

Abstract
Small-molecule inhibitors of neurofibrillary lesion formation may have utility for treatment of Alzheimer’s disease and certain forms of frontotemporal lobar degeneration. These lesions are composed largely of tau protein, which aggregates to form intracellular fibrils in affected neurons. Previously it was shown that chronic overexpression of human tau protein within identified neurons (anterior bulbar cells) of the sea lamprey induced a phenotype resembling tauopathic neurodegeneration, including the formation of tau filaments, fragmentation of dendritic arbors, and eventual cell death. Development of this neurodegenerative phenotype was blocked by chronic administration of a benzothiazole derivative termed N3 ((E)-2-[[4-(dimethylamino)phenyl]azo]-6-methoxybenzothiazole) to lamprey aquaria. Here we examined the mechanism of action of N3 and an alkene analog termed N4 ((E)-2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methoxybenzothiazole) in vitro and in the lamprey model. Results showed that although both compounds entered the lamprey central nervous system, only N3 arrested tauopathy. On the basis of in vitro aggregation assays, neither compound was capable of directly inhibiting tau filament formation. However N3, but not N4, was capable of partially antagonizing the binding of Thioflavin S to synthetic tau filaments. The results suggest that occupancy of N3 binding sites on nascent tau filaments may significantly retard the progressive degeneration accompanying tau overexpression in lamprey.
Keywords: Alzheimer’s disease, tau protein, tauopathy, neurofibrillary lesions
Introduction
Alzheimer’s disease (AD) is defined in part by the appearance of neurofibrillary lesions within neuronal cell bodies (neurofibrillary tangles) and processes (neuropil threads and dystrophic neurites). Each manifestation of neurofibrillary pathology is composed of abnormally hyperphosphorylated and aggregated forms of the microtubule-associated protein tau (Buee et al., 2000; Lee et al., 2001). Adult human tau consists of six splice variants, all of which are capable of aggregating in AD (Goedert et al., 1992). Tau lesion formation correlates with cognitive decline in affected individuals (Ghoshal et al., 2002), and is accompanied by the loss of axons, higher order dendrites, dendritic microtubules, and synapses in affected neurons (Kowall and Kosik, 1987; Ihara, 1988; McKee et al., 1989; Yamaguchi et al., 1990), implicating tau aggregation as a marker and potential mediator of AD neurodegeneration (Honson and Kuret, 2008).
To test this hypothesis, individual human tau isoforms have been expressed in biological model systems and examined for their ability to replicate neurofibrillary pathology and associated neurodegenerative changes (Gotz et al., 2007). A particularly robust cellular model has been created in the sea lamprey, Petromyzon marinus, by injecting vectors encoding fluorescently tagged human tau isoforms into the giant anterior bulbar cells (ABCs) of lamprey hindbrain (Hall et al., 1997; Hall et al., 2000; Hall et al., 2001). Injected ABCs express human tau over a period of months, during which time the normal cytoskeleton is gradually replaced by filamentous tau aggregates (Hall et al., 1997; Hall et al., 2000). Simultaneously, the cells progress through a series of degenerative stages that recapitulate in detail the development of AD cytopathology in authentic human tissue, including loss of synapses and formation of dendritic abnormalities (Braak and Braak, 1995; Hall et al., 2001). The pathological cascade can be induced using either the shortest (htau23) or longest (htau40) isoforms of human tau, suggesting that induction of tau aggregation and neurodegeneration is tau isoform independent in this overexpression model (Hall et al., 2001).
Because of its fidelity to the cytopathology of human neurofibrillary disease, its accessibility to in vivo experimental manipulation, and its ability to resolve events at the single cell level, the lamprey model is an excellent biological paradigm for testing the activity of compounds that interfere with tauopathic neurodegeneration. Using this system, a benzothiazole derivative termed N3 ((E)-2-[[4-(dimethylamino)phenyl]azo]-6-methoxybenzothiazole) was found to prevent the progressive degeneration ordinarily produced by chronic human tau overexpression, whereas N4 ((E)-2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methoxybenzothiazole, a closely related structural analog, was ineffective in the same animal model (Hall et al., 2002).
These data illustrate the potential of small molecules to protect against tau-mediated neurodegeneration, but raise the issue of mechanism of action. N3 has been proposed to act at micromolar concentrations as a direct inhibitor of tau fibrillization on the basis of in vitro assays (Kuret and Khatami, 2002). Consistent with this mechanism, other benzothiazole derivatives have been found to directly antagonize aggregation of polyglutamine-containing proteins associated with Huntington’s disease at high concentrations (Heiser et al., 2002). Recently, however, we found that compounds closely related to N3 were capable of displacing the binding of synthetic tau filaments to Thioflavin S (ThS), a probe for cross-β-sheet structure (Friedhoff et al., 1998), but did not directly antagonize tau fibrillization in aggregation assays under identical conditions (Honson et al., 2007). Here we investigate this issue using highly purified preparations of N3 and N4. The results confirm the neuroprotective activity of N3, but suggest that its mechanism of action is not mediated by direct inhibition of tau aggregation.
Experimental Procedures
Materials
Recombinant polyhistidine-tagged htau40 and htau40C291A/C322A proteins were prepared as described previously (Carmel et al., 1996; Gamblin et al., 2000). Dithiothreitol was from Acros Organics (Morris Plains, NJ), octadecyl sodium sulfate (ODS) was from Lancaster (Pelham, NH), and ThS, arachidonic acid, and the ExtrAvidin®/biotin/horseradish peroxidase immunohistochemistry kit were from Sigma-Aldrich (St. Louis, MO). DMSO, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and NaCl were from Fisher Scientific (Pittsburgh, PA). Glutaraldehyde, uranyl acetate, and 300 mesh carbon-coated copper grids were from Electron Microscopy Sciences (Ft. Washington, PA).
Stock solutions of arachidonic acid and ODS were prepared just prior to use in ethanol or 1:1 isopropanol/ddH2O, respectively, whereas the fluorescence reporter ThS was prepared in ddH2O. N3 and N4 were prepared in DMSO as 10 mM and 1 mM stock solutions, respectively, and stored frozen at −20°C until used.
Chemical synthesis
Compounds N3 and N4 were custom synthesized by Medichem, Inc.(Lemont, IL). Compound N3 (Fig. 1) was prepared by adding (10 min at 0°C) concentrated H2SO4 (69.6 ml, 1.30 mol) in aliquots to a suspension of 2-amino-6-methoxybenzothiazole (45.3 g, 0.251 mol) in water (500 ml), followed by drop-wise addition (30 min at 0°C) of NaNO2 (16.4 g, 0.238 mol) in water (157 ml). After 15 min, the resultant precipitate was treated with N,N-dimethylaniline (31.8 ml, 0.251 mol) added dropwise over 1 h with frequent stops for agitating the mixture by hand. The mixture was swirled 15 min and a solution of NaOAc (43.2 g, 0.527 mol) in water (200 ml) was added dropwise over 30 min. The reaction mixture was swirled/stirred for 1.5 h, then basified by addition of 25% (w/v) NaOH (700 ml) over 1 h. The reaction mixture was warmed to room temperature, stirred for 2 h, and then filtered. Collected solids were washed with ice water (3 x 400 ml) and allowed to air dry overnight. Crude product was combined with material from a pilot reaction (~5.5 g) and the resulting solids were dissolved in acetone (300 ml) and coated onto silica gel. The crude product eluted with CH2Cl2 to afford 20.8 g (29% yield) of N3 as a deep red solid, mp 236–239 °C. 1H NMR (500 MHz, CDCl3) δ 3.13 (s, 6H), 3.89 (s, 3H), 6.74 (d, J = 9.0 Hz, 1H), 7.06 (dd, J = 2.5, 8.5 Hz, 1H), 7.27 (dd, J = 2.5, 8.5 Hz, 1H), 7.96 (dd, J = 5.5, 9.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 40.23, 55.74, 104.53, 111.54, 115.47, 124.72, 127.02, 135.70, 142.87, 147.41, 153.84, 158.59, 175.10. IR (ATR) 3074-2830, 1597, 1496, 1359, 1338, 1258, 1228 cm−1. MS (ESII+); [M+H]+ 313.0. Anal. Calcd. for C16H16N4OS: C, 61.52; H, 5.16; N, 17.93; S, 10.26. Found: C, 61.28; H, 5.18; N, m17.99; S, 9.87.
Fig. 1. Synthetic schemes for N3 and N4.
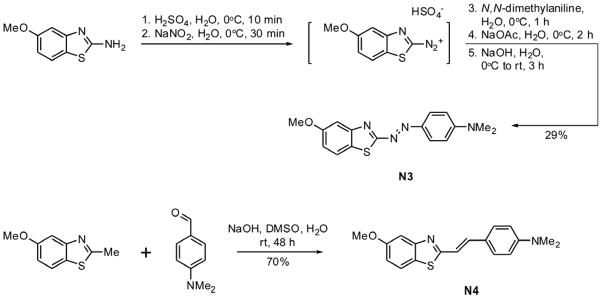
See text for details.
Compound N4 (Fig. 1) was prepared by adding 50% (w/v) NaOH (50.0 ml) to a solution of 6-methoxy-2-methylbenzothiazole (16.2 g, 9.4 mmol) and 4-(dimethylamino)benzaldehyde (12.8 g, 85.9 mmol) in DMSO (80.0 ml). The reaction mixture was stirred for 48 h at room temperature, then poured into water (1.0 liter). The resulting solution was filtered and the solids washed with water (2 x 500 ml), and thoroughly dried in a vacuum oven at 55°C. Solids were then dissolved in 500 mL CHCl3 (the minimum volume necessary for complete dissolution) and the solution was filtered through a bed of silica gel (~500 g) that had been pre-moistened with CHCl3. The silica was rinsed with CHCl3 (~3 liters). The combined filtrates were concentrated and the solid residue thoroughly dried under vacuum to yield 18.6 g (70% yield) of pure N4 as a yellow solid, mp 204–206 °C. 1H NMR (500 MHz, CDCl3) δ 3.00 (s, 6H), 3.86 (s, 3H), 6.69 (d, J = 8.5 Hz, 1H), 7.02 (dd, J = 2.5, 8.5 Hz, 1H), 7.16 (d, J = 16.0 Hz, 1H), 7.27 (d, J = 2.5 Hz, 1H), 7.34 (d, J = 16.0 Hz, 1H), 7.44 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 40.16, 55.74, 104.21, 112.06, 115.07, 117.44, 122.86, 123.48, 128.65, 135.41, 137.23, 148.50, 151.00, 157.50, 165.83. IR (ATR) 3191-2808, 1599, 1527, 1457, 1278, 1197, 1060, 803 cm−1. MS (APCI+); [M+H]+ 311.1. Anal. Calcd. for C18H18N2OS: C, 69.65; H, 5.84; N, 9.02; S, 10.33. Found: C, 69.42; H, 5.92; N, 8.98; S, 10.17.
Chemical properties (molecular mass and volume, ClogP, topological polar surface area) were calculated using Chem 3D Ultra (Cambridge Software, Cambridge, MA).
Lamprey methods
Larval lampreys (Petromyzon marinus) were collected and maintained in 500 ml aquaria as described previously (Hall 2001, Hall 2002). Neurodegeneration of large bulbar reticulospinal neurons located at the level of the 8th nerve (anterior bulbar cells, or ABCs) was induced by microinjection of plasmid DNA encoding htau23 tagged with green fluorescent protein, and was assessed immunohistochemically on serial transverse 10 μm sections with anti-tau monoclonal antibody Tau5 as described previously (Hall et al., 2000).
Central nervous system (CNS) uptake of N3 and N4
Lampreys were maintained in 1 μM compound (and 0.2% DMSO vehicle) for 5 d, with the media (including compound) changed on the second and fourth days. On the fifth day, brains were dissected in lamprey Ringers solution (Hall et al., 1989; Hall et al., 1997) and stored at −20°C until used. Frozen CNS tissue was suspended in ten volumes (v/w) of homogenization buffer (10 mM HEPES, pH 7.4, 100 mM NaCl) and sonicated for 1 min on ice (setting 3; Sonic Dismembrator Model 100, Fisher Scientific). Brain homogenates were then extracted with an equal volume of either isobutanol (for N3 extractions) or ethyl acetate (for N4 extractions), vortexed for 1 min, and centrifuged (16,000g for 4 min). The organic layer was then removed to a clean vial and subjected to absorbance spectroscopy (Varian Cary 50 Bio) at 505 or 388 nm for N3 and N4, respectively. Standard curves relating absorbance to ligand concentration were prepared by spiking lamprey CNS homogenates with known amounts of N3 or N4, then extracting and measuring absorbance as described above. CNS tissue measurements were corrected by subtracting the absorbance from negative control animals not exposed to N3 or N4. Amounts of ligand (pmol/mg wet tissue) were then determined by interpolation of standard curves.
Tau aggregation assays
Htau40 or htau40C291A/C322A were incubated without agitation in assembly buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM dithiothreitol) at 37°C for up to 24 h in the presence of varying concentrations of N3 or N4 and fibrillization inducers arachidonic acid (75 μM) or ODS (50 μM). Resultant filaments were analyzed by electron microscopy as described in (Necula and Kuret, 2004) or by ThS fluorescence (λex = 440 nm; λem = 485 nm) as described in (Honson et al., 2007).
Data analysis
Decreases in ThS fluorescence owing to its displacement by varying concentrations of N3 and N4 were modeled by the sigmoidal function:
| (eq. 1) |
where F and Fmax are blank-adjusted ThS fluorescence in the presence and absence of compound (at concentration x), respectively, Fmin represents fluorescence at infinite compound concentration, logAC50 is the log of the compound concentration producing 50% inhibition of the ThS fluorescence, and n is the Hill coefficient. All parameters estimated by regression methods are reported ± standard error of the estimate.
Results
Compounds N3 and N4 are two closely related benzothiazole derivatives that differ only in the structure of the bridge (diazene in N3, alkene in N4) that links each nucleus to its dimethylaniline moiety (Fig. 1). Their predicted molecular properties are summarized in Table 1. The diazene linkage of N3 decreases hydrophobic character (i.e., it decreases ClogP and confers a ~2-fold higher topological polar surface area relative to N4), and contributes two additional hydrogen bond acceptors. Otherwise, the compounds have very similar calculated molecular volumes and differ in mass by only two Da.
Table 1.
Calculated molecular properties
| Property | N3 | N4 |
|---|---|---|
| Molecular mass (Da) | 312.4 | 310.4 |
| Molecular volume (Å3) | 248 | 256 |
| a ClogP | 4.68 | 5.24 |
| b TPSA (Å2) | 50.7 | 28.1 |
| H-bond acceptors | 5 | 3 |
| H-bond donors | 0 | 0 |
| Rotatable bonds | 4 | 4 |
Calculated log of octanol/water partition coefficient
Topological polar surface area
N3 and N4 were synthesized from commercially available starting materials as shown in Fig. 1. The diazene N3 was prepared by diazotization of 2-amino-6-methoxybenzothiazole followed by coupling with N,N-dimethylaniline (Caprathe et al., 1999), whereas the related alkene N4 was synthesized by base catalyzed aldol condensation of 6-methoxy-2-methylbenzothiazole and 4-(dimethylamino)benzaldehyde (Sanfilippo et al., 1988). Both compounds were purified on silica gel and characterized by melting point determination, elemental analysis, 500 MHz 1H NMR, 125 MHz 13C NMR, IR, and mass spectroscopy. Results indicated that both preparations were highly purified and amenable to experimentation.
To determine whether purified N3 and N4 could protect against tau-mediated neurodegeneration, each was administered (1 μM final concentration) to aquaria housing lamprey overexpressing a GFP-human htau23 fusion protein in single neurons (ABCs) of the lamprey CNS. Cellular morphology was examined 30 d after induction of tauopathy. Control animals were maintained in 0.2% DMSO vehicle alone. These conditions were previously shown to be adequate for assessing the activity of N3 (Hall et al., 2002). Immunocytochemical analysis of transverse sections through the soma and dendritic fields of injected ABCs showed that animals maintained in 0.2% DMSO vehicle alone entered stage III neurodegeneration (characterized by extensive tau aggregation, beading or distortion of all dendrites, and dendritic fragmentation extending up to tertiary and secondary branches; (Hall et al., 2001)) at this time point (Fig. 2A). In contrast, the presence of 1 μM N3, but not N4, led to marked reductions in dendritic abnormalities and tau aggregates (Fig. 2BC). These data confirmed that N3, but not N4, had neuroprotective activity in this cellular tauopathy model.
Fig. 2. N3 but not N4 protects lamprey neurons from dendritic fragmentation induced by human tau overexpression.
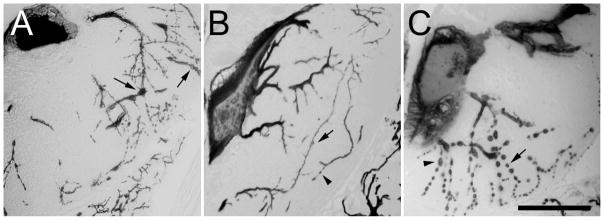
Lampreys microinjected with plasmid DNA encoding htau23 were maintained in aquaria containing (A), 0.5% (v/v) DMSO vehicle, (B), 1 μM N3, or (C), 1 μM N4 with media changes every second day, then fixed in 10% formalin, 10% acetic acid and 80% ethanol 30 d post-plasmid injection. Serial transverse sections cut at the level of ABC somata and dendrites were then prepared and immunostained with the monoclonal antibody Tau5 to reveal ABC dendritic morphology and the extent of degeneration. The figure shows representative samples from each data set. Note that tau-expressing ABCs treated with DMSO vehicle (A) or N4 (C) usually show extensive dendritic beading (arrows), especially in distal dendrites, but also in some proximal dendrites (C, caret). By contrast, N3-treated ABCs (B) typically exhibit relatively more intact distal dendrites, with beading (caret) being relatively rare (compare with distal dendritic fields in A and C). Scale bar: 100 μm.
Why was N3 neuroprotective whereas the closely related N4 was not? One possibility was that N3, which had a greater topological polar surface area than N4 (Table 1), and a higher aqueous solubility, also had greater bioavailability in lamprey. To test this hypothesis, non-injected lampreys were maintained in aquaria containing either 1 μM N3 or N4 for 5 d, then sacrificed and analyzed for ligand uptake into the CNS. Freshly dissected CNS samples were homogenized as described in Experimental Procedures, then extracted with organic solvents. Both N3 and N4 partitioned almost completely into the organic layer under these conditions, consistent with their high ClogP values (Table 1). Standard curves constructed from lamprey CNS homogenates spiked with known amounts of N3 or N4 were linear, with the amounts of ligand extracted being directly proportional to the absorbance measured at their respective absorbance wavelength maxima (λ = 505 nm for N3 extracted in isobutanol, and 388 nm for N4 extracted in ethyl acetate) (Fig. 3A). The amounts of N3 and N4 present in lamprey CNS were then determined from standard curves by interpolation as 10.6 ± 2.8 and 7.9 ± 3.7 pmol/mg (± S.D.; n = 4), respectively (Fig. 3B). Thus both N3 and N4 enter lamprey CNS and accumulate to levels that do not differ at p < 0.05 within 5 days of administration. Assuming lamprey CNS density resembles that of mammalian brain (1.06 g/ml; (Torack et al., 1976)), both N3 and N4 enter and accumulate in CNS to ~10 μM concentrations. These data suggest that differences in N3 and N4 neuroprotective activites do not result from differential bioavailability.
Fig. 3. Both N3 and N4 enter lamprey CNS.
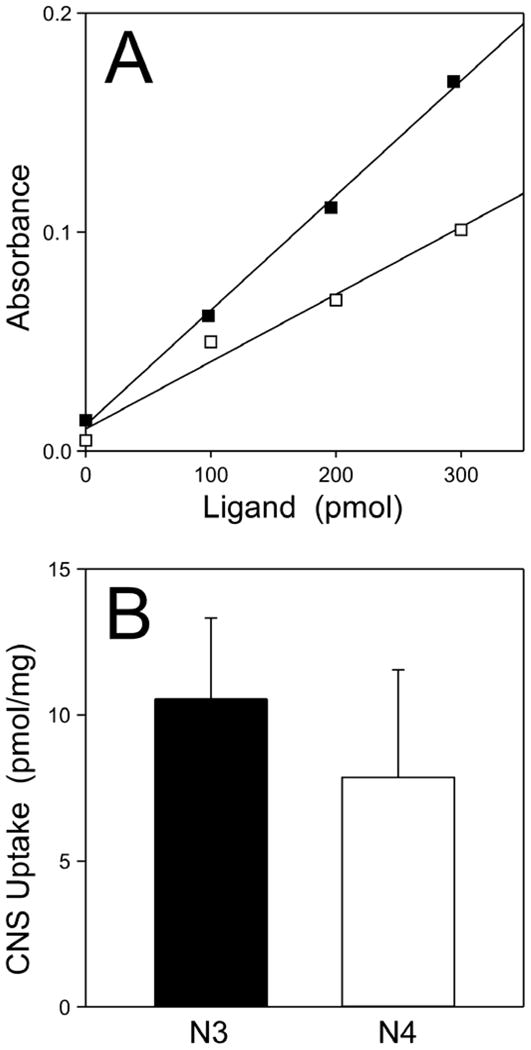
Uninjected lampreys were maintained in aquaria containing 0.2% (v/v) DMSO vehicle or 1 μM N3 or N4 for 5 d (with media changes on the second and fourth day). DMSO treated CNS were pooled, homogenized, spiked with varying amounts of N3 or N4, and finally extracted with either isobutanol (for isolation of N3) or ethyl acetate (for isolation of N4). The organic layers were then subjected to absorbance spectroscopy at 505 or 388 nm for N3 (■) and N4 (□), respectively. (A), resultant standard curves relating ligand amounts to absorbance intensity, where each line represents best fit of the data to linear regression. (B), levels ± S.D. of N3 (n = 4) and N4 (n = 4) in treated lamprey CNS. Both N3 and N4 entered lamprey CNS and attained levels that were not statistically different at p < 0.05.
A previous study found that N3 (obtained as part of a screening library) could inhibit aggregation of recombinant tau induced by arachidonic acid in vitro (Kuret and Khatami, 2002), suggesting that N3 protected degenerating ABCs by directly interfering with tau aggregation. In an effort to reproduce this observation, htau40 was incubated under reducing conditions with arachidonic acid inducer and varying concentrations of purified N3, N4, or DMSO vehicle alone and then analyzed for filament formation by transmission electron microscopy. Htau40 was used for this experiment because it aggregates readily at low micromolar concentrations in anionic surfactants whereas three repeat isoforms, such as htau23, perform poorly under these conditions (King et al., 2000). As expected, N4 did not inhibit tau aggregation relative to DMSO vehicle control at up to 10 μM concentrations (Fig. 4A). Surprisingly, however, neither did N3 (Fig. 4A). To ensure that this result was not specific to arachidonic acid inducer, the ability of N3 and N4 to antagonize htau40 aggregation induced by ODS, an alkyl sulfate surfactant (Chirita et al., 2003), was investigated in parallel. Results showed again that neither compound inhibited tau aggregation under these conditions (data not shown). Finally, because wild-type htau40 can oxidize to form disulfide bonded aggregates (Sahara et al., 2007), which may interfere with filament formation, the mutant htau40C291A/C322A was analyzed in parallel with wild-type tau. Htau40C291A/C322A readily forms filaments in the presence of anionic surfactants, but cannot form disulfide bonded aggregates because it lacks Cys residues (Gamblin et al., 2000). Once again, however, neither N3 (Fig. 4BC) nor N4 (data not shown) was capable of directly inhibiting the aggregation reaction relative to DMSO vehicle control reactions. These data indicate that N3 and N4 lack direct tau aggregation antagonist activity at neuroprotective concentrations, and that this mechanism cannot rationalize the neuroprotective efficacy of N3 in the lamprey tauopathy model.
Fig. 4. Effects of N3 and N4 on tau fibrillization in vitro.
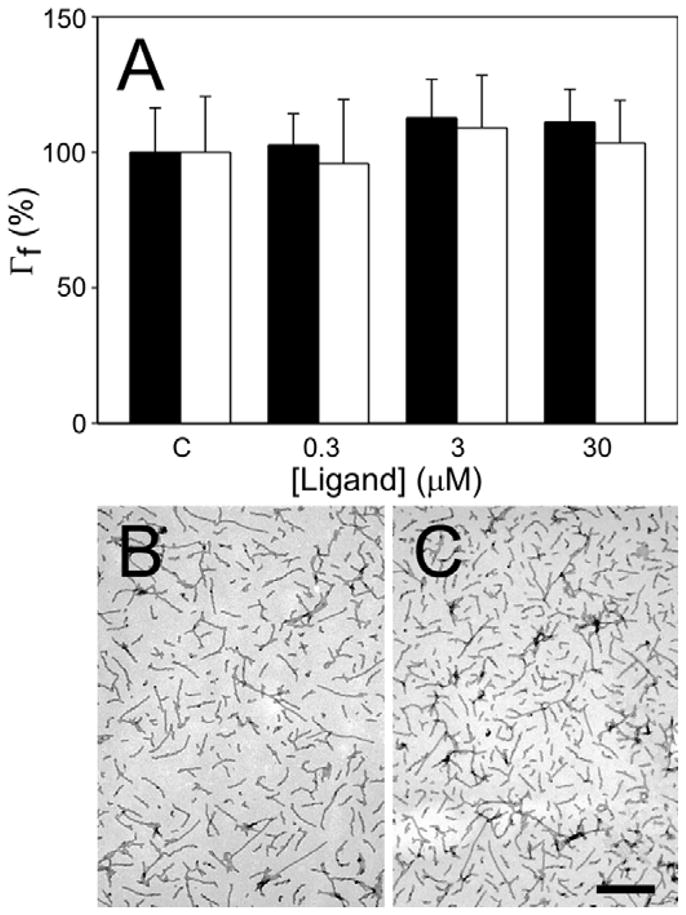
(A), htau40 was incubated (24 h at 37°C) with arachidonic acid inducer (75 μM) in the presence of varying concentrations of N3 (black bars) or N4 (white bars), then examined by electron microscopy to assess tau filament formation. Filament length (Γf) for each condition was recorded as the percent filament length observed in the presence of DMSO vehicle alone (defined as 100%). Error bars indicate S.D (n = 3). Neither N3 nor N4 modulated htau40 fibrillization at any tested concentration. To confirm these findings, arachidonic acid-induced fibrillization of htau40C291A/C322A was examined in the absence (B) and presence (C) of N3 by electron microscopy. N3 did not inhibit fibrillization of htau40C291A/C322A. Scale bar indicates 500 nm.
Because N3 binds with high affinity to synthetic tau filaments (Honson et al., 2007), a second possibility was that N3 and N4 interacted differentially with tau aggregates once formed. To test this hypothesis, the ability of N3 and N4 to displace ThS binding to synthetic htau40 aggregates prepared in the presence of arachidonic acid was assessed using fluorescence spectroscopy (Fig. 5). As found previously, N3 inhibited ThS fluorescence in a concentration dependent manner without evident cooperativity (Hill coefficient = 1.1 ± 0.2), indicating that the concentration of binding sites was low relative to the concentrations of ligand (Goldstein, 1944). These observations are consistent with only a portion of bulk tau forming aggregates (Chirita et al., 2003), and with multiple tau protomers contributing to each thioflavin dye-like binding site as found in other cross-β-sheet aggregates (Krebs et al., 2005; Lockhart et al., 2005). ThS fluorescence was inhibited with an AC50 of 36 ± 6 nM, and a maximal inhibition of 73 ± 2 %. Thus N3 was a partial antagonist of ThS binding with a potentially complex binding equilibrium. In contrast, N4 did not displace ThS binding at any concentration tested (Fig. 5). These data show that N3 and N4 differ markedly in ThS probe displacement activity despite their close similarity in structure.
Fig. 5. N3, but not N4, displaces ThS binding to synthetic tau filaments.
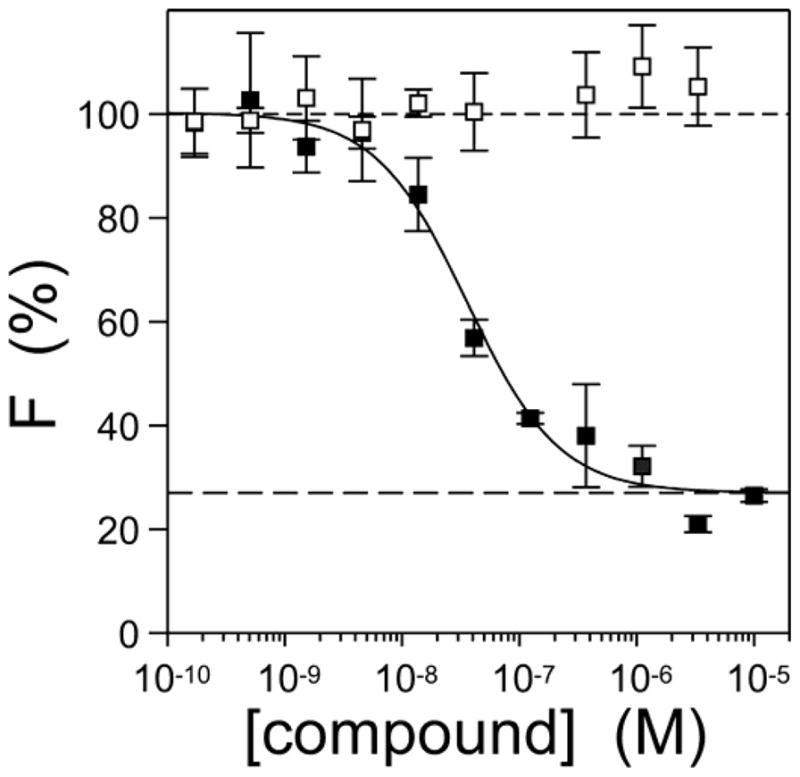
Htau40 (5 μM) was incubated (37°C for 18 h) with arachidonic acid inducer (50 μM) in the presence of 10 μM ThS reporter and varying concentrations of N3 (■) or N4 (□), then subjected to fluorescence spectroscopy (λex = 440 nm; λem = 485 nm). Each point represents mean net ThS fluorescence (F) ± S.D. (expressed as % fluorescence in the presence of DMSO vehicle alone; n = 3) whereas the solid lines reflect best fit of data points to Eq. 1. N3, but not N4 antagonized ThS fluorescence under these conditions.
To characterize the mechanism of N3 binding in greater detail, N3 and ThS concentrations were varied simultaneously in the presence of fixed tau aggregate concentrations and subjected to fluorescence spectroscopy. When the resultant fluorescence intensities were plotted versus ThS concentration in double reciprocal format, a family of converging lines that intersected in the second quadrant was obtained (Fig. 6A). The pattern implicated a mixed-type inhibition mechanism, where the inhibitor affects both the apparent maximum binding (i.e., maximal fluorescence, corresponding to the ordinate intercept) and binding affinity (the abscissa intercept) of the ThS probe (Segel, 1975). Mixed-type inhibition implied that N3 bound tau aggregates in both free and ThS-bound states. To further characterize N3 binding, the slopes and ordinate intercepts of the double reciprocal plots were replotted versus N3 concentration (Fig. 6B). Both replots were parabolic, indicating that the relationship between double-reciprocal plot parameters and inhibitor concentration was in part a power function of the latter, and therefore implying the existence of multiple N3 binding sites. The excellent fit of the replot data points to a second order polynomial was most consistent with the existence of two N3 binding sites that at least partially overlapped with the ThS site. Together these data indicate that N3 and N4 differ markedly in their ability to bind tau filaments, with N3 binding to at least two sites on tau filaments at therapeutic concentrations, whereas N4 binds none.
Fig. 6. N3 binds multiple sites on tau aggregates.
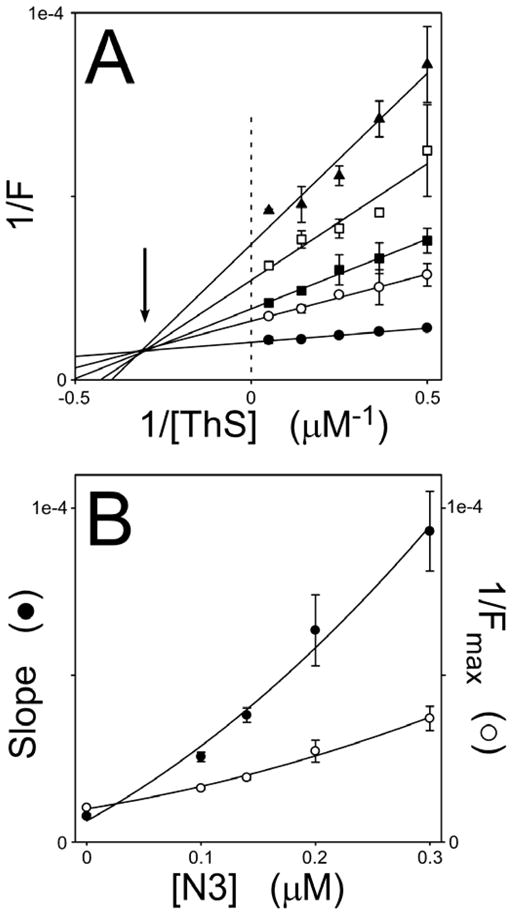
Htau40 (5 μM) was incubated (37°C for 18 h) with arachidonic acid inducer (50 μM) in the presence of varying concentrations of ThS reporter and N3 inhibitor, then subjected to fluorescence spectroscopy (λex = 440 nm; λem = 485 nm). (A), double reciprocal plots of ThS fluorescence (F) versus ThS concentration (n = 3) at 0 (●), 0.1 (○), 0.14 (■), 0.2 (□), and 0.3 (▲) μM N3. Each line represents best fit of the data points to a linear regression. Intersection of the regression lines in the second quadrant (arrow) was consistent with N3 acting as a mixed-type inhibitor of ThS binding. (B), slopes and Fmax values ± S.E.E. from panel A were replotted as a function of N3 concentration and fitted to a second order polynomial. The parabolic dependence of these parameters on N3 concentration suggests the presence of multiple N3 binding sites.
Discussion
These data confirm that the benzothiazole N3, but not its closely related analog N4, protects neurons against tau aggregation and neurodegeneration in a biological model of tauopathy. Previous characterization of an impure N3 preparation obtained from high-throughput screening plates found that it possessed direct anti-tau fibrillization activity (Kuret and Khatami, 2002), suggesting that N3 protected lamprey neurons by directly interfering with tau aggregate formation and accumulation. However, here it was found that custom purified N3 could not antagonize the aggregation of full-length htau40 or mutant htau40C291A/C322A induced by arachidonic acid or ODS in vitro at concentrations that were neuroprotective in lamprey. It is likely, therefore, that mechanisms other than direct inhibition of aggregation mediate the neuroprotective action of N3 in lamprey ABCs. These may include formation of N3 metabolites with potent aggregation inhibitory activity, or differential interaction of N3 and N4 with cellular factors that modulate the tau aggregation reaction.
With respect to the latter possibility, the data indicate that N3 and N4 differ in at least one key respect: N3 but not N4 binds tau aggregates with submicromolar affinity at sites that at least partially overlap with those bound by ThS. The observed mixed-type displacement pattern suggested that N3 bound both free and ThS-bound aggregates, and was consistent with the inability of N3 to completely displace ThS when present at high concentration. The differential ability of benzothiazole derivatives including N3 and N4 to bind proteins in cross-β-sheet conformation was observed previously (Caprathe et al., 1999). Small substitutions and/or additions to the benzothiazole nucleus can greatly change its affinity for such aggregates. For example, addition of either a methoxy group to the 6-position of the benzothiazole ring or methylation to yield a quaternary nitrogen dramatically increases the affinity of both diazene and alkene analogs for Aβ1-40 filaments (Caprathe et al., 1999). These data suggest that binding affinity may depend on the number of specific binding interactions between ligand and filament. The lone electron pairs of the diazene moiety may contribute to these interactions by acting as hydrogen bond acceptors within the N3-filament complex. In contrast, the alkene moiety of N4 lacks lone electron pairs and so may not possess the required interactions for binding at the N3 binding site. These considerations may explain why N4 binds poorly to Aβ1-40 (Caprathe et al., 1999), insulin (Caprathe et al., 1999), and tau (herein) filaments relative to N3. Nonetheless, it cannot be ruled out that N3 binding sites are merely present at higher densities than putative N4 sites, or that putative N4 sites do not overlap with the binding sites for the ThS probe.
How could differential occupancy of binding sites on filaments protect against tauopathy? Inbar and Yang have proposed a sequestration model where nascent amyloid aggregates bind and mislocalize key enzymes, leading to enzyme-mediated gain- or loss-of-function toxicity (Inbar et al., 2006; Inbar and Yang, 2006). Coating of filaments with benzothiazole compounds can inhibit these interactions, and potentially reduce associated neurotoxicity. The model may be especially relevant for tau aggregates, which form in neuronal cytoplasm (Gray et al., 1987) and bind enzymes including protein kinases (Kuret et al., 1997; Ghoshal et al., 1999), proline isomerase-1 (Voneche et al., 1992), and proteasomes (Keck et al., 2003). In each of these examples, tau aggregation appears to gain binding function relative to monomeric tau in normal tissue (Kuret, 2007). For example, association of authentic tau filaments with the proteasome can inhibit proteasome-mediated protein degradation (Keck et al., 2003), potentially initiating a positive feedback loop that leads to increased levels of misfolded proteins and further promotion of aggregation (Goldbaum and Richter-Landsberg, 2004). The link between proteasome function and tau turnover is still tentative (Brown et al., 2005; Delobel et al., 2005; Feuillette et al., 2005), but serves to illustrate how interference with a putative sequestration–mediated pathway at the step of filament binding could potentially facilitate filament turnover and minimize aggregate accumulation. A similar mechanism also may underlie small-molecule mediated neuroprotection against aggregates composed of other proteins. For example, compounds related to benzothiazoles, including pyridothiazoles, antagonize Huntingtin aggregation in cells indirectly (Zhang et al., 2005).
The above model predicts that ThS binding to mature aggregates in lamprey ABCs should be displaced by N3. It was not possible to test this prediction directly, however, because lamprey filaments do not induce a Stokes shift in ThS fluorescence despite being composed primarily of human tau protein (Hall et al., 2001). This result may stem from the coaggregation of small amounts of endogenous lamprey tau with overexpressed human tau in ABC inclusions. It has been shown, for example, that spiking of human Aβ1-40 filaments with small amounts of rodent Aβ1-40 can disrupt the ability of certain ligands, including benzothiazoles, to bind some sites but not others (Ye et al., 2006). Similarly, mixtures of lamprey and human tau could affect ligand binding properties of co-polymer filaments formed in vivo. Nonetheless, it cannot be excluded that other factors limit ThS reactivity, such as that the GFP tag used to label tau protein selectively interferes with the binding of large molecules like ThS to tau filaments, or that the filaments produced in the model differ markedly from both synthetic filaments formed in vitro and authentic tau filaments derived from AD brain in their ability to bind ThS. It is clear, however, that the GFP tag does not modulate tau-induced neurotoxicity in ABCs (Hall et al., 2000).
In summary, small molecules can protect against tau fibrillization and neurodegeneration in a biological model of tauopathy. The mechanism most likely does not involve direct inhibition of tau aggregation, but may depend on high-affinity binding to tau aggregates once formed. The lamprey model may be useful for identifying cellular factors that mediate tauopathic neurodegeneration.
Acknowledgments
We thank Xian Yu for assistance with ThS fluorescence assays, Carmen Chirita and Mihaela Necula for electron microscopy images, and Jun Yao for immunohistochemical processing of lamprey samples. This research was supported by grants from the National Institutes of Health (AG14452 and MH077621; J.K.; AG018661; A.A.) and the Alzheimer’s Association (IIRG-05-14288; J.K.).
References
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Brown MR, Bondada V, Keller JN, Thorpe J, Geddes JW. Proteasome or calpain inhibition does not alter cellular tau levels in neuroblastoma cells or primary neurons. J Alzheimers Dis. 2005;7:15–24. doi: 10.3233/jad-2005-7103. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Caprathe BW, Gilmore JL, Hays SJ, Jaen JC, LeVine H. Method of imaging amyloid deposits. 6,001,331. US Patent. 1999
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization. in vitro J Biol Chem. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- Delobel P, Leroy O, Hamdane M, Sambo AV, Delacourte A, Buee L. Proteasome inhibition and Tau proteolysis: an unexpected regulation. FEBS Lett. 2005;579:1–5. doi: 10.1016/S0014-5793(05)01501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillette S, Blard O, Lecourtois M, Frebourg T, Campion D, Dumanchin C. Tau is not normally degraded by the proteasome. J Neurosci Res. 2005;80:400–405. doi: 10.1002/jnr.20414. [DOI] [PubMed] [Google Scholar]
- Friedhoff P, Schneider A, Mandelkow EM, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Kuret J, Berry RW, Binder LI. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry. 2000;39:14203–14210. doi: 10.1021/bi001876l. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, Smiley JF, DeMaggio AJ, Hoekstra MF, Cochran EJ, Binder LI, Kuret J. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am J Pathol. 1999;155:1163–1172. doi: 10.1016/S0002-9440(10)65219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal N, Garcia-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol. 2002;177:475–493. doi: 10.1006/exnr.2002.8014. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- Goldbaum O, Richter-Landsberg C. Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive aggregates in oligodendrocytes in culture. J Neurosci. 2004;24:5748–5757. doi: 10.1523/JNEUROSCI.1307-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A. Mechanism of enzyme-inhibitor-substrate reactions. Cholinesterase-eserine-acetylcholine system. J Gen Physiol. 1944;27:529–580. doi: 10.1085/jgp.27.6.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Deters N, Doldissen A, Bokhari L, Ke Y, Wiesner A, Schonrock N, Ittner LM. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007;17:91–103. doi: 10.1111/j.1750-3639.2007.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EG, Paula-Barbosa M, Roher A. Alzheimer’s disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol. 1987;13:91–110. doi: 10.1111/j.1365-2990.1987.tb00174.x. [DOI] [PubMed] [Google Scholar]
- Hall GF, Poulos A, Cohen MJ. Sprouts emerging from the dendrites of axotomized lamprey central neurons have axonlike ultrastructure. J Neurosci. 1989;9:588–599. doi: 10.1523/JNEUROSCI.09-02-00588.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall GF, Yao J, Selzer ME, Kosik KS. Cytoskeletal changes correlated with the loss of neuronal polarity in axotomized lamprey central neurons. J Neurocytol. 1997;26:733–753. doi: 10.1023/a:1018562331003. [DOI] [PubMed] [Google Scholar]
- Hall GF, Chu B, Lee G, Yao J. Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci. 2000;113:1373–1387. doi: 10.1242/jcs.113.8.1373. [DOI] [PubMed] [Google Scholar]
- Hall GF, Lee VM, Lee G, Yao J. Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol. 2001;158:235–246. doi: 10.1016/S0002-9440(10)63962-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall GF, Lee S, Yao J. Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci. 2002;19:253–260. doi: 10.1385/jmn:19:3:251. [DOI] [PubMed] [Google Scholar]
- Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, Nordhoff E, Lurz R, Schugardt N, Rautenberg S, Herhaus C, Barnickel G, Bottcher H, Lehrach H, Wanker EE. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington’s disease by using an automated filter retardation assay. Proc Natl Acad Sci U S A. 2002;99:16400–16406. doi: 10.1073/pnas.182426599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol Dis. 2007;363:229–234. doi: 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honson NS, Kuret J. Tau aggregation and toxicity in tauopathic neurodegenerative diseases. J Alzheimers Dis. 2008 doi: 10.3233/jad-2008-14409. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y. Massive somatodendritic sprouting of cortical neurons in Alzheimer’s disease. Brain Res. 1988;459:138–144. doi: 10.1016/0006-8993(88)90293-4. [DOI] [PubMed] [Google Scholar]
- Inbar P, Li CQ, Takayama SA, Bautista MR, Yang J. Oligo(ethylene glycol) derivatives of thioflavin T as inhibitors of protein-amyloid interactions. ChemBioChem. 2006;7:1563–1566. doi: 10.1002/cbic.200600119. [DOI] [PubMed] [Google Scholar]
- Inbar P, Yang J. Inhibiting protein-amyloid interactions with small molecules: a surface chemistry approach. Bioorg Med Chem Lett. 2006;16:1076–1079. doi: 10.1016/j.bmcl.2005.10.067. [DOI] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000;74:1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x. [DOI] [PubMed] [Google Scholar]
- Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- Krebs MR, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Kuret J, Johnson GS, Cha D, Christenson ER, DeMaggio AJ, Hoekstra MF. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer’s disease brain. J Neurochem. 1997;69:2506–2515. doi: 10.1046/j.1471-4159.1997.69062506.x. [DOI] [PubMed] [Google Scholar]
- Kuret J, Khatami K. Methods for inhibiting or reversing tau filament formation. 6,479,528. US Patent. 2002
- Kuret J. Detection and Reduction of Neurofibrillary Lesions. In: Smith HJ, Sewell RDE, Simons C, editors. Protein Folding Diseases: Enzyme Inhibitors and Other Agents as Prospective Therapies. CRC Press, Taylor & Francis Books; Boca Raton, FL: 2007. pp. 287–324. [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong G, Gee AD, Brown J. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on β-amyloid peptide fibrils. J Biol Chem. 2005;280:7677–7684. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- McKee AC, Kowall NW, Kosik KS. Microtubular reorganization and dendritic growth response in Alzheimer’s disease. Ann Neurol. 1989;26:652–659. doi: 10.1002/ana.410260511. [DOI] [PubMed] [Google Scholar]
- Necula M, Kuret J. Electron microscopy as a quantitative method for investigating tau fibrillization. Anal Biochem. 2004;329:238–246. doi: 10.1016/j.ab.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- Sanfilippo PJ, Urbanski M, Press JB, Dubinsky B, Moore JB., Jr Synthesis of (aryloxy)alkylamines. 2. Novel imidazo-fused heterocycles with calcium channel blocking and local anesthetic activity. J Med Chem. 1988;31:2221–2227. doi: 10.1021/jm00119a026. [DOI] [PubMed] [Google Scholar]
- Segel IH, editor. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. New York: Wiley; 1975. [Google Scholar]
- Torack RM, Alcala H, Gado M, Burton R. Correlative assay of computerized cranial tomography CCT, water content and specific gravity in normal and pathological postmortem brain. J Neuropathol Exp Neurol. 1976;35:385–392. doi: 10.1097/00005072-197607000-00001. [DOI] [PubMed] [Google Scholar]
- Voneche V, Portetelle D, Kettmann R, Willems L, Limbach K, Paoletti E, Ruysschaert JM, Burny A, Brasseur R. Fusogenic segments of bovine leukemia virus and simian immunodeficiency virus are interchangeable and mediate fusion by means of oblique insertion in the lipid bilayer of their target cells. Proc Natl Acad Sci USA. 1992;89:3810–3814. doi: 10.1073/pnas.89.9.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Nakazato Y, Shoji M, Ihara Y, Hirai S. Ultrastructure of the neuropil threads in the Alzheimer brain: their dendritic origin and accumulation in the senile plaques. Acta Neuropathol (Berl) 1990;80:368–374. doi: 10.1007/BF00307689. [DOI] [PubMed] [Google Scholar]
- Ye L, Morgenstern JL, Lamb JR, Lockhart A. Characterisation of the binding of amyloid imaging tracers to rodent Aβ fibrils and rodent-human Aβ co-polymers. Biochem Biophys Res Commun. 2006;347:669–677. doi: 10.1016/j.bbrc.2006.06.126. [DOI] [PubMed] [Google Scholar]
- Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci USA. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]