

Abstract
High-throughput genomic technologies are increasingly being used to identify therapeutic targets and risk factors for specific diseases. Using 116 independent liver samples, we identified 793 probe sets that demonstrated a significant association in the frequency of absent calls as tissues progressed from normal to pre-neoplastic to neoplastic, followed by a bioinformatic approach which identified that 78.9% of the significant probe sets contained at least one CpG island in the gene promoter region compared with 58.9% of the remaining genes examined. Our results indicate that further high-throughput methylation studies to more fully characterize molecular events involved in hepatocarcinogenesis are warranted.
Keywords: HCC, hepatocellular carcinoma, HCV, hepatitis C virus, methylation, microarray, hepatocarcinogenesis
1 Introduction
High-throughput genomic technologies are increasingly being used in science and industry to identify therapeutic targets and risk factors for specific diseases. As some notable examples, gene expression microarrays have been successfully used in differentiating two types of leukaemia (Golub et al., 1999), and as prognostic meta-signatures for breast cancer (van de Vijver et al., 2002; van’t Veer et al., 2002, 2003), lung adenocarcinoma (Beer et al., 2002) and diffuse large B-cell lymphoma (Rosenwald et al., 2002). As in other disease areas, through the use of high-throughput technologies, a vast amount of information regarding gene expression is being accumulated for the study of Hepatocellular Carcinoma (HCC). For example, some researchers have conducted microarray gene expression studies and have examined gene expression of resected or explanted livers with HCC and the association with histologic grade (Nam et al., 2005) or impact on recurrence or survival (Iizuka et al., 2003; Mas et al., 2006).
Most high-throughput genomic research in HCC has emphasised gene expression levels (Nam et al., 2005; Iizuka et al., 2003; Mas et al., 2006). However, of increasing importance in the field of cancer research is DNA methylation. DNA methylation occurs when a methyl group attaches to a 5′ cytosine in a CpG dinucleotide. Dense methylation of CpG islands in the gene promoter region affects gene transcription, and is often termed epigenetic silencing (Jones and Laird, 1999; Eng et al., 2000). Hence, aberrant DNA methylation more likely plays a direct role in altered protein expression levels. It is thought that epigenetic events such as methylation of gene promoter regions, specifically, methylation of Tumour Suppressor Genes (TSGs) as well as global hypomethylation of oncogenes, may be implicated in carcinogenesis (Jones and Laird, 1999). It may therefore be of interest to identify a unique combination of genes specific to a phenotype that are either expressed or not expressed.
In most studies of DNA methylation in HCC, only one or a few specific promoter regions were examined (Hirasawa et al., 2006; Jicai et al., 2006; Qiu et al., 2007; Uematsu et al., 2006; Wong et al., 1999, 2000). Furthermore, DNA methylation studies among HCC patients have largely been conducted among HBV-infected patients (Jicai et al., 2006; Wong et al., 1999, 2000; Yang et al., 2005; Wang et al., 2006) or among patients with heterogeneous etiologies (Tchou et al., 2000; Yang et al., 2003; Hsu et al., 2006; Zhang et al., 2006; Shih et al., 2006). It has been established that patients with HBV + HCC likely have different malignant transforming mechanisms compared with patients with HCV + HCC (Poon et al., 2006; Moinzadeh et al., 2005; Iizuka et al., 2004). This point was emphasised in a more recent methylation study in Western patients that examined the promoter regions of 9 TSGs (SOCS-1, GSTP, APC, E-cadherin, pl5, pl6, RAR-J3, pl4 and p73). However, the investigators enrolled a mixture of 51 HCC patients that included 9 HBV+, 18 HCV+ and 24 HBV−/HCV− patients (Yang et al., 2003). The largest methylation study conducted in HCC patients to date examined 105 TSGs among 60 HCC tissues and their surrounding non-tumourous tissues and in 10 normal liver tissues (Calvisi et al., 2007). These researchers showed that patterns of promoter hypermethylation can be of prognostic relevance for HCC. However, similar to other research in this area, the patients examined in this study differed with respect to etiology (32 HBV, 14 HCV, 4 EtOH, 1 Cryptogenetic, 9 etiology not available) and only 25 of 60 had cirrhosis.
Since methylation of CpG islands in gene promoter regions has been associated with gene silencing (Jones and Laird, 1999) and additionally, since DNA methylation has been demonstrated to lead to chromosomal instability hence genomic aberrations (Baylin et al., 2000, 2001) including genetic damage in colorectal tumours (Jones and Laird, 1999), we postulated that methylation may play an important role in the pathogenesis of HCC due to HCV infection. Further, since methylation is most relevant when it results in gene silencing (Esteller, 2002), in this study we explored the differences in the frequencies of genes present vs. absent in liver tissues representing the progression to HCC by examining a homogeneous population with underlying etiology of HCV-infection.
2 Materials and methods
2.1 Patients and samples
Tumour tissue samples were collected from patients at the Hume-Lee Transplant Center, Medical College of Virginia Hospitals from 40 HCV + HCC patients, representing the neoplastic condition. Tumour samples with more than 85% of Tumour cell content were used for the microarray studies. In addition, 19 normal donor liver samples were obtained from explanted liver donors chosen for isolated hepatocyte preparation for transplantation. Liver function and histopathology for these liver donors were shown to be normal. All 19 normal patients were seronegative for HCV Ab. The cirrhotic liver has been described as being pre-malignant or a pre-neoplastic condition (McCaughan et al., 2002), therefore, we included 57 independent HCV + cirrhosis non-HCC samples representing the pre-neoplastic condition. Each normal, pre-neoplastic and neoplastic liver sample was hybridised to an Affymetrix HG-U133A 2.0 Array. The Institutional Review Board approved the study protocol at Virginia Commonwealth University.
2.2 Laboratory methods
The sample preparation protocol followed the Affymetrix GeneChip® Expression Analysis Manual (Santa Clara, CA). Total RNA was extracted from tissue samples using TRIzol (Life Technologies, Rockville, MD). Integrity of RNA was checked using Agilent 2100 Bioanalyser. Briefly, total RNA was reverse-transcribed using T7-polydT primer and converted into double-stranded cDNA (One-Cycle Target Labelling and Control Reagents, Affymetrix, Santa Clara, CA), with templates being used for an in vitro transcription reaction to yield biotin-labelled antisense cRNA. The labelled cRNA was chemically fragmented and made into the hybridisation cocktail according to the Affymetrix GeneChip protocol, which was then hybridised to U133A 2.0 GeneChips. The array image was generated by the high-resolution GeneChip® Scanner 3000 by Affymetrix® (Affymetrix, Santa Clara, CA).
2.3 Statistical methods
Quality assessment was performed by assessing the average background, scaling factor, percent present calls and 3′ : 5′ ratios of GAPDH and β-actin. Prior to conducting statistical analyses, all control probe sets were removed. Unlike previous research that sought to identify aberrantly methylated genes in pancreatic carcinoma using gene expression microarrays and focusing on genes with >5-fold change (Sato et al., 2003), we focused our analysis on whether the gene was present or absent, since differential expression may not indicate methylation but simply identify genes expressed at different levels. Therefore, probe-level intensities were used to compute the probe set level present, marginally present, and absent calls using the Affymetrix Detection Call algorithm (Liu et al., 2002). The Detection Call algorithm uses the Perfect Match (PM) and mismatch (MM) intensities for probe pairs within a probe set and applies a Wilcoxon signed rank test for assessing whether the probe set is Present (P), Marginally Present (M), or Absent (A). For probes i in probe set j, the p-value obtained from testing H0j: median (Rij) = τ vs. HAj: median(Rij) > τ, where
| (1) |
is compared with the following thresholds: If pj < α1, then probe set j is declared Present; if α1 < pj < α2, then probe set j is declared Marginally Present; if Pj > α2, then probe set j is declared Absent. The current defaults in the Affymetrix GeneChip Operating Software are α1 = 0.05 and α2 = 0.065. All data processing and statistical analysis procedures were conducted in the R programming environment (R Development Core Team, 2008) using appropriate Bioconductor packages (Gentleman et al., 2004; Gautier et al., 2004).
Procedure for filtering genes
First, all control probe sets were excluded leaving 22,215 probe sets. Prior to hypothesis testing, the resulting matrix containing the detection call results per probe set was filtered by excluding those probe sets declared absent among all 116 samples (N = 4129) and excluding those probe sets declared either marginally present or present among all 116 samples (N = 4834), leaving 13,252 probe sets for analysis.
Class comparisons
To assess whether a probe set is increasingly silenced as one moves from normal, to pre-neoplastic, to neoplastic liver tissue, it was of interest to determine whether there is a significant monotonic decreasing trend among the proportion of present calls in these diagnostic groups. For each probe set, a linear-by-linear association test was used to determine if there is a monotonic trend among patients cross-classified by diagnosis and detection call (Table 1). Specifically, for scores (um and vn) assigned to variables X (rows) and Y (columns) indicating the ordered nature of disease (normal, pre-neoplastic, neoplastic) and detection calls (absent, marginally present, present), for each probe set the Poisson log-linear model
| (2) |
was fit where emn represents the expected cell count for the mth row and nth column and remaining parameters represent the overall (λ), row and column effects (Simonoff, 2003). Of specific interest is the parameter estimate for θ. That is, if θ > 0, the two factors are directly associated, if θ < 0 the two factors are inversely associated, and if θ = 0 the two factors are independent. The p-value associated with the parameter estimate for θ was obtained for each probe set specific model. Thereafter, to control for multiple hypothesis testing using the FDR, we restricted attention to probe sets with a significantly negative estimate using a q-value < 0.05, such that among the list of candidate genes we identify, 5% are expected to be false positives (Storey and Tibshirani, 2003).
Table 1.
Probe set level Poisson log-linear models were fit to the cross-tabulation where scores (um and vn) were assigned to rows and columns indicating the ordered nature of tissue type and detection calls
| Normal (v1) | Pre-neoplastic (v2) | Neoplastic (v3) | |
|---|---|---|---|
| Absent (u1) | n 11 | n 12 | n 13 |
| Marginally present (u2) | n 21 | n 22 | n 23 |
| Present (u3) | n 31 | n 32 | n 33 |
Bioinformatic verification
Since gene silencing due to methylation has only been conclusively demonstrated when the promoter region of the gene is methylated (Jones and Laird, 1999), we used a bioinformatic approach to identify whether any of the significant genes have CpG islands in their promoter region, to further refine our list of candidate methylated genes. Annotation data for significant probe sets were obtained for identifying whether the gene represented by the probe set had a CpG island in its promoter region. Specifically, the Bioconductor AnnotationDbi (version 1.6.0) and hgul33a2.db (version 2.2.11) packages were used to look up the gene symbol and Entrez ID associated with each probe set ID. Probe sets without corresponding Entrez IDs were removed from the bioinformatic analysis. Sequences and annotations of these candidate genes were retrieved from the NCBI (http://www.ncbi.nlm.nih.gov/). The definition of the promoter region in a gene is an unsolved issue, but in most cases it resides in a short region (e.g., 500 bp) immediately upstream of the transcription start site (Waterston et al., 2002; Yamashite et al., 2006). We identified CpG islands in the promoter regions based on two popular algorithms, Takai and Jones (2002) and Gardiner-Garden and Prommer (1987), using the data-processing procedure described in Jiang et al. (2007).
Clustering
Prior to clustering, we reduced the dimensionality of the data set by excluding probe sets consistently declared present or absent among the 116 samples. Specifically, probe sets called absent in at least 80% of the samples as well as probe sets called present in at least 80% of the samples were excluded, leaving 4650 probe sets for clustering. Agglomerative hierarchical clustering using Ward’s method (Kaufman and Rousseeuw, 1990) was performed using the Euclidean distance of the detection calls (Present and Marginally Present = 1, Absent = 0) as the distance measure.
3 Results
3.1 Quality control
Prior to statistical analysis, all hybridised GeneChips were examined with respect to quality. Average background, scaling factor, percent present calls and the 3′ : 5′ ratios for GAPDH and β-actin were similar among all GeneChips. No chip was eliminated due to quality concerns.
3.2 Differences in detection call profiles
From the probe-set-level Poisson log-linear models, 793 probe sets demonstrated a significant association in the frequency of absent calls as tissues progressed from normal to pre-neoplastic to HCC, using an FDR of 5% (Supplemental Table 1). Interestingly, ADRA1A, CRHBP, CXCL14, ECM1, EPO, FCN2, FCN3 and GREM2 were also identified in another high-throughput gene expression study as being an important discriminator between dysplastic liver tissue and HCC (Wurmbach et al., 2007). This conservative FDR suggests that among the 793 probe sets, approximately 40 are expected to be false positives. When controlling for the FDR at 1%, 339 probe sets were significant. Thus, many genes exhibited a trend of being more frequently absent as tissue moved from normal to preneoplastic to neoplastic.
3.3 Bioinformatic analysis of CpG sequences in the promoter regions of genes
Among the 793 significant probe sets, 55 had poor gene annotation information, largely due to recent updates of gene databases. Thus, the bioinformatic analysis was restricted to 738 probe sets. These 738 probe sets matched 644 possible genes, including 70 genes, each of which had two significant probe sets, and 12 genes, each of which had three significant probe sets. Of these 644 genes, 577 were found in a full set of human genes processed from all the human genes deposited in NCBI using the previously described pipeline (Jiang et al., 2007). According to the Takai and Jones (T&J) algorithm (Takai and Jones, 2002), 455 of these 577 genes (78.9%) contained at least one CpG island in their promoter region (Supplemental Table 1), which is significantly greater in comparison with 58.9% of the remaining distinct genes on the Affymetrix HG-U133A 2.0 Array included in the analysis that contained a CpG island via the same algorithm (p = 1.9 × 10−21). As expected, the Gardiner-Garden and Frommer algorithm (Gardiner-Garden and Frommer, 1987), which used the less restrictive criteria in searching CpG islands, identified a higher percent of genes containing a CpG island (483 genes, 83.7%). Of the 125 probe sets significant at the Q-value < 0.001 level, 94 had complete annotation information, and of these 94, the 69 having a CpG island in the gene promoter region according to the T&J algorithm are listed in Table 2.
Table 2.
Gene symbol, percent of present probe sets in each group, and q-value from the Poisson log-linear models for probe sets significant at the Q-value < 0.001 level and having a CpG island in the gene promoter region
| % Present | % Present | % Present | CpG Island | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Probe set | Gene symbol | Normal | Pre-neoplastic | Neoplastic | q-value | Takai & Jones |
| 218434_s_at | AACS | 79 | 19 | 18 | 0.0002 | Yes |
| 213245_at | ADCY1 | 79 | 18 | 23 | 0.00084 | Yes |
| 207589_at | ADRA1B | 53 | 2 | 0 | <0.00001 | Yes |
| 210678_s_at | AGPAT2 | 95 | 49 | 30 | 0.0001 | Yes |
| 218444_at | ALG12 | 79 | 14 | 8 | 0.00001 | Yes |
| 216563_at | ANKRD12 | 89 | 86 | 10 | <0.00001 | Yes |
| 39249_at | AQP3 | 68 | 51 | 13 | 0.00047 | Yes |
| 210727_at | CALCA | 53 | 0 | 3 | 0.00003 | Yes |
| 212886_at | CCDC69 | 100 | 65 | 35 | 0.00004 | Yes |
| 201913_s_at | COASY | 89 | 65 | 35 | 0.00032 | Yes |
| 205471_s_at | DACH1 | 89 | 82 | 38 | 0.00018 | Yes |
| 212649_at | DHX29 | 95 | 98 | 48 | 0.00001 | Yes |
| 204840_s_at | EEA1 | 89 | 77 | 40 | 0.00038 | Yes |
| 201026_at | EIF5B | 100 | 82 | 45 | 0.00003 | Yes |
| 214153_at | ELOVL5 | 95 | 75 | 35 | 0.00002 | Yes |
| 58696_at | EXOSC4 | 74 | 40 | 20 | 0.00086 | Yes |
| 210627_s_at | GCS1 | 68 | 9 | 5 | 0.00002 | Yes |
| 20272 l_s_at | GFPT1 | 79 | 16 | 10 | 0.00001 | Yes |
| 210964_s_at | GYG2 | 74 | 9 | 15 | 0.00047 | Yes |
| 210331_at | HECW1 | 37 | 2 | 3 | 0.00069 | Yes |
| 210045_at | IDH2 | 74 | 0 | 3 | <0.00001 | Yes |
| 204786_s_at | IFNAR2 | 89 | 86 | 30 | 0.00001 | Yes |
| 212439_at | IHPK1 | 47 | 2 | 3 | 0.00054 | Yes |
| 202809_s_at | INTS3 | 84 | 54 | 33 | 0.00089 | Yes |
| 203682_s_at | IVD | 100 | 75 | 48 | 0.00019 | Yes |
| 212496_s_at | JMJD2B | 68 | 11 | 8 | 0.00006 | Yes |
| 220116_at | KCNN2 | 100 | 18 | 23 | 0.00005 | Yes |
| 212101_at | KPN A 6 | 84 | 93 | 55 | 0.00093 | Yes |
| 212103_at | KPN A 6 | 68 | 18 | 8 | 0.00015 | Yes |
| 203713_s_at | LLGL2 | 100 | 74 | 48 | 0.00051 | Yes |
| 203514_at | MAP3K3 | 89 | 44 | 23 | 0.00004 | Yes |
| 214056_at | MCL1 | 53 | 75 | 10 | 0.00083 | Yes |
| 203644_s_at | MON1B | 63 | 4 | 8 | 0.00008 | Yes |
| 209450_at | OSGEP | 84 | 37 | 20 | 0.00023 | Yes |
| 219737_s_at | PCDH9 | 79 | 96 | 45 | 0.00072 | Yes |
The Entrez IDs for the 793 significant probe sets and their corresponding q-values were then uploaded into the Ingenuity Pathway Analysis software. The top three Diseases and Disorders associated with the significant probe sets were cancer (221 molecules, P-value range 9.03 × 10−7 – 5.64 × 10−3), gastrointestinal disease (71 molecules, P-value range 9.03 × 10–7 – 4.25 × 10−3) and inflammatory response (6 molecules, P-value range 5.98 × 10−5 – 4.59× 10−3). There were many significant molecular and cellular functions, including cellular growth and proliferation (182 molecules), gene expression (141 molecules), cell death (162 molecules), cellular development (122 molecules) and cell cycle (70 molecules) (Figure 1). The top two canonical pathways were RAR Activation (P = 0.0005) and IGF-l Signalling (P = 0.0006). Significant genes previously associated with hepatotoxicity, including liver dysplasia, liver necrosis or cell death, liver cirrhosis, liver hepatitis, liver proliferation and liver enlargement, are listed in Table 3.
Figure 1.
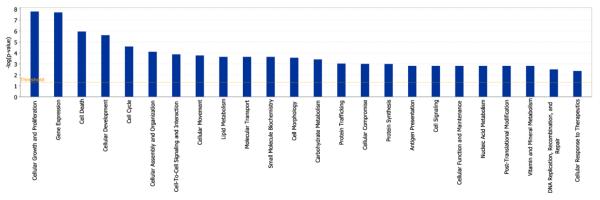
Molecular and cellular functions corresponding to the 793 significant probe sets and sorted by p-value according to ingenuity pathway analysis software (see online version for colours)
Table 3.
Gene symbol, percent of present probe sets in each group, and q-value from the Poisson log-linear models for significant probe sets corresponding to genes associated with hepatotoxicity, and whether the gene has a CpG island according to the Takai & Jones (T&J) and Gardiner-Garden and Frommer (G-G&F) algorithm
| Gene symbol |
% Present | % Present | % Present | CpG Island | CpG Island | ||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Probe set | Normal | Pre-neoplastic | Neoplastic | q-value | T&J | G-G&F | |
| ADRA1A | 211492_s_at | 26.3 | 5.3 | 0.0 | 0.0060 | Y | Y |
| ADRA1B | 207589_at | 52.6 | 1.8 | 0.0 | <0.0001 | Y | Y |
| AVPR1A | 206251_s_at | 89.5 | 17.5 | 40.0 | 0.0218 | N | Y |
| E2F1 | 2028_s_at | 26.3 | 7.0 | 0.0 | 0.0041 | Y | Y |
| EPO | 207257_at | 47.4 | 22.8 | 7.5 | 0.0055 | Y | Y |
| EPOR | 37986_at | 94.7 | 78.9 | 55.0 | 0.0079 | Y | Y |
| FGF2 | 204422_s_at | 78.9 | 50.9 | 40 | 0.0280 | Y | Y |
| GGT1 | 211417_x_at | 100.0 | 100.0 | 80.0 | 0.0039 | N | N |
| GGT1 | 208284_x_at | 100.0 | 100.0 | 87.5 | 0.0188 | N | N |
| GGT1 | 209919_x_at | 100.0 | 100.0 | 85.0 | 0.0162 | N | N |
| GSK3B | 209945_s_at | 100.0 | 98.2 | 82.5 | 0.0177 | N | N |
| HMOX1 | 203665_at | 100.0 | 93.0 | 67.5 | 0.0016 | Y | Y |
| HNF1A | 210515_at | 94.7 | 5.3 | 35.0 | 0.0066 | NA | NA |
| HNF1A | 216930_at | 36.8 | 0.0 | 5.0 | 0.0060 | NA | NA |
| IFNAR2 | 204786_s_at | 89.5 | 86.0 | 30.0 | <0.0001 | Y | Y |
| IGF1 | 211577_s_at | 94.7 | 96.5 | 65.0 | 0.0012 | N | N |
| IGF1 | 209542_x_at | 100.0 | 94.7 | 57.5 | <0.0001 | N | N |
| IL4 | 207538_at | 78.9 | 0.0 | 0.0 | <0.0001 | N | N |
| IL6R | 217489_s_at | 15.8 | 1.8 | 0.0 | 0.0167 | Y | Y |
| JUN | 213281_at | 94.7 | 100.0 | 80.0 | 0.0464 | Y | Y |
| MMP14 | 160020_at | 100 | 94.7 | 75.0 | 0.0014 | Y | Y |
| NR1I2 | 207203_s_at | 21.1 | 3.5 | 2.5 | 0.0189 | N | N |
| PML | 211014_s_at | 26.3 | 17.5 | 2.5 | 0.0318 | N | Y |
| PPARA | 206870_at | 63.2 | 5.3 | 10.0 | 0.0009 | Y | Y |
| RARA | 203749_s_at | 89.5 | 70.2 | 52.5 | 0.0049 | N | N |
| SLC4A2 | 202111_at | 68.4 | 28.1 | 25.0 | 0.0156 | Y | Y |
| SLCO1A2 | 211481_at | 31.6 | 1.8 | 0.0 | 0.0008 | Y | Y |
| SLCO1A2 | 211480_s_at | 15.8 | 0.0 | 0.0 | 0.0156 | Y | Y |
| SLCO1A2 | 207308_at | 42.1 | 7.0 | 10.0 | 0.0088 | Y | Y |
| SMPD1 | 209420_s_at | 100.0 | 100.0 | 75.0 | 0.0014 | Y | Y |
| SOD2 | 215078_at | 52.6 | 82.5 | 25.0 | 0.0057 | Y | Y |
| SREBF1 | 202308_at | 100.0 | 91.2 | 62.5 | 0.0006 | Y | Y |
| TNFSF14 | 207907_at | 52.6 | 10.5 | 10.0 | 0.0055 | N | Y |
| XIAP | 206536_s_at | 94.7 | 31.6 | 27.5 | 0.0012 | NA | NA |
3.4 Clustering
Agglomerative hierarchical clustering using the 4650 probe sets, which remained after applying an unsupervised filter, revealed that samples belonging to the same tissue type clustered together using present/absent calls alone (Figure 2).
Figure 2.
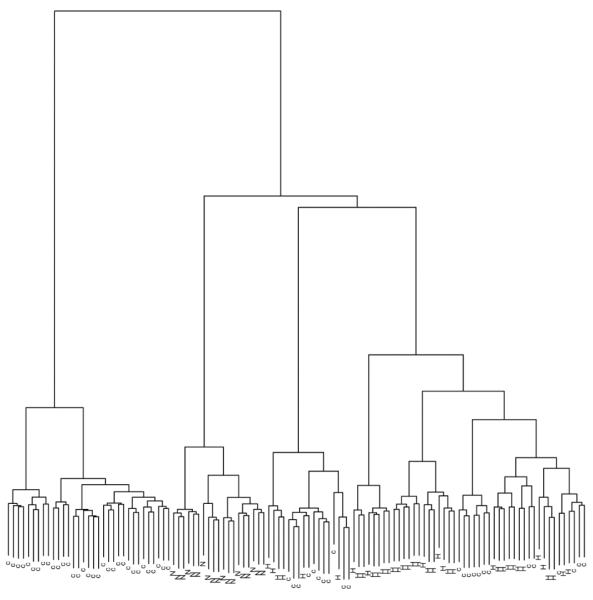
Agglomerative hierarchical clustering using Ward’s method on the 4650 probe sets retained after filtering. Labels indicate underlying tissue of origin as N = normal, c = pre-neoplastic tissue (HCV + cirrhosis) and H = neoplastic tissue (HCV + HCC)
4 Discussion
In this study, we identified genes that are progressively absent in pre-neoplastic and neoplastic liver tissue compared with normal liver tissue, as a proxy representing hypermethylation of gene promoter regions. The Affymetrix HG-U133A 2.0 Array and the detection call algorithm, which results in a gene being labelled as Present, Marginally Present, or Absent, were used in our assessment, thereby permitting us to examine the status (presence or absence) of thousands of genes simultaneously. To our knowledge, this is the first exploratory attempt at examining genome-wide methylation in a homogeneous etiology of HCV + HCC patients that did not restrict attention to known Tumour suppressor and oncogenic genes.
For lung cancer, the percent of genes with CpG islands in their promoter regions thought to be silenced by DNA methylation has been estimated to be between 0.5% and 3% (Rauch et al., 2006). Our results using a very conservative FDR of 5% identified 793 (5.98%) of 13,252 probe sets examined to have a significant monotonic increasing frequency of absent calls in pre-neoplastic and neoplastic liver tissues. Considering that the absence of such genes may be linked to promoter methylation, our estimate for the percent of genes significant and with CpG islands in their promoter regions thought to be silenced by DNA methylation in HCC is quite similar.
Methylation events are of particular interest in cancer research, since methylation has been shown to be reversed by demethylating agents. In fact, one approach in the study of methylation has been to analyse mRNA samples from cell lines treated and untreated with 5-Aza-2′-deoxycytidine (5-aza-dC – a demethylating agent) and Trichostatin A (TSA – a potent inhibitor of histone deacetylase) and identify differentially expressed genes. For example, expression of genes in HepG2 cell line was examined using the Affymetrix HG-U133 Plus 2.0 GeneChip before and after treatment with 5-aza-dC, TSA, and the combination of 5-aza-dC and TSA (Dannenberg and Edenberg, 2006). A larger effect on re-expression was observed for 5-aza-dC in comparison with TSA, implying that methylation plays a stronger role in gene silencing than histone deacetylation. Though the HCC cell line experiments indicate that methylation may play an important role in hepatocellular carcinogenesis, cell lines have been found to yield inconsistent results in comparison with results obtained from primary tumours. This may be due to progression of methylation in cultured cells (Jones and Laird, 1999). Further, only a small number of independent cell lines are available for study and often these cell lines differ widely from one another with respect to stage of disease and etiology. Therefore, primary tissue sample studies are preferred.
Another group has used Affymetrix HG-U133A 2.0 Array GeneChips to study methylation in pancreatic cancer (Sato et al., 2003). They declared a gene to be methylated if cell lines treated with 5-Aza-dC or TSA (or the combination) experienced a >5-fold increase in gene expression. Others have investigated methylation in glioma by treating glioma cell lines with 5-Aza-dC and TSA and then profiling the samples using Affymetrix GeneChip (Kim et al., 2006). In this same paper, the authors hybridised both normal brain and glioma tumour samples to Affymetrix GeneChips and identified genes differentially expressed comparing normal with tumour. While these authors confirmed that some genes were indeed methylated by performing follow-up studies using Methylation-Specific PCR (MSP) and bisulphite sequencing, we believe that genes identified as differentially expressed are not necessarily aberrantly methylated. Since promoter methylation is clinically relevant when it results in gene silencing (Eng et al., 2000), the statistical method used herein sought to identify genes frequently expressed (Present) in normal tissues and more frequently silenced (Absent) as tissues moved to pre-neoplastic and neoplastic states. However, in all such studies that have used the Affymetrix GeneChip technology to assess methylation, a limitation is that the hybridised material (cRNA) does not directly assess promoter methylation. Technologies for assessing methylation are relatively new, with MSP only being described approximately 10 years ago (Herman et al., 1996). Restriction Landmark Genomic Scanning (RLGS) was an early high-throughput technique for studying methylation (Eng et al., 2000) with many others having been more recently described (Rauch et al., 2006, 2007; Lippman et al., 2005; Nouzova et al., 2004; Adrien et al., 2006; Gebhard et al., 2006). Nevertheless, our results using the Affymetrix technology as an exploratory assessment yielded important findings and demonstrate that high-throughput methylation studies in HCC are warranted. Such studies may identify methylated regions that will lead to novel therapeutic interventions and prognostic indicators.
Supplementary Material
Acknowledgement
This research was supported in part by National Institute of Diabetes & Digestive & Kidney Diseases R01DK069859.
Biographical notes
Kellie J. Archer received her PhD in Biostatistics from the School of Public Health at The Ohio State University, Columbus, Ohio, in 2001. Currently, she is an Associate Professor in the Departments of Biostatistics and Surgery at Virginia Commonwealth University. Her research interests include developing and applying statistical methods for analysing high-throughput genomic data.
Zhongming Zhao received his PhD in Human and Molecular Genetics from the University of Texas and MD from Anderson Cancer Center, Houston, in 2000. Currently, he is an Associate Professor in the Departments of Biomedical Informatics, Psychiatry, and Cancer Biology at Vanderbilt University Medical Center. His research interests include bioinformatics and systems biology approaches to studying complex diseases, genome-wide or large-scale analysis of genetic variation and methylation patterns, next-gen sequence analysis, comparative genomics and biomedical informatics.
Tobias Guennel is a Graduate Research Assistant in the Department of Biostatistics at Virginia Commonwealth University. He is a PhD student with an interest in genomic biostatistics.
Daniel G. Maluf completed his MD at the University of Cordoba, Argentina, in 1993. He is currently an Associate Professor in the Department of Surgery at Virginia Commonwealth University. His research interests pertain to identifying variables capable of predicting outcomes in liver and kidney transplant recipients.
Robert A. Fisher completed his MD at Baylor College of Medicine, Texas, in 1980. He is currently a Professor in the Department of Surgery at Virginia Commonwealth University. His research interests include identifying short and long-term risks of morbidity and mortality for the adult donor to determine the efficacy of living donor liver transplant for the adult recipient and in comparing outcomes of living donor liver transplant recipients with recipients who received a cadaveric liver.
Valeria R. Mas received her PhD in Biochemistry from the National University of San Luis, Argentina, in 2002. Currently, she is an Associate Professor in the Departments of Surgery and Pathology at Virginia Commonwealth University. Her research interests pertain to identifying gene expression signatures and molecular markers capable of predicting outcomes in transplant recipients.
Contributor Information
Kellie J. Archer, Department of Biostatistics, Virginia Commonwealth University, Richmond, Virginia 23298-0032 USA.
Zhongming Zhao, Vanderbilt University Medical Center, Departments of Biomedical Informatics, Psychiatry, and Cancer Biology, Memphis, Tennessee 37232, USA zhongming.zhao@Vanderbilt.Edu.
Tobias Guennel, Department of Biostatistics, Virginia Commonwealth University, Richmond, Virginia 23298-0032, USA tguennel@vcu.edu.
Daniel G. Maluf, Department of Surgery, Virginia Commonwealth University, Richmond, Virginia 23298, USA dmaluf@vcu.edu
Robert A. Fisher, Department of Surgery, Virginia Commonwealth University, Richmond, Virginia 23298, USA rafisher@vcu.edu
Valeria R. Mas, Departments of Surgery and Pathology, Virginia Commonwealth University, Richmond, Virginia 23298, USA vmas@mcvh-vcu.edu
References
- Adrien L, Schlecht N, Kawachi N, Smith R, Brandwein-Gensler M, Massimi A, Chen S, Prystowsky M, Childs G, Belbin T. Classification of DNA methylation patterns in Tumour cell genomes using a CpG island microarray. Cytogenet. Genome. Res. 2006;Vol. 114(1):16–23. doi: 10.1159/000091923. [DOI] [PubMed] [Google Scholar]
- Baylin S, Belinsky S, Herman J. Aberrant methylation of gene promoters in cancer-concepts, misconcepts, and promise. Journal of the National Cancer Institute. 2000;Vol. 92(18):1460–1461. doi: 10.1093/jnci/92.18.1460. [DOI] [PubMed] [Google Scholar]
- Baylin S, Esteller M, Roundtree M, Bachman K, Schuebel K, Herman J. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Human Molecular Genetics. 2001;Vol. 10(7):687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- Beer D, Kardia S, Huang C, Giordano T, Levin A, Misek D, Lin L, Chen G, Gharib T, Thomas D, Lizyness M, Kuick R, Hayasaka S, Taylor J, Iannettoni M, Orringer M, Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Medicine. 2002;Vol. 8(8):816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Calvisi D, Ladu S, Gorden A, Farina M, Lee J, Conner E, Schroeder I, Factor V, Thorgeirsson S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007;Vol. 117(9):2713–2722. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg L, Edenberg H. Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics. 2006;Vol. 7:181. doi: 10.1186/1471-2164-7-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C, Herman J, Baylin S. A bird’s eye view of global methylation. Nature Genetics. 2000;Vol. 24:101–102. doi: 10.1038/72730. [DOI] [PubMed] [Google Scholar]
- Esteller M. CpG island hypermethylation and Tumour suppressor genes: a booming present, a brighter future. Oncogene. 2002;Vol. 21:5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;Vol. 196(2):261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad B, Irizarry R. Affy-analysis of affymetrix genechip data at the probe level. Bioinformatics. 2004;Vol. 20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gebhard C, Schwarzfischer L, Pham T, Schilling E, Klug M, Andreesen R, Rehli M. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 2006;Vol. 66(12):6118–6128. doi: 10.1158/0008-5472.CAN-06-0376. [DOI] [PubMed] [Google Scholar]
- Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004;Vol. 5(10):R80.l–R80.16. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub T, Slonim D, Tamayo P, Huard C, Gaasenbeek M, Mesirov J, Coller H, Loh M, Downing J, Caligiuri M, Bloomfield C, Lander E. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;Vol. 286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Herman J, Graff J, Myohanen S, Nelkin B, Baylin S. Methylation-specific per: a novel per assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences. 1996;Vol. 93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa Y, Arai M, Imazeki F, Tada M, Mikata R, Fukai K, Miyazaki M, Ochiai T, Saisho H, Yokosuka O. Methylation status of genes upregulated by demethylating agent 5-aza-2′-deoxycytidine in hepatocellular carcinoma. Oncology. 2006;Vol. 71:77–85. doi: 10.1159/000100475. [DOI] [PubMed] [Google Scholar]
- Hsu L, Lee H, Chau G, Yin P, Chi C, Lui W. Aberrant methylation of ednrb and pl6 genes in hepatocellular carcinoma (hec) in Taiwan. Oncol. Rep. 2006;Vol. 15(2):507–511. [PubMed] [Google Scholar]
- Iizuka N, Hamamoto Y, Oka M. Predicting individual outcomes in hepatocellular carcinoma. Lancet. 2004;Vol. 364(9448):1837–1839. doi: 10.1016/S0140-6736(04)17455-2. [DOI] [PubMed] [Google Scholar]
- Iizuka N, Oka M, Yamada-Okabe H, Nishida M, Maeda Y, Mori N, Takao T, Tamesa T, Tangoku A, Tabuchi H, Hamada K, Nakayama H, Ishitsuka H, Miyamoto T, Hirabayashi A, Uchimura S, Hamamoto Y. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. The Lancet. 2003;Vol. 361:923–929. doi: 10.1016/S0140-6736(03)12775-4. [DOI] [PubMed] [Google Scholar]
- Jiang C, Han L, Su B, Li W, Zhao Z. Features and trend of loss of promoter-associated CpG islands in the human and mouse genomes. Mol. Biol. Evol. 2007;Vol. 24(9):1991–2000. doi: 10.1093/molbev/msm128. [DOI] [PubMed] [Google Scholar]
- Jicai Z, Zongtao Y, Jun L, Haiping L, Jianmin W, Lihua H. Persistent infection of hepatitis b virus is involved in high rate of pl6 methylation in hepatocellular carcinoma. Mol. Carcinog. 2006;Vol. 45(7):530–536. doi: 10.1002/mc.20188. [DOI] [PubMed] [Google Scholar]
- Jones P, Laird P. Cancer epigenetics comes of age. Nature Genetics. 1999;Vol. 21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw P. Finding Groups in Data: An Introduction to Cluster Analysis. John Wiley & Sons; New York: 1990. [Google Scholar]
- Kim T, Zhong S, Fields C, Kim J, Robertson K. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006;Vol. 66(15):7490–7501. doi: 10.1158/0008-5472.CAN-05-4552. [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel A, Colot V, Martienssen R. Profiling DNA methylation patterns using genomic tiling microarrays. Nat. Methods. 2005;Vol. 2(3):219–224. doi: 10.1038/nmeth0305-219. [DOI] [PubMed] [Google Scholar]
- Liu W-M, Mei R, Di X, Ryder T, Hubbell E, Dee S, Webster T, Harrington C, Ho M, Baid J, Smeekens S. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics. 2002;Vol. 18(12):1593–1599. doi: 10.1093/bioinformatics/18.12.1593. [DOI] [PubMed] [Google Scholar]
- Mas V, Maluf D, Archer K, Yanek K, Williams B, Fisher R. Differentially expressed genes between early and advanced hepatocellular carcinoma (hec) as a potential tool for selecting liver transplant recipients. Molecular Medicine. 2006;Vol. 12:97–104. doi: 10.2119/2006-00032.Mas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaughan G, Koorey D, Strasser S. Hepatocellular carcinoma: current approaches to diagnosis and management. Internal Medicine Journal. 2002;Vol. 32:394–400. doi: 10.1046/j.1445-5994.2002.00227.x. [DOI] [PubMed] [Google Scholar]
- Moinzadeh P, Breuhahn K, Stutzer H, Schirmacher P. Chromosomealterations in human hepatocellular carcinomas correlate with aetiology and histological grade – results of an explorative cgh meta-analysis. British Journal of Cancer. 2005;Vol. 92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam S, Park J, Ramasamy A, Shevade S, Islam A, Long P, Park C, Park S, Kim S, Lee S, Park W, Yoo N, Liu E, Miller L, Lee J. Molecular changes from dysplastic nodule to hepatocellular carcinoma through gene expression profiling. Hepatology. 2005;Vol. 42(4):809–818. doi: 10.1002/hep.20878. [DOI] [PubMed] [Google Scholar]
- Nouzova M, Holtan N, Oshiro M, Isett R, Munoz-Rodriguez J, List A, Narro M, Miller S, Merchant N, Futscher B. Epigenomic changes during leukemia cell differentiation: analysis of histone acetylation and cytosine methylation using CpG island microarrays. J. Pharmacol. Exp. Ther. 2004;Vol. 311(3):968–981. doi: 10.1124/jpet.104.072488. [DOI] [PubMed] [Google Scholar]
- Poon T, Wong N, Lai P, Rattray M, Johnson P, Sung J. A tumour progression model for hepatocellular carcinoma: bioinformatic analysis of genomic data. Gastroenterology. 2006;Vol. 131:1262–1270. doi: 10.1053/j.gastro.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Qiu G, Xie H, Wheelhouse N, Harrison D, Chen G, Salto-Tellez M, Lai P, Ross J, Hooi S. Differential expression of hdab2ipa and hdab2ipb in normal tissues and promoter methylation of hdab2ipa in hepatocellular carcinoma. J. Hepatol. 2007;Vol. 46(4):655–663. doi: 10.1016/j.jhep.2006.11.012. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- Rauch T, Li H, Wu X, Pfeifer G. Mira-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;Vol. 66(16):7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau S, Kernstine K, Riggs A, Pfeifer G. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc. Natl. Acad. Sci. USA. 2007;Vol. 104(13):5527–5532. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan W, Connors J, Campo E, Fisher R, Gascoyne R, Muller-Hermelink H, Smeland E, Giltnane J, Hurt E, Zhao H, Averett L, Yang L, Wilson W, Jaffe E, Simon R, Klausner R, Powell J, Duffey P, Longo D, Greiner T, Weisenburger D, Sanger W, Dave B, Lynch J, Vose J, Armitage J, Montserrat E, Lopez-Guillermo A, Grogan T, Miller T, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt L. The use of molecular profiling to predict survival after chemotherapy for diffuse large-b-cell lymphoma. New England Journal of Medicine. 2002;Vol. 346(25):1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo C, Cameron J, Hruban R, Goggins M. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;Vol. 63(13):3735–3742. [PubMed] [Google Scholar]
- Shih Y, Shyu R, Hsieh C, Lai H, Liu K, Chu T, Lin Y. Promoter methylation of the secreted frizzled-related protein 1 gene sfrpl is frequent in hepatocellular carcinoma. Cancer. 2006;Vol. 107(3):579–590. doi: 10.1002/cncr.22023. [DOI] [PubMed] [Google Scholar]
- Simonoff J. Analyzing Categorical Data. Springer; New York: 2003. [Google Scholar]
- Storey J, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;Vol. 100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones P. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA. 2002;Vol. 99(6):3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchou J, Lin X, Freije D, Isaacs W, Brooks J, Rashid A, De Marzo A, Kanai Y, Hirohashi S, Nelson W. Gstpl CpG island DNA hypermethylation in hepatocellular carcinomas. International Journal of Oncology. 2000;Vol. 16:663–676. doi: 10.3892/ijo.16.4.663. [DOI] [PubMed] [Google Scholar]
- Uematsu F, Takahashi M, Yoshida M, Igarashi M, Nakae D. Methylation of neutral endopeptidase 24.11 promoter in rat hepatocellular carcinoma. Cancer Sci. 2006;Vol. 97(7):611–617. doi: 10.1111/j.1349-7006.2006.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vijver M, He Y, van’t Veer L, Dai H, Hart A, Voskuil D, Schreiber G, Peterse J, Roberts C, Marton M, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers E, Friend S, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. New England Journal of Medicine. 2002;Vol. 347(25):1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- van’t Veer L, Dai H, van de Vijver M, He Y, Hart A, Bernards R, Friend S. Expression profiling predicts outcome in breast cancer. Breast Cancer Res. 2003;Vol. 5(1):57–58. doi: 10.1186/bcr562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Veer L, Dai H, van de Vijver M, He Y, Hart A, Mao M, Peterse H, van der Kooy K, Marton M, Witteveen A, Schreiber G, Kerkhoven R, Roberts C, Linsley P, Bernards R, Friend S. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;Vol. 415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang J, Qin Y, Li B, Sun Z, Yang B. Detection of aberrant promoter methylation of gstpl in the Tumour and serum of Chinese human primary hepatocellular carcinoma patients. Clin. Biochem. 2006;Vol. 39(4):344–348. doi: 10.1016/j.clinbiochem.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Waterston R, Lindblad-Toh K, Birney E. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;Vol. 420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wong I, Lo Y, Yeo W, Lau W, Johnson P. Frequent pl5 promoter methylation in Tumour and peripheral blood from hepatocellular carcinoma patients. Clin. Cancer Res. 2000;Vol. 6(9):3516–3521. [PubMed] [Google Scholar]
- Wong I, Lo Y, Zhang J, Liew C-T, Ng M, Wong N, Lai P, Lau W, Hjelm N, Johnson P. Detection of aberrant pl6 methylation in the plasma and serum of liver cancer patients. Can. Res. 1999;Vol. 59:71–73. [PubMed] [Google Scholar]
- Wurmbach E, Chen Y, Khitrov G, Zhang W, Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J, Bottinger E, Friedman S, Waxman S, JM L. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;Vol. 45(4):938–947. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- Yamashite R, Suzuki Y, Wakaguir H. Dbtss: Database of human transcription start sites, progress report 2006. Nucleic Acids Res. 2006;Vol. 34:D86–D89. doi: 10.1093/nar/gkj129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Gao Y, Du Z, Zhao L, Song W. Methylation-based molecular margin analysis in hepatocellular carcinoma. Biochem Biophys Res Commun. 2005;Vol. 338(3):1353–1358. doi: 10.1016/j.bbrc.2005.10.095. [DOI] [PubMed] [Google Scholar]
- Yang B, Guo M, Herman J, Clark D. Aberrant promoter methylation profiles of Tumour suppressor genes in hepatocellular carcinoma. Am. J. Pathol. 2003;Vol. 163(3):1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rossner P, J., Chen Y, Agrawal M, Wang Q, Wang L, Ahsan H, Yu M, Lee P, Santella R. Aflatoxin bl and polycyclic aromatic hydrocarbon adducts, p53 mutations and pl6 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int. J. Cancer. 2006;Vol. 119(5):985–991. doi: 10.1002/ijc.21699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.