyzed borylations12 not only utilize the aforementioned wasteful borylating reagents (e.g., B2Pin2 and neopentylglycolborane), but they also require the use of additives such as zinc12b, 12e and trimethyl(2,2,2-trifluoroethoxy)silane,12g and proceed only at high reaction temperatures (100 – 110 °C). Additionally, although room temperature copper-catalyzed borylations have been reported,11a,11b the substrate scope examined, especially for heteroaryls and reactants with sensitive functional groups, has been extremely limited. Furthermore, these methods not only use inefficient B2Pin2, but they also have been employed largely for aryl iodides. Only 4 examples of aryl bromides have been reported, and aryl chlorides have not proven to be competent substrates in copper-promoted protocols communicated to date. Herein, we report a mild, cost efficient nickel-catalyzed borylation of aryl and heteroaryl halides and pseudo-halides utilizing tetrahydroxydiboron (BBA).
Results and Discussion
We began the optimization of the reaction conditions for the nickel-catalyzed borylation of aryl halides using BBA with 4-bromoanisole. Using microscale high throughput experimentation (HTE), an array of nickel catalysts, ligands, bases and solvents was examined. Extensive screening revealed that a combination of NiCl2(dppp), PPh3, and diisopropylethylamine (DIPEA) in ethanol at 80 °C was efficient, affording the desired boronic acid in good yield as evidenced by conversion to the pinacol boronate and analysis by HPLC (Scheme 2).
Scheme 2.

Microscale HTE screening for optimal reaction conditions with 4-bromoanisole
The optimal HTE conditions performed on microscale were scaled up and repeated on the benchtop. Because boronic acids are known to be relatively unstable tricoordinate boron species,19 to determine the isolated yields the crude reaction mixture was treated with aqueous KHF2 to afford the more robust potassium trifluoroborate salts without purification of the intermediate boronic acid. Thus, in a very straightforward and simple procedure the reaction of 4-bromoanisole (3 mmol) with BBA (1.5 equiv), 1 mol % of NiCl2(dppp), 2 mol % of PPh3, and 3 equivalents of DIPEA in 10 mL of degassed ethanol at 80 °C, followed by aqueous KHF2 addition, yielded potassium trifluoro(4-methoxyphenyl)borate (1a, Table 1, entry 1) in 91% yield. Importantly, all reagents utilized in this method are inexpensive and bench stable, avoiding the use of glovebox techniques and dry solvents. Furthermore, when compared to other Ni-catalyzed borylations, the reaction occurs in a short period of time (as indicated by GC) without the use of metal additives. With optimal conditions in hand, the substrate scope for aryl bromides containing electron-donating and electron-neutral groups was subsequently investigated (Table 1). The indicated reaction time was determined by GC analysis. Aryl bromides containing a methyl ether group in the para (entry 1), meta (entry 2) and ortho (entry 3) position afforded the desired trifluoroborates in good yield in only 2 h at 80 °C. The same trend was observed for simple methyl substituted arenes (entries 6 and 7). When the reaction of electron neutral aryl bromides was performed at 80 °C, significant amounts of undesired homocoupled product was observed by GC/MS, leading to lower yields. Carrying out the reaction at room temperature diminished this problem, and excellent yields were achieved for those substrates (entries 8 – 10). The presence of alcohol or free amine functional groups in the molecule required the use of 3 mol % of the Ni catalyst. However, the reactions proceeded at room temperature in only 6 h, and the desired trifluoroborates were obtained in good yields. Furthermore, the reaction of 2-bromonaphthalene was performed on a 48 mmol scale (~10 g), providing the product 1j in 81% yield after 8 h at room temperature (entry 10). The catalyst loading for this large-scale reaction was reduced to 0.1 mol % of NiCl2(dppp), 0.2 mol % of PPh3 in 90 mL of ethanol (0.5 M). Unfortunately, sterically hindered 2,6-dimethylbromobenzene did not perform well under the reaction conditions, and only 16% conversion was observed by GC/MS (data not shown).
Table 1.
Ni-catalyzed Borylation of Electron-Rich and Electron-Neutral Aryl Bromides with BBA
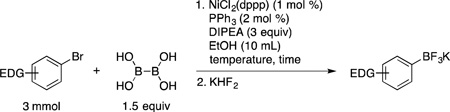 | |||||
|---|---|---|---|---|---|
| entry | product | time | temperature | yield (%) | |
| 1 | 1a | 2 h | 80 °C | 91 | |
| 2 | 1b | 2 h | 80 °C | 89 | |
| 3 | 1c | 2 h | 80 °C | 77 | |
| 4a | 1d | 6 h | rt | 78 | |
| 5a | 1e | 6 h | rt | 67 | |
| 6 | 1f | 4 h | 80 °C | 84 | |
| 7 | 1g | 3 h | 80 °C | 72 | |
| 8 | 1h | 6 h | rt | 90 | |
| 9 |  |
1i | 4 h | rt | 93 |
| 10 | 1j | 4 h 8 h |
rt | 90 81b |
|
3 mol % NiCl2(dppp) and 6 mol % PPh3
48 mmol scale using 0.1 mol % NiCl2(dppp), 0.2 mol % PPh3 in EtOH (90 mL)
Next, aryl bromides containing electron-withdrawing groups were tested using the developed borylation protocol (Table 2). The broad functional group compatibility of this method is illustrated with the set of substrates utilized. Aryl bromides containing nitrile, ketone, ester and aldehyde functional groups at the para and meta positions (entries 1 – 6) were efficiently borylated at room temperature, affording the corresponding trifluoroborates in good to excellent yields. Remarkably, because of the very mild reaction conditions, compounds 2c, 2d, 2e and 2f were obtained without reduction of the carbonyl group. Previously developed palladium-catalyzed borylations with BBA delivered up to 30% of the reduced alcohol side product.18b Of note, borylation of aryl halides containing aldehydes were not included in any previously reported nickel-catalyzed borylation methods. para- and ortho-Substituted fluorine-containing substrates were also borylated (entries 7 – 10). These molecules provide easy access to fluorinated aryl compounds via cross-coupling reactions and are of increasing interest in medicinal chemistry.20 As a limitation of the method, ortho substituted aryl bromides containing electron-withdrawing groups other than fluorine and trifluoromethyl did not provide the desired borylated product, and only protodehalogenation was observed. Furthermore, aryl bromides containing nitro groups afforded only the reduced amine product (eq 1), along with unreacted starting material, as indicated by GC/MS analysis.
Table 2.
Ni-catalyzed Borylation of Electron-Poor Aryl Bromides with BBA
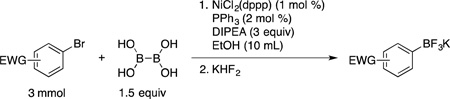 | |||||
|---|---|---|---|---|---|
| entry | product | time | temperature | yield (%) | |
| 1 | 2a | 4 h | rt | 91 | |
| 2 | 2b | 4 h | rt | 78 | |
| 3 | 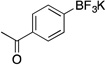 |
2c | 4 h | rt | 93 |
| 4 | 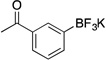 |
2d | 4 h | rt | 81 |
| 5 | 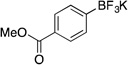 |
2e | 6 h | 50 °C | 92 |
| 6 | 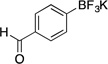 |
2f | 6 h | rt | 84 |
| 7 | 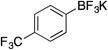 |
2g | 6 h | rt | 86 |
| 8 | 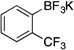 |
2h | 12 h | rt | 71 |
| 9 | 2i | 2 h | rt | 96 | |
| 10 | 2j | 12 h | rt | 75 | |
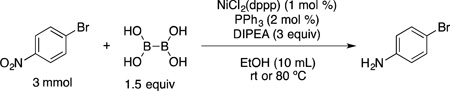 |
(1) |
The scope of the reaction was further expanded to heteroaryl bromides (Table 3). Under the developed reaction conditions, a variety of heteroaryl trifluoroborates such as thiophene, furan, benzofuran, benzothiophene, pyrazole, indole, pyridine, quinoline, and azaindole systems were successfully borylated in good to excellent yields. Nitrogen-containing heterocycles required higher catalyst loading and temperatures. Furthermore, after addition of KHF2 a mixture of potassium and internal salt was obtained for these substrates, and full conversion to the potassium salt required treatment of the crude mixture with K2CO3 in acetonitrile.21 To the best of our knowledge, the examples illustrated on Table 3 represent the largest and most diverse substrate scope for borylation of heteroaryl systems in the current literature. Unfortunately, heteroaryls such as pyrimidine, isoxazole and thiazole did not undergo borylation, and only halide starting material was recovered in these cases.
Table 3.
Ni-catalyzed Borylation of Heteroaryl Bromides with BBA
 | |||||
|---|---|---|---|---|---|
| entry | product | time | temperature | yield (%) | |
| 1 | 3a | 4 h | rt | 94 | |
| 2a | 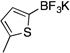 |
3b | 4 h | rt | 85 |
| 3a | 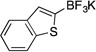 |
3c | 6 h | rt | 74 |
| 4 | 3d | 4 h | rt | 92 | |
| 5 | 3e | 4 h | rt | 91 | |
| 6b | 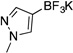 |
3f | 12 h | rt | 82 |
| 7 | 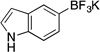 |
3g | 6 h | rt | 86 |
| 8b | 3h | 4 h | 80 °C | 72 | |
| 9b | 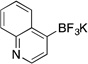 |
3i | 4 h | 80 °C | 74 |
| 10b | 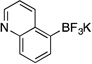 |
3j | 4 h | 80 °C | 81 |
| 11b | 3k | 4 h | 80 °C | 83 | |
| 12b | 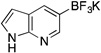 |
3l | 4 h | 80 °C | 83 |
NiCl2(dppp) (0.5 mol %), PPh3 (1 mol %)
NiCl2(dppp) (5 mol %), PPh3(10 mol %)
As mentioned previously, one of the advantages in utilizing BBA as a borylating agent is that it provides direct access to boronic acids as well as a variety of boronate esters. Thus, 3-bromothiophene was subjected to the nickel-catalyzed borylation protocol with BBA followed by different workups to provide diverse boron derivatives (Table 4). Boronic acid was obtained in good yield after a simple hexane wash of the crude mixture. Various boronate esters were accessed after acid workup followed by addition of the corresponding diol reagent and purification by column chromatography.
Table 4.
Direct Synthesis of Boron Derivatives
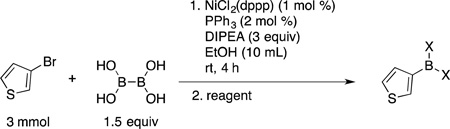 | ||||
|---|---|---|---|---|
| entry | reagent | product | yield (%) | |
| 1 | hexane wash | 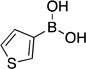 |
4a | 87 |
| 2 | 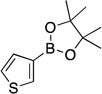 |
4b | 90 | |
| 3 |  |
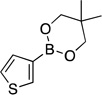 |
4c | 96 |
Additionally, we were interested in utilizing the same developed set of conditions for the more commercially available aryl and heteroaryl chlorides (Table 5). Aryl chlorides containing nitrile, ester, ketone, morpholine, piperazine, ether, fluorine and pyrrole subunits embedded within their structure provided the organotrifluoroborates in moderate to good yields. Unfortunately, because a longer reaction time and heating was required for full conversion, 4-chlorobenzaldehyde afforded the desired potassium trifluoro(4-formylphenyl)borate along with 30% of the product resulting from borylation and concomitant reduction of the aldehyde (data not shown). Heteroaryl chlorides such as quinolinyl and thienyl chloride were also borylated. The use of chlorides as the electrophile of choice required, in general, longer reaction times and higher temperatures than when the corresponding bromide was used. Nevertheless, the same set of conditions could be used for the borylation of both halides.
Table 5.
Ni-catalyzed Borylation of Aryl and Heteroaryl Chlorides with BBA
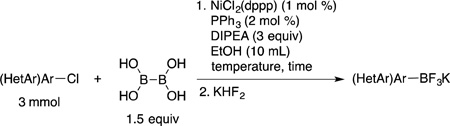 | |||||
|---|---|---|---|---|---|
| entry | Product | time | temperature | yield (%) | |
| 1 | 2a | 4 h | rt | 92 | |
| 2 | 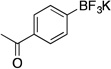 |
2c | 4 h | rt | 89 |
| 3 | 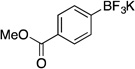 |
2e | 4 h | 80 °C | 89 |
| 4 | 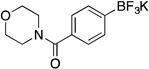 |
5a | 12 h | rt | 64 |
| 5a | 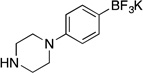 |
5b | 8 h | 80 °C | 52 |
| 6 | 2i | 12 h | rt | 83 | |
| 7 | 1a | 12 h | 80 °C | 66 | |
| 8 | 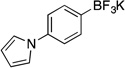 |
5c | 2 h | 80 °C | 69 |
| 9a | 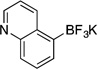 |
3j | 12 h | 80 °C | 75 |
| 10a | 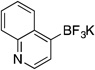 |
3i | 12 h | 80 °C | 58 |
| 11 | 3a | 12 h | rt | 86 | |
NiCl2(dppp) (5 mol %), PPh3 (10 mol %)
Because aryl bromides generally reacted under milder conditions, we were interested in examining the selectivity of the method for molecules containing both bromide and chloride within the molecule (eq 2). Thus, 4-bromochlorobenzene was chosen as a test substrate. Unfortunately, after 30 minutes the major product obtained was the diborylated compound, as identified after addition of pinacol and GC/MS analysis. The remaining 1-bromo-4-chlorobenzene starting material was also recovered, along with small amounts of monoborylated product containing the chloride group. The result indicates that although the reaction should proceed slightly faster for bromides, this preference was insignificant under the developed conditions.
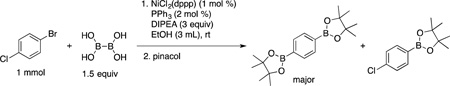 |
(2) |
To compare the efficiency of the method for different borylation partners, the developed conditions were applied to the same aryl bromide, chloride, iodide, triflate, tosylate, mesylate, sulfamate, carbamate and pivalate (Table 6). When compared as a group, bromides are the best electrophilic partners for this reaction. The aryl iodide gave a lower yield compared to other halide electrophiles, mostly because of the large amount of homocoupling product observed. Aryl triflates, tosylates, mesylates and sulfamates were borylated in good yields (entries 4 – 6), while carbamates and pivalates failed to provide borylated product under this set of conditions, and only starting material was recovered.
Table 6.
Ni-catalyzed Borylation of Different Electrophiles
 | |||||
|---|---|---|---|---|---|
| entry | X | time | yield (%) | ||
| 1 | Br | 4 h | 93 | ||
| 2 | Cl | 4 h | 89 | ||
| 3 | I | 2 h | 75 | ||
| 4 | OTf | 6 h | 88 | ||
| 5a | OTs | 2 h | 76 | ||
| 6 | OMs | 12 h | 81 | ||
| 7a | OSO2NMe2 | 12 h | 71 | ||
| 8a | OC(O)NEt2 | 24 h | 0 | ||
| 9a | OPiv | 24 h | 0 | ||
reaction run at 80 °C
Because the palladium-catalyzed borylation with BBA failed to afford the desired borylated product when aryl mesylates were used,18b we were interested in investigating the scope of the reaction for this class of electrophiles (Table 7). Aryl mesylates containing electron-donating, electron-withdrawing and electron-neutral groups underwent borylation under the developed conditions. The products were obtained in good yields without the use of additives as required using previously developed nickel-catalyzed borylation methods.12e
Table 7.
Ni-catalyzed Borylation of Aryl and Heteroaryl Mesylates with BBA
 | |||||
|---|---|---|---|---|---|
| entry | product | time | temperature | yield (%) | |
| 1 | 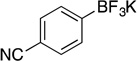 |
2a | 4 h | 50 °C | 82 |
| 2 | 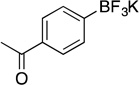 |
2c | 4 h | 50 °C | 88 |
| 3 | 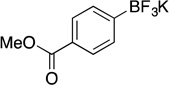 |
2e | 4 h | 80 °C | 71 |
| 4 | 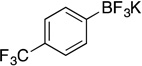 |
2g | 12 h | 50 °C | 81 |
| 5 | 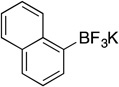 |
1i | 8 h | 50 °C | 87 |
| 6a | 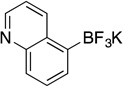 |
3j | 8 h | 80 °C | 76 |
NiCl2(dppp) (5 mol %), PPh3 (10 mol %)
The proposed reaction mechanism is analogous the one proposed for the Pd-catalyzed borylation with BBA (Scheme 3). Based on this mechanism, PPh3 is necessary to help stabilize the in situ formed Ni(0) catalyst. Because homocoupling products were observed for some substrates, we envisioned the possibility of a catalytic cycle occurring parallel to the borylation cycle. The formed boronic acid can compete with BBA in transmetalation with the oxidative addition complex. As observed in our studies, lower catalyst loading and reaction temperature minimized the side product formation and afforded the desired boronic acid in good yields.
Scheme 3.

Proposed Reaction Mechanism
In conclusion, a nickel-catalyzed borylation using tetrahydroxydiboron has been developed. The same set of conditions was efficient to borylate a wide array of aryl and heteroaryl bromides, chlorides, mesylates, tosylates, triflates and sulfamates containing diverse functional groups. All reagents utilized in this method are stable and can be stored on the benchtop. The low cost of nickel compared to that of palladium, combined with the ability to use non-proprietary ligands, makes the method economically attractive for industrial purposes. The use of BBA allows access to different boron derivatives, and most importantly this approach provides direct access to boronic acids and also to the more robust trifluoroborates. To the best of our knowledge, the examples herein that proceeded at room temperature are the first effective Ni-catalyzed Miyaura-borylations to be carried out under such mild condition. Finally, the substrate scope for heteroaryls is the largest found in the currently available literature.
Experimental Section
Analytical Methods
Melting points (°C) are uncorrected. All known compounds were characterized by 1H, 13C, 19F, and 11B NMR spectra, and melting point determination (for solids) and compared with literature values. All new compounds were characterized by 1H, 13C, 19F, and 11B NMR spectra, IR spectroscopy, high-resolution mass spectrometry (HRMS) and melting point determination (for solids). Compounds reported in Tables 5 and 7 that were previously characterized on Tables 1–3, were characterized by 1H NMR spectra and the values compared. 1H, 13C, 19F and 11B NMR spectra were recorded at 500.4, 125.8, 470.8 and 128.4 MHz, respectively. 19F NMR chemical shifts were referenced to external CFCl3 (0.0 ppm). 11B NMR chemical shifts were referenced to external BF3•OEt2 (0.0 ppm) with a negative sign indicating an upfield shift. HRMS (CI) data were obtained in Pos mode, using methane as the ionizing gas. HRMS (ESI) data were obtained in Pos or Neg mode.
General Procedure for Parallel Microscale Experimentation
In a glovebox, Ni catalysts (0.4 µmol) were dosed into the 96-well reactor vial as solutions (50 µL of a 0.008 M solution in CH3CN or THF depending upon the solubility of the catalyst). Ligand (0.8 µmol, 50 µL of a 0.016 M solution in CH3CN or THF depending upon the solubility of the catalyst) was then added to the reaction vials, and this was evacuated to dryness. In the case of solid bases, they were added to the ligand/catalyst mixture (60 µmol, 100 µL of a 0.6 M slurry solution in THF), and this was evacuated to dryness. A parylene stir-bar was then added to each reaction. The 4-bromoanisole (20 µmol/reaction), (HO)2B-B(OH)2 (30 µmol/reaction), liquid bases (60 µmol/reaction) and tert-butylbiphenyl (1 µmol/reaction, used as an internal standard to measure HPLC yield) were then dosed together in the desired reaction solvents using a single-tip pipettor. The reactions were then sealed, taken outside the glovebox, and heated at 80 °C for 18 h. After cooling to ambient temperature, pinacol (100 µL of a 1.8 M solution in CH3CN) was added to the reactions, and the plate was diluted with 500 µL of MeCN. A silicon-rubber storage mat was added, and the contents were shaken to homogenize. Into a separate 96-well-plate LC plate with 1 mL vials was then added 750 µL of MeCN, and then 20 µL of the diluted reaction mixtures. The 96-well plate LC block was then sealed with a silicon rubber storage mat. The reactions were then analyzed using an HPLC modified with a 96-well plate auto-sampler.
General Procedure for Ni-catalyzed Borylation of Aryl and Heteroaryl Halides and Mesylates
To a glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %) and (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv). The vessel was capped and then evacuated and backfilled with Ar (process repeated 3 x). EtOH (10 mL, degassed) was added via syringe followed by the addition of DIPEA (1.6 mL, 9 mmol, 3 equiv) and the halide (3 mmol) in a similar manner (solid halides were added with the other solid reagents before sealing). The reaction was then heated at determined temperatures until the starting material was consumed (as monitored by GC). After the required time, the reaction was cooled to rt and transferred to a 250 mL round bottom flask and concentrated under reduced pressure. The concentrated crude reaction was taken up in MeOH (~ 15 mL or enough to make a free-flowing solution) and cooled to 0 °C. To this solution was added 7.5 equivalents of a 4.5 M aqueous KHF2 (5 mL), and the reaction was stirred for 10 min at 0 °C. The ice bath was removed, and the reaction was stirred at rt for 20 min (or until full conversion to the corresponding trifluoroborate as determined by 11B NMR). The resulting mixture was concentrated and then lyophilized overnight to remove any traces of H2O. The compound was purified with continuous Soxhlet extraction (overnight) with acetone (150 mL). The collected solvent was concentrated until a minimal volume of acetone remained (~5 mL). The addition of Et2O (~25 mL) led to the precipitation of the desired product. The collected solid was washed with Et2O. In cases where the trifluoroborate was obtained with a trace amount of the protonated base and for nitrogen-containing trifluoroborates (internal salt formation), the crude mixture was concentrated after Soxhlet extraction and dissolved in acetonitrile (~ 15 mL). To this solution was added K2CO3 (4 g, 10 equiv) and the reaction was stirred for 4 h (quinoline and pyridine derivatives required 16 h). The slurry was concentrated, and acetone was added to the solid mixture followed by filtration (process repeated 3 x). The collected solvent was concentrated until a minimal volume of acetone remained (~5 mL). The addition of Et2O (~25 mL) led to the precipitation of the desired product. The collected solid was washed with Et2O.
Potassium Trifluoro(4-methoxyphenyl)borate (1a).18b
Following the general procedure, a mixture of 4-bromoanisole (0.56 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 2 h. The title compound was obtained in 91% yield (0.58 g, 2.73 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.28 (d, J = 7.3 Hz, 2H), 6.69 (d, J = 7.3 Hz, 2H), 3.67 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 158.0, 132.9, 112.5, 55.1; 19F NMR (470.8 MHz, DMSO-d6) δ – 138.2; 11B NMR (128.4 MHz, DMSO-d6) δ 2.6.
Potassium Trifluoro(3-methoxyphenyl)borate (1b).22
Following the general procedure, a mixture of 3-bromoanisole (0.56 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 2 h. The title compound was obtained in 89% yield (0.57 g, 2.67 mmol) as a white solid, mp 179 – 181 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.05 (t, J = 7.1 Hz, 1H), 6.97 (d, J = 6.6 Hz, 1H), 6.93 (s, 1H), 6.62 (d, J = 7.3 Hz, 1H) 3.69 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 158.5, 127.6, 124.2, 116.8, 111.1, 54.8; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.2; 11B NMR (128.4 MHz, DMSO-d6) δ 3.7.
Potassium Trifluoro(2-methoxyphenyl)borate (1c).18b
Following the general procedure, a mixture of 2-bromoanisole (0.56 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 2 h. The title compound was obtained in 77% yield (0.44 g, 2.31 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.34 (d, J = 6.0 Hz, 1H), 7.07 (t, J = 7.0 Hz, 1H), 6.72 (d, J = 8.0 Hz, 2H), 3.65 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 162.8, 133.5, 127.2, 119.6, 110.0, 55.1; 19F NMR (470.8 MHz, DMSO-d6) δ – 136.8; 11B NMR (128.4 MHz, DMSO-d6) δ 2.5.
Potassium Trifluoro(4-hydroxyphenyl)borate (1d).18b
Following the general procedure, a mixture of 4-bromophenol (0.52 g, 3 mmol), NiCl2(dppp) (48.8 mg, 0.09 mmol, 3 mol %), PPh3 (47.2 mg, 0.18 mmol, 6 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 78% yield (0.47 g, 2.34 mmol) as light pink solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.56 (s, 1H), 7.10 (d, J = 8.1 Hz, 2H), 6.50 (d, J = 7.9 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 155.2, 132.6, 113.7; 19F NMR (470.8 MHz, DMSO-d6) δ – 138.0; 11B NMR (128.4 MHz, DMSO-d6) δ 3.6.
Potassium (4-Aminophenyl)trifluoroborate (1e).18b
Following the general procedure, a mixture of 4-bromoaniline (0.52 g, 3 mmol), NiCl2(dppp) (48.8 mg, 0.09 mmol, 3 mol %), PPh3 (47.2 mg, 0.18 mmol, 6 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 67% yield (0.40 g, 2.01 mmol) as a brown solid (treatment with K2CO3 needed), mp = 200 °C (decomposed). 1H NMR (500 MHz, DMSO-d6) δ 6.99 (d, J = 7.9 Hz, 2H), 6.35 (d, J = 7.7 Hz, 2H), 4.38 (s, 2H).; 13C NMR (125.8 MHz, DMSO-d6) δ 145.8, 132.3, 113.3; 19F NMR (470.8 MHz, DMSO-d6) δ – 137.7; 11B NMR (128.4 MHz, DMSO-d6) δ 3.7.
Potassium Trifluoro(p-tolyl)borate (1f).18b
Following the general procedure, a mixture of 4-bromotoluene (0.51 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 84% yield (0.50 g, 2.52 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.21 (d, J = 7.4 Hz, 2H), 6.89 (d, J = 7.3 Hz, 2H), 2.20 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 158.0, 132.9, 112.5, 55.1; 19F NMR (470.8 MHz, DMSO-d6) δ – 138.6; 11B NMR (128.4 MHz, DMSO-d6) δ 3.5.
Potassium Trifluoro(o-tolyl)borate (1g).18b
Following the general procedure, a mixture of 2-bromotoluene (0.51 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 3 h. The title compound was obtained in 72% yield (0.43 g, 2.16 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.32 (d, J = 6.8 Hz, 1H), 7.03 – 6.78 (m, 3H), 2.29 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 140.8, 132.0, 128.5, 125.4, 123.7, 22.0; 19F NMR (470.8 MHz, DMSO-d6) δ – 137.5; 11B NMR (128.4 MHz, DMSO-d6) δ 4.4.
Potassium [1,1'-Biphenyl]-4-yltrifluoroborate (1h).23
Following the general procedure, a mixture of 4-bromo-1,1'-biphenyl (0.7 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 90% yield (0.70 g, 2.7 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.60 (d, J = 7.8 Hz, 2H), 7.45 – 7.40 (m, 6H), 7.29 (t, J = 7.3 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 142.1, 137.5, 132.6, 129.4, 127.1, 126.9, 125.3; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.0; 11B NMR (128.4 MHz, DMSO-d6) δ 2.8.
Potassium Trifluoro(naphthalen-1-yl)borate (1i).23
Following the general procedure, a mixture of 1-bromonaphthalene (0.62 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 93% yield (0.65 g, 2.79 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.39 (d, J = 8.1 Hz, 1H), 7.70 (m, 1H), 7.58 – 7.53 (m, 2H), 7.36 – 7.18 (m, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 137.0, 133.4, 130.7, 128.9, 127.8, 125.6, 125.3, 124.3, 123.8; 19F NMR (470.8 MHz, DMSO-d6) δ – 135.2; 11B NMR (128.4 MHz, DMSO-d6) δ 3.6.
Potassium Trifluoro(naphthalen-2-yl)borate (1j).24
Following the general procedure, a mixture of 2-bromonaphthalene (10.0 g, 48.3 mmol), NiCl2(dppp) (27 mg, 0.05 mmol, 0.1 mol %), PPh3 (26 mg, 0.10 mmol, 0.2 mol %), (HO)2B-B(OH)2 (6.5 g, 72.5 mmol, 1.5 equiv) and DIPEA (25.2 mL, 144.9 mmol, 3 equiv) in EtOH (97 mL) was stirred at rt for 8 h. The title compound was obtained in 81% yield (9.2 g, 39.1 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.78 (s, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.38 – 7.28 (m, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 133.4, 132.5, 131.2, 130.1, 127.9, 127.6, 125.5, 125.0, 124.5; 19F NMR (470.8 MHz, Acetone-d6) δ – 141.9; 11B NMR (128.4 MHz, Acetone -d6) δ 4.4.
Potassium (4-Cyanophenyl)trifluoroborate (2a).18b
Following the general procedure, a mixture of 4-bromobenzonitrile (0.55 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 91% yield (0.57 g, 2.73 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.53 (brs, 4 H); 13C NMR (125.8 MHz, DMSO-d6) δ 132.4, 132.4, 130.4, 120.4, 108.2; 19F NMR (470.8 MHz, DMSO-d6) δ – 140.5; 11B NMR (128.4 MHz, DMSO-d6) δ 2.9.
Potassium (3-Cyanophenyl)trifluoroborate (2b). 18b
Following the general procedure, a mixture of 3-bromobenzonitrile (0.55 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 78% yield (0.49 g, 2.34 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1H), 7.58 (s, 1H), 7.48 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 136.5, 135.1, 129.4, 127.9, 120.6, 109.9; 19F NMR (470.8 MHz, DMSO-d6) δ – 140.3; 11B NMR (128.4 MHz, DMSO-d6) δ 2.8.
Potassium (4-Acetylphenyl)trifluoroborate (2c). 18b
Following the general procedure, a mixture of 4’-bromoacetophenone (0.60 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 93 % yield (0.63 g, 2.79 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.74 (d, J = 7.6 Hz, 2H), 7.50 (d, J = 7.7 Hz, 2H), 2.51 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 198.6, 134.7, 131.8, 126.7, 26.9; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.8; 11B NMR (128.4 MHz, DMSO-d6) δ 3.0.
Potassium (3-Acetylphenyl)trifluoroborate (2d).25
Following the general procedure, a mixture of 3’-bromoacetophenone (0.60 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 81% yield (0.55 g, 2.43 mmol) as a white solid, mp 180 – 182 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.94 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.58 (d, J = 7.1 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 2.51 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 199.5, 137.0, 135.8, 131.9, 127.2, 125.7, 27.2; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.5; 11B NMR (128.4 MHz, DMSO-d6) δ 3.2.
Potassium Trifluoro(4-(methoxycarbonyl)phenyl)borate (2e). 18b
Following the general procedure, a mixture of methyl 4-bromobenzoate (0.65 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 50 °C for 6 h. The title compound was obtained in 92 % yield (0.67 g, 2.76 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.75 (d, J = 7.0 Hz, 2H), 7.50 (d, J = 6.8 Hz, 2H), 3.80 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 167.5, 131.8, 127.6, 126.9, 52.0; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.9; 11B NMR (128.4 MHz, DMSO-d6) δ 3.1.
Potassium Trifluoro(4-formylphenyl)borate (2f).26
Following the general procedure, a mixture of 4-bromobenzaldehyde (0.56 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 84% yield (0.53 g, 2.52 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H), 7.67 (d, J = 7.3 Hz, 2H), 7.58 (d, J = 7.2 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 193.8, 134.4, 132.2, 128.2; 19F NMR (470.8 MHz, DMSO-d6) δ – 140.0; 11B NMR (128.4 MHz, DMSO-d6) δ 3.2.
Potassium Trifluoro(4-(trifluoromethyl)phenyl)borate (2g). 18b
Following the general procedure, a mixture of 4-bromobenzotrifluoride (0.68 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 86% yield (0.65 g, 2.58 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.57 (d, J = 7.5 Hz, 2H), 7.44 (d, J = 7.5 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 132.1, 126.5, 125.3 (d, J = 238.6 Hz), 123.1 (d, J = 3.7 Hz); 19F NMR (470.8 MHz, DMSO-d6) δ – 60.6, – 140.0; 11B NMR (128.4 MHz, DMSO-d6) δ 3.0.
Potassium Trifluoro(2-(trifluoromethyl)phenyl)borate (2h).12f
Following the general procedure, a mixture of 2-bromobenzotrifluoride (0.68 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 12 h. The title compound was obtained in 71% yield (0.54 g, 2.13 mmol) as a white solid, mp 200 – 202 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.67 (d, J = 7.3 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.34 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 134.5 (d, J = 3.8 Hz), 130.1, 125.8, 124.7, 124.5 (d, J = 6.7 Hz); 19F NMR (470.8 MHz, DMSO-d6) δ – 57.5 (q, J = 9.3 Hz), −137.1; 11B NMR (128.4 MHz, DMSO-d6) δ 2.5.
Potassium Trifluoro(4-fluorophenyl)borate (2i).18b
Following the general procedure, a mixture of 1-bromo-4-fluorobenzene (0.53 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 2 h. The title compound was obtained in 96% yield (0.58 g, 2.88 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.38 (t, J = 7.4 Hz, 2H), 6.90 (t, J = 9.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 161.4 (d, J = 238.8 Hz), 133.2 (d, J = 6.5 Hz), 113.1 (d, J = 18.4 Hz); 19F NMR (470.8 MHz, DMSO-d6) δ – 118.3, – 138.8; 11B NMR (128.4 MHz, DMSO-d6) δ 3.2.
Potassium Trifluoro(2-fluorophenyl)borate (2j).12e
Following the general procedure, a mixture of 1-bromo-2-fluorobenzene (0.53 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 2 h. The title compound was obtained in 75% yield (0.45 g, 2.25 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.34 (dd, J = 12.0, 6.2 Hz, 1H), 7.07 (dd, J = 13.2, 5.9 Hz, 1H), 6.91 (t, J = 7.1 Hz, 1H), 6.79 (t, J = 8.6 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 166.0 (d, J = 238.8 Hz), 134.5 (d, J = 10.9 Hz), 129.1 (d, J = 641.3 Hz), 127.5 (d, J = 8.0 Hz), 122.8, 114.0 (d, J = 25.9 Hz); 19F NMR (470.8 MHz, DMSO-d6) δ – 107.0, – 137.12 (dd, J = 92.8, 39.0 Hz); 11B NMR (128.4 MHz, DMSO-d6) δ 2.51 (dd, J = 99.1, 49.2 Hz).
Potassium Trifluoro(thien-3-yl)borate (3a).19a
Following the general procedure, a mixture of 3-bromothiophene (0.49 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 94% yield (0.54 g, 2.82 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.18 (s, 1H), 7.11 – 6.88 (m, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 132.2, 124.7, 122.9; 19F NMR (470.8 MHz, DMSO-d6) δ – 135.5 (d, J = 58.2 Hz); 11B NMR (128.4 MHz, DMSO-d6) δ 2.6 (d, J = 47.7 Hz).
Potassium Trifluoro(5-methylthien-2-yl)borate (3b)
Following the general procedure, a mixture of 2-bromo-5-methylthiophene (0.53 g, 3 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 0.5 mol %), PPh3 (7.9 mg, 0.03 mmol, 1 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 85% yield (0.52 g, 2.55 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 6.58 – 6.53 (m, 2H), 2.35 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 137.2, 127.1, 125.2, 15.2; 19F NMR (470.8 MHz, DMSO-d6) δ – 134.0 (m); 11B NMR (128.4 MHz, DMSO-d6) δ 2.3; IR (neat) 1472, 1222, 1146, 960, 899, 879, 801 cm−1; HRMS (ESI) m/ z calcd. For C5H5BSF3 (M)− 165.0157, found 165.0152.
Potassium Benzo[b]thien-2-yltrifluoroborate (3c).19a
Following the general procedure, a mixture of 2-bromo-1-benzothiophene (0.64 g, 3 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 0.5 mol %), PPh3 (7.9 mg, 0.03 mmol, 1 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 74% yield (0.53 g, 2.22 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.83 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.19 – 7.17 (m, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 141.9, 141.5, 123.9, 123.4, 122.6, 122.5, 122.3; 19F NMR (470.8 MHz, DMSO-d6) δ – 134.8; 11B NMR (128.4 MHz, DMSO-d6) δ 2.4.
Potassium Trifluoro(furan-3-yl)borate (3d).27
Following the general procedure, a mixture of 3-bromofuran (0.44 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 92% yield (0.48 g, 2.76 mmol) as a light yellow tan solid, mp 175 – 177 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.35 (s, 1H), 7.09 (s, 1H), 6.19 (s, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 143.0, 140.7, 114.1; 19F NMR (470.8 MHz, DMSO-d6) δ –134.6 (d, J = 58.2 Hz); 11B NMR (128.4 MHz, DMSO-d6) δ 3.1.
Potassium Benzofuran-5-yltrifluoroborate (3e)
Following the general procedure, a mixture of 5-bromobenzofuran (0.59 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 91% yield (0.61 g, 2.73 mmol) as an off white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.76 (m, 1H), 7.57 (m, 1H), 7.31 – 7.28 (m, 2H), 6.78 (m, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 153.8, 144.3, 128.4, 126.1, 123.9, 109.1, 106.9; 19F NMR (470.8 MHz, DMSO-d6) δ –138.1; 11B NMR (128.4 MHz, DMSO-d6) δ 3.5; IR (neat) 1545, 1380, 1265, 1152, 904 cm−1; HRMS (ESI) m/ z calcd. For C8H5BOF3 (M)−185.0386, found 185.0381.
Potassium Trifluoro(1-methyl-1H-pyrazol-4-yl)borate (3f)
Following the general procedure, a mixture of 4-bromo-1-methyl-1H–pyrazole (0.48 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 12 h. The title compound was obtained in 82% yield (0.46 g, 2.46 mmol) as a white solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.06 (s, 1H), 7.04 (s, 1H), 3.69 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 142.4, 132.4, 38.1; 19F NMR (470.8 MHz, DMSO-d6) δ –132.9; 11B NMR (128.4 MHz, DMSO-d6) δ 3.0. IR (neat) 1546, 1172, 944, 906, 830 cm−1; HRMS (ESI) m/ z calcd. For C4H5BN2F3 (M)− 149.0498, found 149.0491.
Potassium Trifluoro(1H-indol-5-yl)borate (3g).19a
Following the general procedure, a mixture of 5-bromo-1H-indole (0.59 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 6 h. The title compound was obtained in 86% yield (0.57 g, 2.58 mmol) as a white solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, acetone-d6) δ 9.75 (s, 1H), 7.74 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 7.15 – 7.06 (m, 1H), 6.31 (d, J = 1.9 Hz, 1H); 13C NMR (125.8 MHz, acetone-d6) δ 135.5, 127.5, 125.7, 123.0, 122.6, 109.1, 101.0; 19F NMR (470.8 MHz, Acetone -d6) δ –140.5; 11B NMR (128.4 MHz, acetone -d6) δ 5.3.
Potassium Trifluoro(2-methylpyridin-4-yl)borate (3h)
Following the general procedure, a mixture of 4-bromo-2-methylpyridine (0.52 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 72% yield (0.43 g, 2.16 mmol) as a light yellow tan solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.14 (d, J = 4.4 Hz, 1H), 7.14 (s, 1H), 7.05 (d, J = 4.2 Hz, 1H), 2.36 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 155.3, 147.0, 126.6, 124.4, 24.4; 19F NMR (470.8 MHz, DMSO-d6) δ –140.8; 11B NMR (128.4 MHz, DMSO-d6) δ 2.7; IR (neat) 1537, 1380, 1259, 1170, 966, 833 cm−1; HRMS (ESI) m/ z calcd. For C6H6BNF3 (M)− 160.0545, found 160.0546.
Potassium Trifluoro(quinolin-4-yl)borate (3i)
Following the general procedure, a mixture of 4-bromoquinoline (0.62 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 74% yield (0.52 g, 2.22 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.64 (m, 1H), 8.39 (m, 1H), 7.86 (m, 1H), 7.57 (m, 1H), 7.50 – 7.33 (m, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 149.5, 147.8, 132.4, 130.7, 129.1, 127.8, 124.5, 124.4; 19F NMR (470.8 MHz, DMSO-d6) δ – 136.6; 11B NMR (128.4 MHz, DMSO-d6) δ 3.1; IR (neat) 1238, 1090, 1055, 998, 935, 846, 750 cm−1; HRMS (ESI) m/ z calcd. For C9H6BNF3 (M)− 196.0545, found 196.0540.
Potassium Trifluoro(quinolin-5-yl)borate (3j)
Following the general procedure, a mixture of 5-bromoquinoline (0.62 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 81% yield (0.57 g, 2.43 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.74 – 8.71 (m, 2H), 7.72 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 6.5 Hz, 1H), 7.51 (m, 1H), 7.32 (m, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 148.9, 148.5, 138.4, 131.5, 129.1, 128.7, 126.7, 119.7; 19F NMR (470.8 MHz, DMSO-d6) δ – 135.0; 11B NMR (128.4 MHz, DMSO-d6) δ 3.3; IR (neat) 1546, 1172, 968, 906 cm−1; HRMS (ESI) m/ z calcd. For C9H6 BNF3 (M)− 196.0545, found 196.0538.
Potassium Trifluoro(quinolin-6-yl)borate (3k)
Following the general procedure, a mixture of 6-bromoquinoline (0.62 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 83% yield (0.58 g, 2.49 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.75 (m, 1H), 8.21 (d, J = 8.2 Hz, 1H), 7.88 (s, 1H), 7.85 – 7.76 (m, 2H), 7.38 (m, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 148.9, 147.7, 135.9, 134.6, 130.0, 127.8, 126.5, 120.6; 19F NMR (470.8 MHz, DMSO-d6) δ – 139.1; 11B NMR (128.4 MHz, DMSO-d6) δ 3.3; IR (neat) 1569, 1344, 1170, 984, 836, 650 cm−1; HRMS (ESI) m/ z calcd. For C9H6BF3N (M)− 196.0545, found 196.0543.
Potassium Trifluoro(1H-pyrrolo[2,3-b]pyridin-5-yl)borate (3l)
Following the general procedure, a mixture of 5-bromo7-azaindole (0.59 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 83% yield (0.56 g, 2.49 mmol) as a yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 11.05 (s, 1H), 8.17 (s, 1H), 7.79 (s, 1H), 7.22 (d, J = 3.3 Hz, 1H), 6.26 (d, J = 3.3 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 148.1, 147.1, 131.0, 124.0, 119.3, 99.5; 19F NMR (470.8 MHz, DMSO-d6) δ – 137.3; 11B NMR (128.4 MHz, DMSO-d6) δ 4.0; IR (neat) 1276, 1144, 903, 668 cm−1; HRMS (ESI) m/ z calcd. For C9H6BNF3 (M)− 185.0498, found 185.0498.
Thien-3-ylboronic acid (4a).28
Following the general procedure, a mixture of 3-bromothiophene (0.49 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The crude reaction was transferred to a separatory funnel followed by the addition of EtOAc (10 mL) and then aq HCl (20 mL of a 1 M solution). The layers were separated, and the water layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were dried (Na2SO4), concentrated, and lyophilized overnight. The crude solid was washed with hexane to afford the title compound in 87% yield (0.33 g, 2.61 mmol) as a white solid, mp 125 – 127 °C (lit. 126 – 128 °C). 1H NMR (500 MHz, DMSO-d6) δ 7.96 (d, J = 2.5 Hz, 1H), 7.46 (dd, J = 4.7, 2.7 Hz, 1H), 7.41 (d, J = 4.8 Hz, 1H); 13C NMR (125.8 MHz, DMSO-d6) δ 135.2, 132.8, 125.5; 11B NMR (128.4 MHz, acetone-d6) δ 27.8.
4,4,5,5-Tetramethyl-2-(thien-3-yl)-1,3,2-dioxaborolane (4b). 29
Following the general procedure, a mixture of 3-bromothiophene (0.49 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The crude reaction was transferred to a separatory funnel followed by the addition of EtOAc (10 mL) and then aq HCl (20 mL of a 1 M solution). The layers were separated and the water layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated. To the crude mixture was added CH2Cl2 (10 mL) and pinacol (1.06 g, 9 mmol, 3 equiv). The reaction was stirred for 2 h. The crude mixture was transferred to a separatory funnel followed by the addition of H2O (20 mL). The layers were separated, and the water layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated. The title compound was obtained after column chromatography (hexane/EtOAc, 3:1) in 90% yield (0.57 g, 2.7 mmol) as a colorless oil. 1H NMR (500 MHz, acetone-d6) δ 7.95 (m, 1H), 7.45 (m, 1H), 7.37 (m, 1H), 1.31 (s, 12H); 13C NMR (125.8 MHz, acetone -d6) δ 136.4, 132.1, 125.6, 83.6, 24.5; 11B NMR (128.4 MHz, acetone-d6) δ 28.8.
5,5-Dimethyl-2-(thien-3-yl)-1,3,2-dioxaborinane (4c).30
Following the general procedure, a mixture of 3-bromothiophene (0.49 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The crude reaction was transferred to a separatory funnel followed by the addition of EtOAc (10 mL) and then aq HCl (20 mL of a 1 M solution). The layers were separated and the water layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated. To the crude mixture was added CH2Cl2 (10 mL) and 2,2-dimethyl-1,3-propanediol (0.94 g, 9 mmol, 3 equiv). The reaction was stirred for 2 h. The crude mixture was transferred to a separatory funnel followed by the addition of H2O (20 mL). The layers were separated, and the water layer extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated. The title compound was obtained after column chromatography (hexane/ EtOAc, 3:1) in 96% yield (0.56 g, 2.88 mmol) as a white solid, mp 125 – 127 °C. 1H NMR (500 MHz, CDCl3) δ 7.85 (m, 1H), 7.39 (m, 1H), 7.32 (m, 1H), 3.76 (s, 4H), 1.03 (s, 6H); 13C NMR (125.8 MHz, CDCl3) δ 134.8, 131.6, 124.9, 72.1, 31.8, 21.8; 11B NMR (128.4 MHz, CDCl3) δ 25.1.
Potassium (4-Cyanophenyl)trifluoroborate (2a, Table 5, entry 1).18b
Following the general procedure, a mixture of 4-chlorobenzonitrile (0.41 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 92% yield (0.58 g, 2.76 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.48 (m, 4 H).
Potassium (4-Acetylphenyl)trifluoroborate (2c, Table 5, entry 2).18b
Following the general procedure, a mixture of 4’-chloroacetophenone (0.46 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 89% yield (0.60 g, 2.67 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.69 (d, J = 7.9 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 2.49 (s, 3H).
Potassium Trifluoro(4-(methoxycarbonyl)phenyl)borate (2e, Table 5, entry 3).18b
Following the general procedure, a mixture of methyl 4-bromobenzoate (0.65 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 89% yield (0.65 g, 2.67 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.71 (d, J = 7.7 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H), 3.79 (s, 3H).
Potassium Trifluoro(4-(morpholine-4-carbonyl)phenyl)borate (5a, Table 5, entry 4).18b
Following the general procedure, a mixture of (4-chlorophenyl)(morpholino)methanone (0.68 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 12 h. The title compound was obtained in 64% yield (0.57 g, 1.92 mmol) as an off white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.41 (d, J = 7.0 Hz, 1H), 7.16 (d, J = 7.1 Hz, 1H), 3.58 (brs, 8H); 13C NMR (125.8 MHz, DMSO-d6) δ 170.7, 132.5, 131.5, 125.6, 66.6; 19F NMR (470.8 MHz, DMSO-d6) δ –139.6; 11B NMR (128.4 MHz, DMSO-d6) δ 3.2.
Potassium Trifluoro(4-(piperazin-1-yl)phenyl)borate (5b, Table 5, entry 5)
Following the general procedure, a mixture of 1-(4-chlorophenyl)piperazine (0.59 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 8 h. The title compound was obtained in 52% yield (0.42 g, 1.56 mmol) as an off white solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.16 (d, J = 7.5 Hz, 2H), 6.66 (d, J = 7.5 Hz, 2H), 2.91 (brs, 4H), 2.80 (brs, 4H); 13C NMR (125.8 MHz, DMSO-d6) δ 150.0, 132.2, 114.7, 50.8, 46.2; 19F NMR (470.8 MHz, DMSO-d6) δ –138.2; 11B NMR (128.4 MHz, DMSO-d6) δ 3.6; IR (neat) 1545, 1173, 968, 906 cm−1; HRMS (ESI) m/ z calcd. For C10H13BN2F3 (M)− 229.1124, found 229.1129.
Potassium Trifluoro(4-fluorophenyl)borate (2i, Table 5, entry 6).18b
Following the general procedure, a mixture of 1-chloro-4-fluorobenzene (0.39 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 12 h. The title compound was obtained in 83% yield (0.50 g, 2.49 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.32 (t, J = 7.5 Hz, 1H), 6.85 (dd, J = 9.3, 8.7 Hz, 1H).
Potassium Trifluoro(4-methoxyphenyl)borate (1a, Table 5, entry 7).18b
Following the general procedure, a mixture of 4-chloroanisole (0.43 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 12 h. The title compound was obtained in 66% yield (0.42 g, 1.98 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.20 (d, J = 8.2 Hz, 2H), 6.64 (d, J = 8.0 Hz, 2H), 3.65 (s, 3H).
Potassium (4-(1H-Pyrrol-1-yl)phenyl)trifluoroborate (5c, Table 5, entry 8).18b
Following the general procedure, a mixture of 1-(4-chlorophenyl)-1H–pyrrole (0.53 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 2 h. The title compound was obtained in 69% yield (0.57 g, 2.28 mmol) as white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.42 – 7.38 (m, 2H), 7.27 – 7.22 (m, 4H), 6.23 – 6.19 (m, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 138.0, 132.8, 119.1, 118.1, 110.0; 19F NMR (470.8 MHz, DMSO-d6) δ –139.0; 11B NMR (128.4 MHz, DMSO-d6) δ 3.5.
Potassium Trifluoro(quinolin-5-yl)borate (3j, Table 5, entry 9)
Following the general procedure, a mixture of 5-chloroquinoline (0.49 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 12 h. The title compound was obtained in 75% yield (0.53 g, 2.25 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.75 – 8.70 (m, 2H), 7.70 (d, J = 8.0 Hz, 1H), 7.58 (m, 1H), 7.51 (m, 1H), 7.32 (m, 1H). HRMS (ESI) m/ z calcd. For C9H6BNF3 (M)− 196.0545, found 196.0545.
Potassium Trifluoro(quinolin-4-yl)borate (3i, Table 5, entry 10)
Following the general procedure, a mixture of 4-chloroquinoline (0.49 g, 3 mmol), NiCl2(dppp) (81.3 mg, 0.15 mmol, 5 mol %), PPh3 (78.7 mg, 0.3 mmol, 10 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at 80 °C for 12 h. The title compound was obtained in 58% yield (0.41 g, 1.74 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.61 (d, J = 4.0 Hz, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.53 (m, 1H), 7.43 (d, J = 3.8 Hz, 1H), 7.37 (m, 1H). HRMS (ESI) m/ z calcd. For C9H6BNF3 (M)− 196.0545, found 196.0540.
Potassium Trifluoro(thien-3-yl)borate (3a, Table 5, entry 11).19a
Following the general procedure, a mixture of 3-chlorothiophene (0.36 g, 3 mmol), NiCl2(dppp) (16.3 mg, 0.03 mmol, 1 mol %), PPh3 (15.7 mg, 0.06 mmol, 2 mol %), (HO)2B-B(OH)2 (0.4 g, 4.5 mmol, 1.5 equiv) and DIPEA (1.6 mL, 9 mmol, 3 equiv) in EtOH (10 mL) was stirred at rt for 4 h. The title compound was obtained in 86% yield (0.49 g, 2.58 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.14 (m, 1H), 7.02 – 6.93 (m, 2H).
Potassium (4-Cyanophenyl)trifluoroborate (2a, Table 7, entry 1).18b
Following the general procedure, a mixture of 4-cyanophenyl methanesulfonate (0.30 g, 1.5 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 1 mol %), PPh3 (7.9 mg, 0.03 mmol, 2 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 50 °C for 4 h. The title compound was obtained in 82% yield (0.26 g, 1.23 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.72 – 7.36 (m, 4H).
Potassium (4-Acetylphenyl)trifluoroborate (2c, Table 7, entry 2).18b
Following the general procedure, a mixture of 4-acetylphenyl methanesulfonate (0.32 g, 1.5 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 1 mol %), PPh3 (7.9 mg, 0.03 mmol, 2 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 50 °C for 4 h. The title compound was obtained in 82% yield (0.30 g, 1.32 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.74 (d, J = 7.6 Hz, 2H), 7.50 (d, J = 7.7 Hz, 2H), 2.51 (s, 3H).
Potassium Trifluoro(4-(methoxycarbonyl)phenyl)borate (2e, Table 7, entry 3).18b
Following the general procedure, a mixture of methyl 4-((methylsulfonyl)oxy)benzoate (0.34 g, 1.5 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 1 mol %), PPh3 (7.9 mg, 0.03 mmol, 2 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 80 °C for 4 h. The title compound was obtained in 71% yield (0.26 g, 1.06 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.71 (d, J = 7.4 Hz, 2H), 7.44 (d, J = 7.0 Hz, 2H), 3.79 (s, 3H).
Potassium Trifluoro(4-(trifluoromethyl)phenyl)borate (2g, Table 7, entry 4).18b
Following the general procedure, a mixture of 4-(trifluoromethyl)phenyl methanesulfonate (0.31 g, 1.5 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 1 mol %), PPh3 (7.9 mg, 0.03 mmol, 2 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 50 °C for 12 h. The title compound was obtained in 81% yield (0.31 g, 1.21 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.52 (d, J = 7.4 Hz, 2H), 7.41 (d, J = 7.5 Hz, 2H).
Potassium Trifluoro(naphthalen-1-yl)borate (1i, Table 7, entry 5).23
Following the general procedure, a mixture of naphthalen-1-yl methanesulfonate (0.33 g, 1.5 mmol), NiCl2(dppp) (8.1 mg, 0.015 mmol, 1 mol %), PPh3 (7.9 mg, 0.03 mmol, 2 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 50 °C for 8 h. The title compound was obtained in 87% yield (0.30 g, 1.3 mmol) as a white solid, mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.38 (d, J = 7.8 Hz, 1H), 7.73 – 7.65 (m, 1H), 7.54 (dd, J = 14.6, 7.3 Hz, 2H), 7.38 – 7.21 (m, 3H).
Potassium Trifluoro(quinolin-5-yl)borate (3j, Table 7, entry 6)
Following the general procedure, a mixture of quinolin-5-yl methanesulfonate (0.33 g, 1.5 mmol), NiCl2(dppp) (40.7 mg, 0.075 mmol, 5 mol %), PPh3 (39.4 mg, 0.15 mmol, 10 mol %), (HO)2B-B(OH)2 (0.2 g, 2.25 mmol, 1.5 equiv) and DIPEA (0.8 mL, 4.5 mmol, 3 equiv) in EtOH (5 mL) was stirred at 80 °C for 8 h. The title compound was obtained in 76% yield (0.27 g, 1.14 mmol) as a light yellow solid (treatment with K2CO3 needed), mp > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.71 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.1 Hz, 1H), 7.58 (d, J = 6.1 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.31 (dd, J = 7.8, 4.2 Hz, 1H). HRMS (ESI) m/ z calcd. For C9H6BNF3 (M)− 196.0545, found 196.0548.
Supplementary Material
Acknowledgment
This work was generously supported by the NIGMS (R01 GM081376), NSF (GOALI), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Graduate Research Fellowship to L.N.C. We also acknowledge AllyChem for their donation of tetrahydroxydiboron reagent. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining the HRMS data.
Footnotes
Supporting Information Available: Copies of 1H, 13C, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hall DG, editor. Boronic Acids. Weinheim, Germany: Wiley-VCH; 2011. [Google Scholar]
- 2.Candeias N, Montalbano F, Cal PMSD, Gois P. Chem. Rev. 2010;110:6169. doi: 10.1021/cr100108k. [DOI] [PubMed] [Google Scholar]
- 3.Qiao J, Lam P. Synthesis. 2011;6:829. [Google Scholar]
- 4.Berthon-Gelloz G, Hayashi T. Rhodium- and Palladium-catalyzed Asymmetric Conjugate Addition of Organoboronic Acid. In: Hall, editor. Boronic Acids. Weinheim, Germany: Wiley-VCH; 2011. [Google Scholar]
- 5.For the Nobel Prize lecture see:Suzuki A. Angew. Chem. Int. Ed. 2011;50:6722. doi: 10.1002/anie.201101379.
- 6.Yang W, Gao W, Wang B. Biological and Medicinal Applications of Boronic Acids. In: Hall, editor. Boronic Acids. Weinheim, Germany: Wiley-VCH; 2011. [Google Scholar]
- 7.Suzuki A, Brown HC. Organic Syntheses via Boranes. Vol. 3 Milwaukee: Aldrich Chemical Company; 2003. [Google Scholar]
- 8.For recent examples see:Hartsel JA, Craft DT, Chen Q-H, Ma M, Carlier PR. J. Org. Chem. 2012;77:3127. doi: 10.1021/jo202371c. Chmiel J, Heesemann I, Mix A, Neumann B, Stammler H-G, Mitzel NW. Eur. J. Org. Chem. 2010:3897. Pei T, Tellers DM, Streckfuss EC, Chen C-Y, Davies IW. Tetrahedron. 2009;65:3285.
- 9.For selected examples see:Chen HY, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew. Chem. Int. Ed. 2001;40:2168. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0.
- 10.For selected examples see:Cho JY, Tse MK, Holmes D, Maleczka RE, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649. Ishiyama T, Takag J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2002;124:390. doi: 10.1021/ja0173019. Ishiyama T, Takagi J, Yonekawa Y, Hartwig JF, Miyaura N. Adv. Synth. Catal. 2003;345:1103. Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. Murphy JM, Liao X, Hartwig JF. J. Am. Chem. Soc. J. Am. Chem. Soc. 2005;2007;127129:14263. 15434. doi: 10.1021/ja053433g. Tzschucke CC, Murphy JM, Hartwig JF. Org. Lett. 2007;9:761. doi: 10.1021/ol062902w. Preshlock SM, Ghaffari B, Maligres PE, Krska SW, Maleczka RE, Jr, Smith MR., III J. Am. Chem. Soc. 2013;135:7572. doi: 10.1021/ja400295v.
- 11.(a) Zhu W, Ma D. Org. Lett. 2006;8:261. doi: 10.1021/ol052633u. [DOI] [PubMed] [Google Scholar]; (b) Kleeberg C, Dang L, Lin Z, Marder TB. Angew. Chem. Int. Ed. 2009;48:5350. doi: 10.1002/anie.200901879. [DOI] [PubMed] [Google Scholar]; (c) Yan G, Yang M, Yu J. Lett. Org. Chem. 2012;9:71. [Google Scholar]
- 12.(a) Rosen BM, Huang C, Percec V. Org. Lett. 2008;10:2597. doi: 10.1021/ol800832n. [DOI] [PubMed] [Google Scholar]; (b) Leowanawat P, Resmerita A-M, Moldoveanu C, Liu C, Zhang N, Wilson DA, Hoang LM, Rosen BM, Percec V. J. Org. Chem. 2010;75:7822. doi: 10.1021/jo101718v. [DOI] [PubMed] [Google Scholar]; (c) Wilson DA, Wilson CJ, Rosen BM, Percec V. Org. Lett. 2008;10:4879. doi: 10.1021/ol801972f. [DOI] [PubMed] [Google Scholar]; (d) Moldoveanu C, Wilson DA, Wilson CJ, Corcoran P, Rosen BM, Percec V. Org. Lett. 2009;11:4974. doi: 10.1021/ol902155e. [DOI] [PubMed] [Google Scholar]; (e) Wilson DA, Wilson CJ, Moldoveanu C, Resmerita A-M, Corcoran P, Hoang LM, Rosen BM, Percec V. J. Am. Chem. Soc. 2010;132:1800. doi: 10.1021/ja910808x. [DOI] [PubMed] [Google Scholar]; (f) Moldoveanu C, Wilson DA, Wilson CJ, Leowanawat P, Resmerita A-M, Liu C, Rosen BM, Percec V. J. Org. Chem. 2010;75:5438. doi: 10.1021/jo101023t. [DOI] [PubMed] [Google Scholar]; (g) Yamamoto T, Morita T, Yamakawa T. Org. Lett. 2011;13:5766. doi: 10.1021/ol202267t. [DOI] [PubMed] [Google Scholar]; (h) Huang K, Yu D-G, Zheng S-F, Wu Z-H, Shi Z-J. Chem. Eur. J. 2011;17:786. doi: 10.1002/chem.201001943. [DOI] [PubMed] [Google Scholar]
- 13.For selected examples see:Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508. Ishiyama T, Ishida K, Miyaura N. Tetrahedron. 2001;57:9813. Furstner A, Seidel G. Org. Lett. 2002;4:541. doi: 10.1021/ol0171463. Murata M, Sambommatsu T, Watanabe S, Masuda Y. Synlett. 2006:1867. Billingsley KL, Barder TE, Buchwald SL. Angew. Chem. Int. Ed. 2007;46:5359. doi: 10.1002/anie.200701551. Barder TE, Buchwald SL. J. Org. Chem. 2008;73:5589. doi: 10.1021/jo800727s. Kawamorita S, Ohmiya H, Iwai T, Sawamura M. Angew. Chem. Int. Ed. 2011;50:8363. doi: 10.1002/anie.201103224. Tang W, Keshipeddy S, Zhang Y, Wei X, Savoie J, Patel ND, Yee NK, Senanayake CH. Org. Lett. 2011;13:1366. doi: 10.1021/ol2000556. Lu J, Guan Z-Z, Gao J-W, Zhang Z-H. Appl. Organometal. Chem. 2011;25:537. Murata M, Watanabe S, Masuda Y. J. Org. Chem. 1997;62:6458. doi: 10.1021/jo971143f.
- 14.(a) Jung ME, Lazarova TI. J. Org. Chem. 1999;64:2976. doi: 10.1021/jo9902751. [DOI] [PubMed] [Google Scholar]; (b) Song YL, Morin C. Synlett. 2001:266. [Google Scholar]; (c) Ma D, Wu Q. Tetrahedron Lett. 2001;42:5279. [Google Scholar]; (d) Zaidlewicz M, Wolan A. J. Organomet. Chem. 2002;657:129. [Google Scholar]
- 15.(a) Nakamura H, Fujiwara M, Yamamoto Y. J. Org. Chem. 1998;63:7529. doi: 10.1021/jo980818r. [DOI] [PubMed] [Google Scholar]; (b) Yu S, Saenz J, Srirangam JK. J. Org. Chem. 2002;67:1699. doi: 10.1021/jo016131f. [DOI] [PubMed] [Google Scholar]; (c) Falck JR, Bondlela M, Venkataraman SK. J. Org. Chem. 2001;66:7148. doi: 10.1021/jo015838z. [DOI] [PubMed] [Google Scholar]; (d) Deng H, Jung JK, Liu T, Kuntz KW, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2003;125:9032. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 16.Yang W, He H, Drueckhammer DG. Angew. Chem. Int. Ed. 2001;40:1714. [PubMed] [Google Scholar]
- 17.(a) Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956. [Google Scholar]; (b) Pennington T, Kardiman C, Hutton C. Tetrahedron Lett. 2004;45:6657. [Google Scholar]; (c) Yuen A, Hutton C. Tetrahedron Lett. 2005;46:7899. [Google Scholar]; (d) Sun J, Perfetti MT, Santos WL. J. Org. Chem. 2011;76:571. doi: 10.1021/jo200250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Molander GA, Trice SLJ, Dreher SD. J. Am. Chem. Soc. 2010;132:17701. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J. Am. Chem. Soc. 2012;134:11667. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Molander GA, Canturk B, Kennedy LE. J. Org. Chem. 2009;74:973. doi: 10.1021/jo802590b. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009;131:6961. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For reviews see:Kirsch P, editor. Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications. Weinheim: Wiley-VCH; 2004. Schlosser M. Angew. Chem. Int. Ed. 2006;45:5432. doi: 10.1002/anie.200600449. Isanbor C, Hagan DO. J. Fluorine Chem. 2006;127:303. Müller K, Faeh C, Diederich F. Science. 2007;317:188. doi: 10.1126/science.1131943. Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c.
- 21.Raushel J, Sandrock DL, Josyula KV, Pakyz D, Molander GA. J. Org. Chem. 2011;76:2762. doi: 10.1021/jo2001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson PG, Percy JM, Redmond JM, McCarter AW. J. Org. Chem. 2012;77:6384. doi: 10.1021/jo3011705. [DOI] [PubMed] [Google Scholar]
- 23.Lennox AJJ, Lloyd-Jones GC. Angew. Chem. Int. Ed. 2012;51:9385. doi: 10.1002/anie.201203930. [DOI] [PubMed] [Google Scholar]
- 24.Navarre L, Darses S, Genet J-P. Eur. J. Org. Chem. 2004:69. [Google Scholar]
- 25.Molander GA, Petrillo DE. J. Am. Chem. Soc. 2006;128:9634. doi: 10.1021/ja062974i. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira RA, Silva RO, Molander GA, Menezes PH. Magn. Reson. Chem. 2009;47:873. doi: 10.1002/mrc.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molander GA, Biolatto B. J. Org. Chem. 2003;68:4302. doi: 10.1021/jo0342368. [DOI] [PubMed] [Google Scholar]
- 28.Molander GA, Cavalcanti LN, Canturk B, Po-Shen P, Kennedy LE. J. Org. Chem. 2009;74:7364. doi: 10.1021/jo901441u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Billingsley KL, Buchwald SL. J. Org. Chem. 2008;73:5589. doi: 10.1021/jo800727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ukai K, Aoki M, Takaya J, Iwasawa N. J. Am. Chem. Soc. 2006;128:8706. doi: 10.1021/ja061232m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.