

Abstract
Metformin is the most commonly prescribed drug for type II diabetes and is associated with decreased cancer risk. Previously, we showed that metformin prevented tobacco carcinogen (NNK)-induced lung tumorigenesis in a non-diabetic mouse model, which was associated with decreased IGF-I/insulin receptor signaling but not activation of AMPK in lung tissues, as well as decreased circulating levels of IGF-1 and insulin. Here, we used liver-IGF-1-deficient (LID) mice to determine the importance of IGF-1 in NNK-induced lung tumorigenesis and chemoprevention by metformin. LID mice had decreased lung tumor multiplicity and burden compared to WT mice. Metformin further decreased lung tumorigenesis in LID mice without affecting IGF-1 levels, suggesting that metformin can act through IGF-1-independent mechanisms. In lung tissues, metformin decreased phosphorylation of multiple receptor tyrosine kinases (RTKs) as well as levels of GTP-bound Ras independently of AMPK. Metformin also diminished plasma levels of several cognate ligands for these RTKs. Tissue distribution studies using [14C]-metformin showed that uptake of metformin was high in liver but 4 fold lower in lungs, suggesting that the suppression of RTK activation by metformin occurs predominantly via systemic, indirect effects. Systemic inhibition of circulating growth factors and local RTK signaling are new AMPK-independent mechanisms of action of metformin that could underlie its ability to prevent tobacco carcinogen-induced lung tumorigenesis.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide, and no effective chemopreventive agents currently exist (1). Because a majority of lung cancers are associated with tobacco use (85–90%), the development of chemopreventive agents is a priority for current or former smokers at high-risk for this disease. The most common molecular driver in smoking-related lung cancer is K-ras, which is mutated in 20–30% of lung adenocarcinomas. Currently, no therapies against KRAS exist (2).
To address the need for targeted chemopreventive agents, strategies to inhibit ancillary pathways that cooperate with KRAS to decrease lung tumor formation are being developed. An example of such a strategy includes modulation of the insulin-like growth factor receptor (IGF-1R) pathway, which plays a critical role in cell metabolism, growth and development (3, 4). While elevated levels of circulating IGF-1 are associated with increased risk of breast, prostate and colorectal cancers (5–7), it is unclear whether such an association exists in lung cancer, possibly due to a lower incidence of obesity in smokers at high risk to develop the disease (8). Preclinical studies show that overexpression of IGF-1 in alveolar type II cells in lung tissues increases spontaneous tumor formation and synergizes with tobacco carcinogen (TC) exposure to accelerate lung tumorigenesis (9). Treatment with a specific IGF-1R inhibitor significantly decreased IGF-1/TC–induced lung tumor progression in this model (9). Although lung cancer clinical trials with drugs that inhibit IGF-1R such as figitumumab, an anti-IGF-1R antibody, have been disappointing, high level of circulating IGF-1 were retrospectively identified as a possible predictive biomarker of clinical benefit (10). These studies suggest IGF-1 plays a role in lung carcinogenesis and that genetic and pharmacologic manipulation of IGF-1 in murine models of lung cancer might better define its role.
Metformin (1,1-dimethylbiguanide) is the most commonly prescribed anti-diabetic drug in the world. In diabetics, metformin activates AMP-activated protein kinase (AMPK) in the liver, which inhibits hepatic gluconeogenesis with subsequent decreases in insulin and IGF-1 (11, 12). Population-based studies show that metformin use is associated with decreased cancer risk, suggesting a potential role as an anti-cancer agent (13, 14). The mechanism of metformin action in cancer remains unclear, and recent evidence suggests that metformin exerts antineoplastic effects through pathways independent of AMPK, including inhibition of mammalian target of rapamycin (mTOR) (15, 16). The fact that metformin must be actively transported into cells by transporters such as the organic cation transporter-1 (OCT-1), that are primarily expressed in the liver raises questions as to whether metformin has direct effects in target tissues (17, 18). Therefore, distinguishing between direct cellular actions and indirect systemic effects is critical for determining how metformin prevents cancer.
Our laboratory has shown that metformin prevents tobacco carcinogen (NNK)-induced lung tumorigenesis (19) in a model that is characterized by mutation in KRAS- and increased activation of the mTOR pathway (20–22). Oral metformin reduced lung tumor burden by 40–50%, while intraperitoneal (IP) metformin decreased tumor burden by 72% (19). Despite the fact that AMPK was not activated in the lungs of metformin-treated mice, significant inhibition of IGF-1R/IR and downstream mTOR signaling was observed (19). Metformin also decreased circulating levels of insulin and insulin-like growth factor-1 (IGF-1) by ~35% and ~21% respectively (19). To explain these data, we hypothesized that either modest but prolonged suppression of IGF-1/insulin signaling by metformin is sufficient to inhibit lung tumorigenesis, or that inhibition of additional signaling pathways are involved. In support of the second hypothesis, metformin use has been associated with decreases in several growth factors and hormones in patients with diabetes or polycystic ovarian syndrome (23, 24).
Here, we investigate the role of IGF-I in the ability of metformin to prevent lung tumorigenesis. Liver IGF-1-deficient (LID) mice, which have a 75% reduction in circulating IGF-1 levels (25), exhibited a marked decrease in NNK-induced lung tumorigenesis compared to WT mice. Metformin further prevented tumorigenesis in LID mice, which was associated with inhibition of several RTK signaling networks in lung tissues independently of local AMPK activation. Biodistribution studies showed that lung tissues had low metformin uptake relative to liver, supporting the concept of indirect mechanisms of chemoprevention by metformin in target tissues. These studies not only identify IGF-I as a target for lung cancer chemoprevention, but also describe a novel, systemic mode of metformin action that may underlie its chemopreventive effect in a variety of tissues.
Results
Mice with reduced IGF-1 levels exhibit decreased susceptibility to lung tumorigenesis, which is potentiated by metformin treatment
To specifically determine the role of circulating IGF-1 in NNK-induced lung tumorigenesis, we employed LID mice that were backcrossed 7 generations into the A/J strain, thereby inducing susceptibilty to NNK. After treatment with NNK, lung tumor multiplicity, size and burden were significantly decreased in LID mice compared to WT mice (67%, 40% and 74% decrease, respectively) (Figure 1A, B, C). In WT mice, oral metformin decreased lung tumor multiplicity and burden to a similar extent as our previous study (64% and 77% decrease, respectively). LID mice treated with metformin had further inhibition of lung tumorigenesis. Tumor multiplicity and burden were decreased significantly in LID mice in the metformin group compared to the control group (76% and 85%, respectively). These data indicate that basal levels of IGF-1 determine response to NNK and that metformin further potentiates the protective effect of IGF-1 loss.
Figure 1. Reduced susceptibility to NNK-induced lung tumorigenesis in liver-IGF-1 deficient mice is potentiated by metformin treatment.

(A) Tumor multiplicity, (B) tumor volume and (C) and tumor burden were assessed in WT or LID mice treated with or without 13 weeks of 5 mg/mL oral metformin after NNK exposure (n = 8 mice per group). Each point represents one mouse and lines represent mean values ± SD, *p<0.01. Plasma IGF-1 (D), plasma insulin (E) and blood glucose (F) were measured in mice at the end of the tumorigenesis study (n = 4 mice per group).
Because LID mice have lower but not absent levels of circulating IGF-1, it was possible that the additive effect of metformin could be due to ablation of the remaining circulating IGF-1. Therefore, we measured plasma IGF-1 levels in the four groups at the end of the tumorigenesis study. LID mice had IGF-1 levels 75% lower than WT mice (Figure 1D), consistent with previous studies. In WT mice, metformin decreased levels of IGF-1 by 28%. In LID mice, metformin had no effect on the remaining circulating IGF-1, disproving the hypothesis that metformin works through further decreasing IGF-I levels in LID mice.
Because metformin decreases insulin levels in diabetics, insulin levels were also measured, even though this is not a diabetic model. LID mice had 3-fold higher levels of insulin compared to WT mice (Figure 1E), and all treatment groups were normoglycemic (Figure 1F), which has been previously described. Although there was a trend towards a small decrease in insulin levels in WT mice with metformin, it did not reach statistical significance. Metformin decreased insulin levels by 36% in LID mice, but did not return insulin to basal levels observed in WT mice. Taken together, metformin modestly decreases IGF-I but not insulin in WT mice. In LID mice, it has no effect on IGF-I but modestly decreases insulin levels. These studies show that circulating IGF-1 promotes lung tumorigenesis, and that metformin potentiates the preventive effect of IGF-1 loss, suggesting that other signaling pathways are involved.
Metformin inhibits phosphorylation of multiple receptor tyrosine kinases (RTKs) in lung tissues and decreases circulating levels of their cognate ligands
To investigate other potential targets of metformin, we assessed the activation status of 39 RTKs in lung tissues from mice given three daily injections of metformin (representative RTK-array blots in Figure 2A). 18 of the 39 RTKs exhibited greater than a 40% reduction in phosphorylation with metformin (p<0.05) (Figure 2B). Many of these RTKs have been linked to lung tumorigenesis or have been identified as molecular targets in lung cancer, including EGFRs (26), FGFRs (27, 28), c-Met (29), and VEGFRs (30). To validate the changes in RTK signaling, immunoblotting for specific RTKs was performed based on antibody availability. We confirmed significant decreases in phosphorylation of IGF-1R/IR, EGFR, VEGFR2, cMET and FGFR without changes in total levels of these RTKs (Figure 2C and Supplemental Figure 1). The change in phosphorylation of certain RTKs from the array in Figure 2A, such as Axl and TrkC, could not be confirmed by immunoblotting (Supplemental Figure 2). Therefore, these receptors were not pursued further. As reported previously, no activation of AMPK was observed in lung tissues. To assess whether RTK inhibition by metformin could be attributed to decreases in their respective circulating ligands, quantitative ELISA assays for EGF, HGF, FGF and VEGF were performed on plasma from the same mice treated with metformin (Figure 2D). Metformin modestly, but significantly, decreased circulating plasma levels of EGF, HGF and FGF by 32%, 24% and 31%, respectively. The same circulating growth factors were also decreased by 13 weeks of oral metformin administration in the NNK-treated mice (Supplemental Figure 3). No differences in VEGF levels were observed (data not shown). Together, these data suggest that metformin inhibits multiple RTKs in lung tissues by decreasing levels of their circulating ligands.
Figure 2. Metformin inhibits activation of multiple lung RTKs and decreases levels of their circulating ligands.

(A) A/J mice were treated with metformin (250 mg/kg ip qdX3). One hour after the last injection, lungs were harvested and snap frozen. Lung lysates were analyzed using mouse phospho-RTK arrays (R&D Systems). (B) Densitometry was performed these arrays using Image J software to compare vehicle and metformin treated samples. RTKs listed are those that exhibited at least a 40% decrease in signal and statistical significance (p<0.05, n = 8). (C) To confirm the RTK array results, western blotting was performed on parallel samples. (D) Plasma from these mice was also collected and growth factor levels were measured using the appropriate ELISA kit. (E) A/J mice were exposed to NNK (100 mg/kg ip qdX3) to induce lung tumorigenesis, and then treated with 5 mg/mL metformin in drinking water for 13 weeks. Immunoblotting analysis of lung lysates was performed for RTKs and downstream components of the Ras/Mek/Erk and Akt/mTOR pathways.
To confirm modulation of RTK activation, we analyzed RTK status after oral administration of metformin under conditions where lung tumorigenesis was inhibited (Figure 2E). The same RTKs that were inhibited by short-term injection of metformin were also inhibited by 13 weeks of oral administration. However, the response of RTKs in lung tissues to oral metformin appeared to depend on administration of NNK. For example, oral administration of metformin for 13 weeks without NNK did not yield observable decreases in lung RTK phosphorylation (Supplemental Figure 4). Because RTKs require Ras for signal propagation and NNK induces mutations in KRAS with subsequent mTOR activation, we analyzed the Ras/MEK/ERK and Akt/mTOR pathways in lungs from these mice. Oral metformin suppressed the formation of active, GTP-bound Ras, phosphorylation of MEK and ERK, and phosphorylation of Akt and S6 (a substrate of mTOR) (Figure 2E), indicating broad inhibition of RTK-stimulated signaling networks by metformin. Broad inhibition of local RTK signaling could create an unfavorable environment for the development and progression of NNK-induced lung tumors.
Metformin decreases RTK activation in the absence of AMPK in vitro
AMPK is the best-studied molecular target of metformin; however, no changes in AMPK activation were observed in metformin-treated lungs. To investigate the dependence of RTK inhibition on AMPK, we analyzed RTK phosphorylation and downstream signaling in vitro using AMPK-deficient MEFs. Metformin inhibited phosphorylation of the 5 RTKs that were decreased by metformin in vivo (IGF-1R/IR, EGFR, VEGFR2, cMET and FGFR), The Ras/Mek/Erk and Akt/mTOR pathways were also inhibited in a dose-dependent manner (Figure 3). These studies confirm that RTK inhibition by metformin is independent of AMPK.
Figure 3. Metformin inhibits activation of RTKs and downstream targets independently of AMPK.
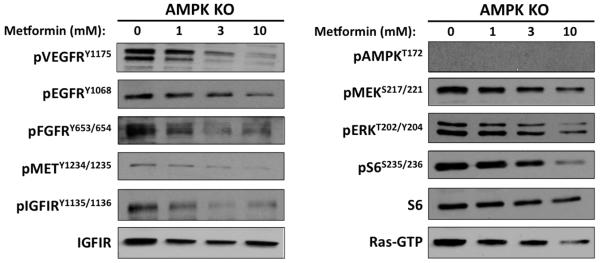
AMPK∝ −/− MEFs were treated for 24h with the indicated doses of metformin. Immunoblots are representative of three independent experiments.
Metformin exhibits tissue specific distribution
Although the in vitro studies confirm inhibition of RTK signaling by metformin, these experiments required millimolar concentrations of metformin that can rarely, if ever, be achieved in patients, as steady-state concentrations of metformin in diabetic patients are 0.5–2 μg/mL (~4–15 μM) (31), Therefore, we hypothesized that systemic effects of metformin are primarily responsible for decreased RTK signaling and decreased lung tumorigenesis, rather than direct effects of metformin in lung tissues. To directly measure metformin uptake, we performed a biodistribution study in mice using 14C-labelled metformin. Three different routes of administration were tested: IP (Figure 4A), gavage (Figure 4B) and oral ad libitum (Figure 4C). For the IP and gavage studies, tissues were harvested one hour after administration, the time at which Cmax occurs in patients. With IP or gavage administration, metformin levels were ~4.5 times higher in liver than in lungs, and the overall distribution patterns were similar. Only kidney and colon had higher metformin levels than liver with IP injection, which could be due to the proximity of injections to these organs that could result in artificially high local concentrations. In mice that were allowed to drink oral metformin ad libitum, absolute levels of metformin were extremely low compared to IP or gavage administration (Supplemental Figure 5A). Uptake in tissues was more uniform in the ad libitum oral group compared to IP and gavage, suggesting that metformin equilibrates across tissues after continuous low-dose exposure (Supplemental Figure 5B). The higher uptake of metformin in livers compared to lung tissues is consistent with higher levels of expression of metformin transporters and the ability of metformin to activate AMPK in liver tissues.
Figure 4. Biodistribution of metformin in mice.

A/J mice were injected IP with metformin (250 mg/kg containing [14C]-metformin at 25 mCi/kg body weight) (A), given metformin by gavage (250 mg/kg containing [14C]-metformin at 25 mCi/kg body weight) (B), or allowed to drink ad libitum metformin dissolved in drinking water (5 mg/mL containing [14C]-metformin at 25 mCi/kg body weight)(C). One hour after injection or gavage, or after five days of oral metformin, organs were harvested and wetweighed. Radioactivity was measured in a gamma-scintillation counter and converted to mol of metformin per kg of tissue weight. Bar graphs represent metformin uptake relative to liver (n = 6 mice for each administration route).
Metformin does not alter energy metabolism in lungs
Metformin administration in mice has been shown to reduce the energy state in the liver, resulting in a 3-fold increase in AMP/ATP ratio compared with control mice (32). It also hampers the mitochondrial complex I in hepatocytes, which inhibits oxidative phosphorylation and induces glycolysis (33). Therefore, we tested whether metformin induces similar metabolic effects in lungs by measuring ATP and lactate levels. No changes in ATP levels were observed in metformin-treated mouse liver or lungs in our study (Figure 5A). Although this data is inconsistent with previous reports showing decreased hepatic ATP levels after metformin treatment (32), this could be due to administration route or mouse strain. For example, Foretz et al. used C57BL/6J mice treated orally for 5 days, whereas our study used A/J mice treated IP for 3 days. Metformin significantly increased lactate levels in liver, however lactate was unchanged in metformin-treated lungs (Figure 5B). These metabolites were also measured in metformin-treated IO33 cells in vitro, a cell line derived from a murine lung adenocarcinoma caused by NNK. In IO33 cells, millimolar concentrations of metformin decreased ATP levels and dose-dependently increased lactate levels (Figure 5C). Immunoblotting confirmed dose-dependent AMPK activation, as well as RTK inhibition (Figure 5D). Together, these results suggest that the ability of metformin to inhibit mitochondrial ATP production and induce glycolysis is not a relevant mechanism in lung tissues in vivo, consistent with the observation that AMPK is not activated in lung tissues after metformin treatment.
Figure 5. Metformin does not affect ATP or lactate production in lungs.

Liver (A) and lungs (B) from A/J mice treated with metformin (250 mg/kg ip qdX3), were harvested and subjected to the Enzylight ATP or lactate assay kits (BioAssay Systems). (C) ATP or lactate assays were performed on IO33 cells treated for 24h with metformin (D) Immunoblotting analysis of lysates from the same IO33 cells as in (C).
Discussion
The mechanism of action of metformin in cancer remains a controversial issue, given the abundant but conflicting data across tissues and cell lines. Initially, it was shown that the primary mode of metformin action was disruption of mitochondrial complex I, leading to a decreases in ATP production and activation of the energy sensor AMPK (33, 34). In this setting, metformin imposes a bio-energetic crisis that forces cells to adapt by shutting down catabolic processes and switching from oxidative phosphorylation to glycolysis (35). Indeed, metformin-induced activation of AMPK affects many metabolic processes in liver, muscle and fat leading to reduced glucose production and increased insulin sensitivity (11, 12, 36). However, these mechanisms may be more relevant for its anti-diabetic properties. In the setting of cancer, it was initially proposed that AMPK activation by metformin leads to inhibition of mTORC1 in a TSC1/2-dependent manner (37). However, metformin can suppress mTORC1 independently of AMPK activation (15, 16)(19).
Our current studies indicate that NNK-induced lung tumorigenesis is dependent upon the IGF-1/insulin signaling pathway. By utilizing LID mice that have decreased circulating IGF-1 levels, we showed that loss of IGF-1 alone is sufficient to markedly inhibit lung tumorigenesis. LID mice are protected from a variety of cancer types (38–40), but this is the first study showing decreased lung tumor susceptibility in this model. Although large phase III trials with drugs targeting IGF-1R such as figitumumab have not shown clinical benefit in advanced stage lung cancer, our study suggests that the IGF-1 pathway plays an important, early role in carcinogenesis, implying that IGF-I signaling may be a better target for cancer prevention. Administration of metformin to LID mice suppressed lung tumorigenesis even further, suggesting that even if IGF-I were an important target of metformin, it does not explain all of its anti-tumor effects. LID mice exhibit high insulin levels as a compensatory mechanism for reduced IGF-1, which maintains normoglycemia. Although it is possible that lowering insulin levels in LID mice by metformin might contribute its anti-tumor effects, we believe this is unlikely for several reasons. First, insulin levels were still 2 fold above baseline after metformin treatment. It is unlikely that this reduction explains the 90% reduction in lung tumor burden in LID mice treated with metformin. If insulin were an important promoter of lung tumorigenesis, we might expect the compensatory hyperinsulinemia observed in LID mice to counteract the loss of IGF-1 and tumorigenesis to be similar between LID and WT mice. This was not the case, as loss of IGF-I alone (and hyperinsulinemia) markedly reduced lung tumorigenesis. Second, activation of IGF-I/IR in lung tissues is much more responsive to exogenously administered IGF-1 than insulin (19). Although a role for insulin cannot be eliminated, it is unlikely that modulation of insulin by metformin significantly contributing to the decreased lung tumorigenesis in this model system.
The fact that metformin further suppressed lung tumorigenesis in LID mice without decreasing the levels of remaining IGF-1 led us to hypothesize that metformin was affecting other growth factor pathways. An unbiased RTK phosphorylation assay showed that metformin suppresses the activation of multiple RTKs, most notably EGFR, cMET, VEGFR and FGFR, all of which are implicated in lung tumorigenesis. Receptor tyrosine phosphorylation plays a critical role in oncogenesis and drug resistance, and aberrant activation of RTKs occurs frequently in cancer (41, 42). The Ras/Mek/Erk and Akt/mTOR pathways represent two intracellular points of convergence for RTK signaling, both of which are inhibited by metformin treatment.
Several reports have shown inhibitory effects of metformin on RTK regulation in breast cancer models, where metformin disrupted erbB2/IGF-1R complexes and inhibited erbB3 activity (43–45). Our study suggests that the effect of metformin on certain RTKs is due to decreased circulation of their respective ligands. The biological origins of these growth factors vary; IGF-I is primarly synthesized in the liver, EGF is secreted primarily by the salivary glands (46), HGF is secreted by mesenchymal and hematopoetic cells (47), and FGFs are produced in most epithelial cells (48). Growth factors are also upregulated and secreted by cancer cells to locally promote growth in an autocrine fashion (49). Based on the high hepatic uptake of metformin, we hypothesize that metformin primarily inhibits liver-derived growth factor production that ultimately inhibits RTK signaling in end organs. The mechanism by which metformin decreases serum levels of growth factors is unexplained. Because trace levels of metformin were observed in the lungs, we cannot rule out other local mechanisms that may contribute to decreased RTK activation, such as induction of RTK phosphatases such as PTP1B and PTPN12 (50). The basis for broad RTK inhibition is under investigation.
Ras mutations are found in approximately 30% of all human tumors (51), with KRAS mutations being the most prevalent in pancreatic (52, 53), colorectal (54) and lung cancers (55). Targeting Ras has become an active area of pharmaceutical development, with efforts focusing on inhibiting Ras expression, inhibiting membrane localization, modulating Ras interaction with GEFs or GAPs, or inhibiting Ras effectors (56–58). Unfortunately, no approved agents currently exist. This report identifies metformin as a well-tolerated, inexpensive FDA-approved drug that dampens RAS signaling through additive inhibition of upstream RTKs. Although NNK induces KRAS mutations in over 90% of lung tumors, the total tumor volume induced by NNK occupies a small portion of lung volume. Therefore, the RAS activity data obtained in these studies with lung lysates likely reflects a small contribution of tumor tissue and is indicative of the ability of metformin to inhibit wildtype Ras. It is unknown if metformin can inhibit the activity of mutant KRas. Nonetheless, the fact that metformin inhibits several RTKs and RAS at steady state levels similar to those observed in diabetic patients suggests that metformin might have activity in a variety of RAS- or RTK-driven cancers. We propose a novel systemic mechanism of action for metformin, whereby metformin creates a suboptimal environment for tumor growth in target tissues by limiting the availability of circulating growth factors that decreases activation of RTK signaling networks. Importantly, these studies show that the chemopreventive efficacy of metformin is not limited to diabetic models. Such mechanisms could be validated in clinical prevention trials, especially those that focus on current or former smokers at high-risk for lung cancer.
Materials and Methods
Materials
LID mice were generated by Dr. Derek LeRoith (Mount Sinai Hospital) as previously described (25) and kindly provided to us by Dr. Stephen Hursting (UT-Austin). NNK was purchased from Toronto Research Chemicals. Primary antibodies for immunoblotting analysis were purchased from Cell Signaling Technologies. Metformin (1,1-Dimethylbiguanide hydrochloride) was obtained from Sigma Chemicals. The Mouse Phospho-RTK Array kit and Growth Factor ELISAs were purchased from R&D Systems. The Ras Activation Assay Kit was purchased from Millipore. Metformin [biguanidine-14C] was obtained from Moravek Biochemicals and Radiochemicals. The Enzylight ATP and Enzylight Lactate Assay Kits were purchased from BioAssay Systems.
Administration of NNK and metformin to A/J Mice
All mice were housed according to the guidelines of the Animal Care and Use Committee under an approved animal protocol. LID mice were backcrossed six times into the A/J background to make them susceptible to NNK and genotyped using PCR for the presence of the Cre transgene and the LoxP sites flanking the fourth exon of the IGF-I gene, as described elsewhere (59). For the tumorigenesis studies, 8-week old mice were given three weekly injections of 100 mg/kg NNK as previously described (19). One week following the last dose of NNK, mice were randomized into a control group or a group that received 5 mg/mL metformin dissolved in drinking water (n = 8 mice per group). For short-term biomarker studies 8-week old mice were treated once daily for three days with 200 μL saline or 250 mg/kg metformin. One hour following the third injection, mice were anesthetized using isofluorane and blood was collected by cardiac puncture using BD vacutainer vials containing EDTA. Plasma was obtained by centrifugation. Lung and liver tissues were harvested and snap-frozen in liquid nitrogen. Tissues were crushed on dry ice and lysed in radioimmunoprecipitation (RIPA) buffer containing protease inhibitors (Complete; Roche Applied Science) and phosphatase inhibitor cocktails I and II (Sigma).
Receptor tyrosine kinase arrays
RTK arrays were performed on lung lysates according to the manufacturer's instructions. Briefly, 250 μg of protein were incubated with the array membrane containing antibodies for 39 different murine RTKs. Next, the arrays were incubated with anti-phospho-tyrosine-HRP detection antibody and then exposed to chemiluminescent reagents. Using Image J software, densitometry was performed on duplicate spots of each antibody, and expressed as a ratio over positive control spots in the upper right-hand corner of each array. Phosphorylation of each RTK was compared between vehicle and metformin treated samples (n=8 per group).
Western blot analysis
Immunoblotting analysis of tissue or cell lysates was done as described previously. Briefly, protein concentrations were determined using the BCA Protein Assay Kit (Pierce Biotechnology) and equal amounts of total protein were resolved on 10% SDS-PAGE gels. Proteins were transferred to nitrocellulose membranes, blocked in PBS containing 5% nonfat dry milk and 0.1% Tween-20, and then incubated overnight at 4°C with the appropriate primary antibody. For p-AMPK, p-IGF-1R/IR, and p-Met, antibodies were used at 1:500 dilutions. All other antibodies were diluted 1:1000. Bands were detected using horseradish peroxidase-labeled secondary antibodies (Cell Signaling Technologies) and enhanced chemiluminescence kit (GE Healthcare).
Cell Culture
IO33 cell lines were generated from A/J mouse lung adenocarcinomas induced by NNK, and kindly provided by Dr. Steven Belinsky in 2005 (60). Kras mutation was verified by sequencing in 2007, and cells used in experiments were within 8 passages of Kras mutation verification. AMPK AMPK∞ −/− MEFs were obtained from Dr. Keith Laderoute and were described previously (61). The genotype of these MEFs was determined by PCR and immunoblotting analysis (61), and confirmed in our laboratory upon receipt. Cells lines were incubated at 37°C in a 5.0% CO2 atmosphere and maintained in RPMI media supplemented with 5% (v/v) fetal bovine serum (FBS). Metformin was dissolved directly into media and cells were treated at the given concentrations for 24 hours. Cells were harvested in cold lysis buffer (50mM Tris, 300mM NaCl, 0.1% NP40) containing protease inhibitors (Complete; Roche Applied Science) and phosphatase inhibitor cocktails I and II (Sigma).
14C-Metformin biodistribution studies
8-week old A/J mice were injected IP with 200μL of metformin (250 mg/kg containing 14C-metformin at 100 μCi/kg body weight) prepared in 0.9% saline. One hour after injection, mice were sacrificed by cervical dislocation and tissues were harvested and wet-weighed. Tissues were solubilized for 4 hours at 50°C in Soluene-350 (PerkinElmer), and then added to 10mL of Ultima Gold (PerkinElmer) scintillation fluid. Radioactivity was measured using a liquid scintillation counter, and CPM was converted to μmol/kg of wet tissue. The amount of metformin uptake in tissues was expressed relative to liver uptake for each mouse. For gavage administration, 200 μL of metformin (5mg/mL containing 14C-metformin at 100 μCi/kg body weight) was administered and mice were sacrificed one hour later. For ad libitum oral administration, mice were given 5 mg/mL metformin containing 14C-metformin dissolved in drinking water for five days before sacrifice. Six mice per group were used for each administration route.
Growth factor ELISAs
Levels of various growth factors (EGF, FGF, HGF, VEGF) were analyzed from plasma using the appropriate Quantikine ELISA kits (R&D Systems) according to the manufacturer's protocol.
ATP and Lactate Assays
IO33 cells treated with varying concentrations of metformin for 24 hours, or mouse liver and lungs treated with metformin (250 mg/kg ip qdX3), were harvested and subjected to the Enzylight ATP or Lactate Assay Kits (BioAssay Systems), according to the manufacturer's protocol.
Statistics
Data are presented as mean ± SD. Statistical significance was determined between groups using an unpaired 2-tailed Student's t test. A p value less than 0.05 was considered significant.
Supplementary Material
Footnotes
Conflict of interest:
The authors declare that no conflict of interest exists.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10(4):241–53. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 3.Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene. 2009;28(34):3009–21. doi: 10.1038/onc.2009.172. [DOI] [PubMed] [Google Scholar]
- 4.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 5.Stattin P, Rinaldi S, Biessy C, Stenman UH, Hallmans G, Kaaks R. High levels of circulating insulin-like growth factor-I increase prostate cancer risk: a prospective study in a population-based nonscreened cohort. J Clin Oncol. 2004;22(15):3104–12. doi: 10.1200/JCO.2004.10.105. [DOI] [PubMed] [Google Scholar]
- 6.Key TJ, Appleby PN, Reeves GK, Roddam AW. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. The Lancet Oncol. 2010;11(6):530–42. doi: 10.1016/S1470-2045(10)70095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, et al. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Nat Can Inst. 1999;91(7):620–5. doi: 10.1093/jnci/91.7.620. [DOI] [PubMed] [Google Scholar]
- 8.Cao H, Wang G, Meng L, Shen H, Feng Z, Liu Q, et al. Association between circulating levels of IGF-1 and IGFBP-3 and lung cancer risk: a meta-analysis. PloS One. 2012;7(11):e49884. doi: 10.1371/journal.pone.0049884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim WY, Jin Q, Oh SH, Kim ES, Yang YJ, Lee DH, et al. Elevated epithelial insulin-like growth factor expression is a risk factor for lung cancer development. Cancer Res. 2009;69(18):7439–48. doi: 10.1158/0008-5472.CAN-08-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gualberto A, Hixon ML, Karp DD, Li D, Green S, Dolled-Filhart M, et al. Pretreatment levels of circulating free IGF-1 identify NSCLC patients who derive clinical benefit from figitumumab. Br J Cancer. 2010;104(1):68–74. doi: 10.1038/sj.bjc.6605972. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res. 2010;3(11):1451–61. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 14.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–72. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Dresser MJ, Gray AT, Yost SC, Terashita S, Giacomini KM. Cloning and functional expression of a human liver organic cation transporter. Mol Pharm. 1997;51(6):913–21. doi: 10.1124/mol.51.6.913. [DOI] [PubMed] [Google Scholar]
- 18.Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302(2):510–5. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- 19.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res. 2010;3(9):1066–76. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13(10):433–42. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, et al. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007;13(7):2281–9. doi: 10.1158/1078-0432.CCR-06-2570. [DOI] [PubMed] [Google Scholar]
- 22.Belinsky SA, Devereux TR, Maronpot RR, Stoner GD, Anderson MW. Relationship between the formation of promutagenic adducts and the activation of the K-ras protooncogene in lung tumors from A/J mice treated with nitrosamines. Cancer Res. 1989;49(19):5305–11. [PubMed] [Google Scholar]
- 23.Ersoy C, Kiyici S, Budak F, Oral B, Guclu M, Duran C, et al. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabetes Res Clin Pract. 2008;81(1):56–60. doi: 10.1016/j.diabres.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Hamet P, Sugimoto H, Umeda F, Lecavalier L, Franks DJ, Orth DN, et al. Abnormalities of platelet-derived growth factors in insulin-dependent diabetes. Metabolism. 1985;34(12 Suppl 1):25–31. doi: 10.1016/s0026-0495(85)80006-8. [DOI] [PubMed] [Google Scholar]
- 25.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci. 1999;96(13):7324–9. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 27.Riess JW, Neal JW. Targeting FGFR, ephrins, Mer, MET, and PDGFR-alpha in non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6(11 Suppl 4):S1797–8. doi: 10.1097/01.JTO.0000407562.07029.52. [DOI] [PubMed] [Google Scholar]
- 28.Semrad TJ, Mack PC. Fibroblast growth factor signaling in non-small-cell lung cancer. Clin Lung Cancer. 2012;13(2):90–5. doi: 10.1016/j.cllc.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 30.Herbst RS, Johnson DH, Mininberg E, Carbone DP, Henderson T, Kim ES, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23(11):2544–55. doi: 10.1200/JCO.2005.02.477. [DOI] [PubMed] [Google Scholar]
- 31.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 32.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–69. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 34.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 35.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–9. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 38.Moore T, Carbajal S, Beltran L, Perkins SN, Yakar S, Leroith D, et al. Reduced susceptibility to two-stage skin carcinogenesis in mice with low circulating insulin-like growth factor I levels. Cancer Res. 2008;68(10):3680–8. doi: 10.1158/0008-5472.CAN-07-6271. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res. 2002;62(4):1030–5. [PubMed] [Google Scholar]
- 40.Lashinger LM, Malone LM, McArthur MJ, Goldberg JA, Daniels EA, Pavone A, et al. Genetic reduction of insulin-like growth factor-1 mimics the anticancer effects of calorie restriction on cyclooxygenase-2-driven pancreatic neoplasia. Cancer Prev Res. 2011;4(7):1030–40. doi: 10.1158/1940-6207.CAPR-11-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature reviews Cancer. 2004;4(5):361–70. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 42.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21(2):140–6. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8(13):2031–40. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10(17):2959–66. doi: 10.4161/cc.10.17.16359. [DOI] [PubMed] [Google Scholar]
- 45.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40(8–9):685–93. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Ohlsson B, Jansen C, Ihse I, Axelson J. Epidermal growth factor induces cell proliferation in mouse pancreas and salivary glands. Pancreas. 1997;14(1):94–8. doi: 10.1097/00006676-199701000-00014. [DOI] [PubMed] [Google Scholar]
- 47.Zarnegar R, Michalopoulos GK. The many faces of hepatocyte growth factor: from hepatopoiesis to hematopoiesis. J Cell Bio. 1995;129(5):1177–80. doi: 10.1083/jcb.129.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 49.Sporn MB, Roberts AB. Autocrine growth factors and cancer. Nature. 1985;313(6005):745–7. doi: 10.1038/313745a0. [DOI] [PubMed] [Google Scholar]
- 50.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–18. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochimica et biophysica acta. 2005;1756(2):127–44. doi: 10.1016/j.bbcan.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Wang JY, Lian ST, Chen YF, Yang YC, Chen LT, Lee KT, et al. Unique K-ras mutational pattern in pancreatic adenocarcinoma from Taiwanese patients. Cancer Lett. 2002;180(2):153–8. doi: 10.1016/s0304-3835(01)00818-7. [DOI] [PubMed] [Google Scholar]
- 53.Shibata D, Almoguera C, Forrester K, Dunitz J, Martin SE, Cosgrove MM, et al. Detection of c-K-ras mutations in fine needle aspirates from human pancreatic adenocarcinomas. Cancer Res. 1990;50(4):1279–83. [PubMed] [Google Scholar]
- 54.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327(6120):293–7. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 55.Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131–9. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10(12):842–57. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3(14):1787–808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol ther. 2002;1(6):599–606. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- 59.Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, et al. Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol Endocrinol. 1998;12(9):1452–62. doi: 10.1210/mend.12.9.0162. [DOI] [PubMed] [Google Scholar]
- 60.Jones-Bolin SE, Johansson E, Palmisano WA, Anderson MW, Wiest JS, Belinsky SA. Effect of promoter and intron 2 polymorphisms on murine lung K-ras gene expression. Carcinogenesis. 1998;19(8):1503–8. doi: 10.1093/carcin/19.8.1503. [DOI] [PubMed] [Google Scholar]
- 61.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5'-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26(14):5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.