

Abstract
The administration of high dose synthetic estrogens was the first successful chemical therapy used in the treatment of metastatic breast cancer in postmenopausal women and this approach became the standard of care in postmenopausal women with metastatic breast cancer between 1950s and the end of the 1970s. The most recent analysis of the Women Health Initiative estrogen alone trial in hysterectomised women revealed a persistent significant decrease in the incidence of breast cancer as well as breast cancer mortality. Although estrogens are known to induce proliferation of breast cancer cells, we have shown that physiologic concentrations induce apoptosis in long term estrogen deprived breast cancer cells. We have developed laboratory models that illustrate the new biology of estrogen induced apoptosis or growth to explain the effects of estrogen therapy. The key to the success of estrogen therapy lies in a sufficient period of withdrawal of physiological estrogens (5-10years) and the subsequent regrowth of nascent breast tumor cells that survive under estrogen deprived conditions. These nascent tumors are now vulnerable to estrogen induced apoptosis.
Keywords: Breast cancer, Estradiol, Women Health Initiative, Apoptosis
Despite the extensive progress made in the management of breast cancer, it still remains the most common cause of cancer and the 2nd leading cause of cancer death in women in the United States. An estimated1 230,480 new cases of invasive breast cancer was projected to occur in 2011, as well as an estimated 57,650 cases of breast carcinoma in situ. In addition, approximately 39,520 women were expected to die from breast cancer in 20111.Multiple risk factors has been established for breast cancer and estrogen is a key growth stimulus in the development and progression of the disease. George Beatson2 provided the first medical evidence of the estrogen dependency of breast cancer in 1896. The conclusion that a woman's ovaries provided the fuel that maintained breast cancer was based on the observation of remission of advanced breast tumors in a premenopausal patient that underwent bilateral oophorectomy. Stanley Boyd3 surveyed all known cases in 1900 and concluded a 30% responsive rate, a figure that has stood the test of time for the response rate of breast cancer to any anti-hormone therapy. Animal models provided further evidence on the role of estrogens in breast cancer growth. Lanthrop and Loeb4 observed in 1916, a decrease in the occurrence of mammary carcinomas in castrated immature female mice. Estrogen, an ovarian hormone, was subsequently extracted and purified and induced vaginal cornification in ovarectomised mice5. This advance led to the elucidation of the biological properties of synthetic estrogens using ovariectomised mice, therefore establishing a connection between mitogenic potential of estrogens and breast cancer. The strategy of targeting the estrogen receptor (ER) has led to the discovery of endocrine therapies which function to either block estrogen action by using selective estrogen receptor modulators (SERMS) or depriving the ER of estrogens by using aromatase inhibitors6. Antihormonal therapies remain the gold standard of care in the treatment and prevention of ER positive breast cancer7.
The Women health Initiative: risks and benefits
The use of hormone therapy continues to be a source of controversial debate. The Women Health Initiative (WHI)8, is a set of clinical studies designed to investigate and develop strategies for the prevention and control of common causes of morbidity and mortality in postmenopausal women. The WHI was initiated in 1991 with a tentative end date in 2007 to provide research findings on the effects of postmenopausal hormone therapy, calcium and vitamin D supplements and diet modification on cardiovascular disease, osteoporosis, breast and colorectal cancer. The hormone therapy arm of the study includes random assignment of 27,500 women either to placebo, estrogen plus progestin (hormone therapy) or to estrogen alone (estrogen therapy) in hysterectomised women. The principal outcomes of the study were the incidence of coronary heart disease and osteoporosis, with breast cancer as a potential adverse outcome8. To date, this is the largest randomized placebo controlled trial that conducted parallel studies to assess the outcomes of combined hormone therapy or estrogen alone therapy9
Estrogen plus Progestin therapy
Treatment with hormone therapy (HT) was associated with elevated overall risks. Coronary heart disease(CHD) is a leading cause of death in postmenopausal women and previous animal studies show that estrogen treatment has the potential to prevent the development of coronary atherosclerosis10. Therefore results of the effect of HT on CHD were highly awaited. Patients received 0.625mg/day of conjugated equine estrogens (CEE) plus 2.5mg/day of medroxyprogesterone or placebo. After a mean follow up of 5.2 years, the trial was terminated because not only was the combination therapy not cardio-protective but also HT elevated the risk of CHD[Hazard ratio (HR),1.24;95%confidence interval (CI),1-1.54], which was most apparent at one year of therapy11. Furthermore HT was associated with doubling risk of venous thrombosis12, increased risk of stroke 13 and it did not confer protection against peripheral arterial disease14, dementia and cognitive decline15. Combined hormone therapy increased total (HR, 1.24; P<.001) and invasive (HR, 1.24; P = .003) breast cancers compared with placebo after 5 years of therapy16. The breast cancers in the group receiving HT were diagnosed initially at a slightly lower rate during the first two years of the study but subsequently increased throughout the intervention period. The elevated risk of breast cancer markedly declined soon after stopping the combined hormone therapy17. Short term use of HT was associated with a decrease in colorectal cancer when compared to placebo (p=0.0003) but no protective effect was observed on colorectal cancer mortality over an 8 year intervention and follow up period18. Although HT did not increase lung cancer rates19, more women from the combined therapy group died from lung cancer in particular from non-small cell lung cancer. In addition, there was no significant difference in the incidence of endometrial cancer and ovarian cancer in both treatment arms20. The benefits of HT include a significantly decreased incidence of bone fractures 21. Seven hundred thirty-three women (8.6%) in the estrogen-plus-progestin group and 896 women (11.1%) in the placebo group developed a fracture (HR, 0.76; 95% CI, 0.69-0.83). Total hip bone mineral density increased 3.7% after 3 years of therapy with HT compared with 0.14% in the control group (P<.001). Current recommendations22 are that the use of HT should be individualized. HT can be initiated around the time of menopause to treat menopause-related symptoms and to prevent osteoporosis in high risk patients. Treatment should be considered in conjunction with personal risk factors, such as risk of venous thrombosis, CHD, stroke, and breast cancer.
Estrogen alone Treatment
Between 1993 and 1998, 10,739 postmenopausal women aged 50-79 years with prior hysterectomy were treated with either 0.625mg CEE or placebo23. Despite the early termination of the combined hormone trial, the WHI estrogen therapy (ET) study continued under careful scrutiny. However, in February 2004, the National Institute of Health decided to terminate the intervention phase of the trial prior to the scheduled close out interval of October 2004 to March 2005. The primary outcome of the trial was the rate of CHD, invasive breast cancer incidence as well as stroke, pulmonary embolism (PE), colorectal cancer, hip fractures and death from other causes. After an average of 6.8 years follow-up, no significant effect of ET was observed on CHD rates compared to placebo. During the active intervention period, a reduction in the coronary events occurred in women assigned to ET [HR:0.95; 95% CI:0.79-1.16]24. The reduction was more significant in women aged 50-59 years [HR: 0.63; 95% CI: 0.36-1.08]. However, a 39% increase in the incidence of stroke was observed in the ET group (p=0.07), whereas the risk of venous thromboembolism (VTE), including deep venous thrombosis(DVT) and PE, was increased by 33% in the ET arm but only the increased rate of DVT was statistically significant (p=0.03)23. The increased risk for VTE was most apparent in the first two years and the increased risk is less than that observed for the estrogen plus progestin study25. Therefore, ET provided no overall protection against cardiovascular disease in healthy postmenopausal women. Interestingly invasive breast cancer was diagnosed at a 23% lower rate in the ET group [26 vs. 33 per 10000 person-years], however this did not reach statistical significance (p=0.06)23. No statistical differences were observed in the rates of colorectal cancer or total cancer rates. The major positive finding in the ET trial in 2004 was a 30%-39% reduction in the rates of fractures [HR 0.70, 95% CI 0.63-0.79]. In addition, ET did not significantly affect overall mortality rates or cause-specific mortality. Results from the final analysis of the WHI ET trial26 show that a persistent decrease in the risk of breast cancer was associated with ET and was 0.27% per year compared to 0.35% per year in the placebo arm reaching a statistical significance [HR 0.77, 95% CI 0.62-0.95] after a median follow up of 11.8 years. There was no difference between intervention and post-intervention hazard ratios (p=0.76). The breast cancer risk reduction in the ET arm was most apparent in women without benign breast disease (p=0.01) or a family history of breast cancer (p=0.02). Breast cancer mortality was reduced in the ET group (six deaths, 0.009% per year) compared with controls (16 deaths, 0.024% per year; HR 0.37, 95% CI 0.13–0.91; p=0.03). Fewer women in the ET group died from any cause after a breast cancer diagnosis than did the placebo arm (p=0.04). Although, breast cancer rates and mortality was lower for the women who received ET, beneficial effects are yet to be determined in high risk groups and adverse effects of stroke and VTE remain problematic. HT appeared to have more risks and the only clinical benefit was the reduction of osteoporosis, whereas ET in addition to fracture prevention decreased the incidence and mortality from breast cancer. The question to be addressed is whether it is possible to decipher the paradox that HT and ET produce completely different biological results i.e. HT increases whereas ET reduces the incidence of breast cancer. If clarity is possible then perhaps this knowledge can be used appropriately to help patients.
Chemical therapy for the treatment of breast cancer
The first successful chemical therapy to treat cancer was discovered by Sir Alexander Haddow, a British born physician. Haddow grew up in Broxburn, a small town 10 miles west of Edinburgh, Scotland27. It is said that, he became motivated to study medicine and biology after he was admitted to hospital for a perforated appendix and had the marvelous opportunity to observe daily visits of great Edinburgh surgeons who were inspired to make a difference in an era when public health and hygiene were far from being developed. Upon graduation from medical school he assisted with routine investigation of infections from the entire southeast of Scotland. It was while studying bacterial colony formation that he realized the resemblance to the formation of chemical tumors in higher forms 28. He went on to study the influence of carcinogenic substances on normal and malignant growth as well as drug resistance of cells to resultant tumors. Incidentally, he found that many carcinogenic hydrocarbons also possessed the property of retarding the growth of malignant tumor 29. In order to elucidate the molecular mechanism of these compounds, particular attention was paid to the inhibitory action of synthetic estrogens. In that era, reviews of animal experiment showed that treatment of animals with estrogens induced carcinoma of certain organs such as the cervix, uterus and the breast. The paradoxical action of estrogens showing growth properties, induction of tumors as well as growth retarding effects in certain circumstances led to the first ever reported clinical trial30 in 1944. Seventy three patients with advanced cancer were recruited to the study. Forty postmenopausal women with metastatic breast cancer and thirty cases of malignant disease in other organs received treatment with synthetic estrogens, triphenylchlorethylene, triphenylmethylethylene or stilbestrol. Ten of twenty two women with advanced breast cancer treated with triphenylchlorethylene showed significant regression of the tumors. Breast cancer patients treated with stilbestrol showed that 5 out of 14 cases underwent similar regression of tumors noted with triphenylchlorethylene. Of 4 cases of breast cancer treated with triphenylmethylethylene, only one showed a favorable response. Thirty cases of advanced cancer excluding that of the breast, including cancer of the skin, maxillary anthrum, urinary bladder, ovary, prostate and leukemia, treated with triphenylchlorethylene only carcinomas of the prostate and the bladder showed partial regression of the tumors. Data from the clinical study suggests that the success of estrogen therapy in breast cancer was dependent on the menopausal state of the woman. Haddow31, stated “When the various reports were assembled at the end of that time, it was fascinating to discover that rather general impression, not sufficiently strong from the relatively small numbers in any single group, became reinforced to the point of certainty; namely, the beneficial responses were three times more frequent in women over the age of 60 years than in those under that age; that estrogens may, on the contrary, accelerate the course of mammary cancer in younger women, and that their therapeutic use should be restricted to cases 5 years beyond the menopause. Here was an early and satisfying example of the advantages which may accrue from cooperative clinical trial.” Therefore the longer a woman was postmenopausal, the increased probability of tumor regression in metastatic breast cancer. However “…the extraordinary extent of tumor regression observed in perhaps 1% of post-menopausal cases (with estrogen) has always been regarded as of major theoretical importance, and it is a matter for some disappointment that so much of the underlying mechanisms continues to elude us…”31. Therefore at this point in 1970, the underlying mechanism of estrogen induced tumor regression still remained unanswered.
Time to treatment failure and the transition to tamoxifen
In the 1960's based on the data from clinical trials, high dose stilbestrol became the mainstay of treatment in postmenopausal women with advanced breast cancer. However the estrogen treatment was not without pitfalls. It was imperative that estrogen therapy not be instituted until ovarian secretion has ceased in a woman. The overall objective remission rate from estrogen treatment in 407 patients with advanced breast cancer was 31% 32. The remission rate was associated with increasing number of years after menopause (Table 1). The rate of regression was 9% in women who were less than 5 years postmenopausal, whereas the rate increased to 35% in women who have been postmenopausal for more than 5 years, corresponding with what was observed by Haddow31. A remarkable feature of estrogen therapy observed in this setting was the “withdrawal response”. Stoll 32previously described that when tumor response to estrogen administration is lost, it was found that on treatment withdrawal, 30% of cases underwent a second but shorter period of tumor remission, indicating that patients can be palliated over many years by intermittent estrogen and subsequent withdrawal. The introduction of tamoxifen, a non-steroidal anti-estrogen in the late 1970s revolutionized the clinical practice of endocrine treatment of ER positive breast cancer33. The evidence to support the anti-estrogenic action of tamoxifen was based on its antitumor action using carcinogen induced rat mammary tumor models34, 35 and subsequent athymic mice transplanted with human breast cancer cell lines36. The clinical efficacy of tamoxifen was first evaluated in women with late or recurrent carcinoma of the breast37. Results from this study, was compared to an unpublished data from breast cancer patients treated with diethylstilbestrol (DES) at the same hospital. Although response rates were similar, patients from the DES arm suffered more severe side effects. Similarly, Ingle and colleagues38 directly compared the use of either tamoxifen or DES in the treatment of advanced breast cancer in postmenopausal women. Analysis of the study revealed that there was no statistical significant difference between the efficacies of both treatments but like the Cole study37, toxicity was greater for the patients receiving DES and was severe enough for some patients to drop out of the study. Based on these data, DES fell out of favor for the treatment of metastatic breast cancer and tamoxifen became the preferred agent. Tamoxifen subsequently became the standard of care in the adjuvant treatment and prevention of breast cancer. Several clinical trials investigated the long term benefits of adjuvant tamoxifen therapy. An analysis of an overview 39of 55 randomized trials that compared the use of adjuvant tamoxifen versus no tamoxifen in breast cancer patients worldwide revealed that recurrence reduction for trials of 1 year, 2 years, 5 years during about 10 years of follow up were 21%, 29% and 47% respectively. A highly significant trend was seen towards greater effect based on longer treatment. A corresponding reduction in mortality was 12%, 17% and 26% respectively and this trend was also significant [2p=0.003]. A subsequent report of the meta-analysis 40showed that 5 years of adjuvant tamoxifen decreases the annual breast cancer mortality rate by 31% at fifteen years follow up, irrespective of the use of chemotherapy, age, progesterone receptor status or other tumor characteristics in ER positive breast cancer patients. Furthermore, the reduction seen at 5 years is significantly (2p<0.00001 for recurrence, 2p=0.01 for breast cancer mortality) more effective when compared to 1-2 years of adjuvant tamoxifen. More recently, results from the Adjuvant Tamoxifen- Longer Against Shorter (ATLAS) trial41 show that 10 years adjuvant treatment with tamoxifen produced a further reduction in recurrence and mortality from breast cancer when compared to 5 years of tamoxifen therapy. It is perhaps instructive to point out that the main effect with the decrease in mortality with a decade of tamoxifen occurs in the decade after tamoxifen treatment is stopped. This further suggests the hypothesis originally proposed in the early 1990's, that it was the woman's own estrogen that destroys the appropriately sensitive tamoxifen resistant micrometastasis42. Thus the study of the evolution of anti-hormone drug resistance to tamoxifen (see below) ironically provided an insight into mechanism of estrogen induced apoptosis studied today.
Table 1. Objective response rates in postmenopausal women with metastatic breast cancer using high dose estrogen therapy.
The 407 patients are divided in relation to menopausal status.32. The objective remission rate of breast cancer tumors was higher in women more than 5 years postmenopausal.
| Age Since Menopause | Patient number | % Regression |
|---|---|---|
| Postmenopausal 0-5 Years | 63 | 9% |
| Postmenopausal > 5 Years | 344 | 35% |
Nevertheless, the current recommendation in adjuvant endocrine treatment of ER positive breast cancer is that tamoxifen is a first line treatment for pre or perimenopausal women, while postmenopausal women take aromatase inhibitors as a primary agent for 5 years or for 2 to 3 years after tamoxifen43 for a total of five years of initial anti-hormone therapy. The latter is based on several studies where AIs have shown some superiority to tamoxifen as first-line agents in the treatment of postmenopausal women with breast cancer as well as a significant reduction in endometrial cancer44-46. Furthermore, 5 years of AI therapy have been showed to be highly beneficial as an extended adjuvant treatment in postmenopausal women who had previously received 5 years of tamoxifen therapy, where they show a 2.9% improvement in disease free survival at 4 years (HR 0.68 P= 0.0001) when compared to placebo47, 48.
Evolution of anti-hormone drug resistance
Despite the ability of long term adjuvant tamoxifen to improve survival, some patients develop disease recurrence due to acquired drug resistance. Early laboratory models were created to understand the development of drug resistance and subsequent deployment of second line therapies. Treatment of ovarectomised athymic mice transplanted with ER positive MCF-7 tumors with tamoxifen initially caused tumor regression, but subsequent regrowth of tumors occurred despite continuous tamoxifen treatment49. Re-transplantation of the resistant tumors into athymic mice or rats led to tumor growth in response to tamoxifen and estradiol50. Evaluation of these tumors showed that the tamoxifen stimulated tumors contained twice the estrogen receptor content than that of estradiol(E2) induced tumors50, 51. However, continuous treatment of transplanted MCF-7 tamoxifen resistant tumors with either a pure antiestrogen or no treatment in nude mice results in no tumor growth52. Because AIs deprive the ER of estrogens and fulvestrant degrades the estrogen receptor, the findings from these studies presaged the clinical use of these drugs as second line agents following failure of tamoxifen treatment53. However the early models of drug resistance to SERMS is based on short term treatments and replicates failure of tamoxifen after 1 or 2 years of treatment in advanced breast cancer and this represents phase 1 SERM resistance. In order to mimic 5 years of adjuvant tamoxifen therapy for micrometastatic breast cancer, laboratory models were created to induce phase II resistance to SERMS by serially transplanting tamoxifen stimulated MCF-7 tumors(MCF-7 TAM) into tamoxifen treated athymic mice for more than 5 years 54. Interestingly on stopping tamoxifen, the MCF-7TAM tumors rapidly regressed in response to physiologic estradiol although about 50% of tumors regrew following E2 treatment. The paradoxical E2 induced apoptosis suggests that a woman's own estrogen may produce an antitumor effect on pre-sensitized micrometastatic tumors after 5 years of adjuvant tamoxifen42. Failure of tumor regression after exhaustive anti-hormone therapy with a paradoxical E2 inhibited growth (phase III resistance) indicate a potential treatment plan using E2 as third line endocrine therapy 55. Tumors that regrow after E2 induced apoptosis revert back to the original cancer phenotype and are again sensitive to the antitumor actions of tamoxifen or aromatase inhibitors54.
Estrogen therapy in metastatic breast cancer
In more recent years, the use of estrogens continues to show clinical benefit in postmenopausal women with advanced breast cancer in an estrogen deprived setting. Lonning and colleagues56 treated thirty two women, who had previously taken multiple endocrine therapies, with high dose DES (5mg t.i.d). Four patients achieved complete response, while four patients obtained partial response. In addition five patients had an objective response lasting more than 52 weeks, while 2 patients had stable disease for more than six months. Six patients dropped out of the study due to severe side effects. However, one of the patients who had complete regression of a cytological confirm chest wall relapse and had 5 years of DES therapy, remained disease free for 10 years and six months after starting treatment. A long term follow up of the Ingle38 study that compared DES therapy to tamoxifen showed that the 5 year survival was 35% for DES and 16% for tamoxifen(p=0.039) 57. However DES treatment was associated with nausea, edema, and vaginal bleeding problems whereas hot flushes were more commonly observed with tamoxifen. Another clinical study 58reported in 2009, the findings of the treatment of postmenopausal women who had AI resistant metastatic breast cancer with low dose (6mg) and high dose (30mg) estradiol. Clinical benefit rates in the high dose arm were 28% [95% CI, 18%-41%] and 29% [95%CI, 19%-42%] in the low dose arm but adverse event rate was higher in the 30mg group when compared to the 6mg group. Six patients who were estrogen responsive were re-treated with AIs, among which 2 had partial response and 1 had stable disease. This indicates resensitization to estrogen deprivation and correlate with the hypothesis of the concept of SERM resistance54.
Experimental approach to decipher the mechanism of E2 induced apoptosis
To address the concerns of acquired resistance to long-term estrogen deprivation, a novel cell model59 was developed by our laboratory. An ER+/PR- hormone-independent breast cancer cell line, MCF-7:5C, a variant clone of wild-type MCF-7 cells was obtained by culturing MCF-7 cells continuously in estrogen free media. Treatment with physiologic E2 for six days caused a dramatic 90% reduction in the growth of MCF-7:5C cells60. The growth inhibition observed was confirmed to be apoptosis by Annexin V and DAPI staining. Fulvestrant also reduced the growth of MCF-7:5C cells but the growth inhibition was not due to apoptosis61. Furthermore, these cells were resistant to 4-hydroxytamoxifen. The tumorigenic potential of MCF-7:5C cells was examined by injecting cells into ovarectomised athymic mice and found that these cells, spontaneously grew into tumors in the absence of E261. In contrast, MCF-7:5C tumors in mice treated with E2 regressed in a time dependent manner and became undetectable after 8 weeks of treatment, Similarly, fulvestrant also decreased the growth of the MCF-7:5C tumors but the reduction was statistically significantly less when compared to that of E2 (p<0.001).MCF-7:2A cells62, 63, another long-term estrogen deprived cell line derived from MCF-7 cells is more resistant to estradiol induced apoptosis. Based on clinical data that shows that only about 30% of patients respond to estrogens following anti-hormone resistance, it seemed imperative to see whether E2 induced apoptosis could be enhanced in anti-hormone resistant cells. Overexpression of Bcl-2 elevates cellular glutathione (GSH) level which is associated with increased resistance to chemotherapy-apoptosis64, 65, while restoration of apoptosis occurs in Bcl-2 expressing cells depleted of GSH 66. MCF-7:2A cells express high levels of glutathione synthetase (GS) and glutathione peroxidase 2 (GPx2) which are involved in GSH synthesis67. Exposure of MCF-7:2A cells to a combination therapy of E2 and buthionine sulfoximine (BSO), a GSH inhibitor for 48h to 96h produced a sevenfold increase in apoptosis while the individual treatments had no significant effect on growth. The in vitro findings correlated with in vivo data from a mouse xenograft model in which daily administration of BSO either as a single agent or in combination with E2 significantly decreased tumor growth of MCF-7:2A cells. Thus this provides a potential strategy for future clinical trials involving combination therapy of BSO and low dose estrogen to improve response for patients with anti-hormone resistant advanced breast cancer 68
Conjugated equine estrogens
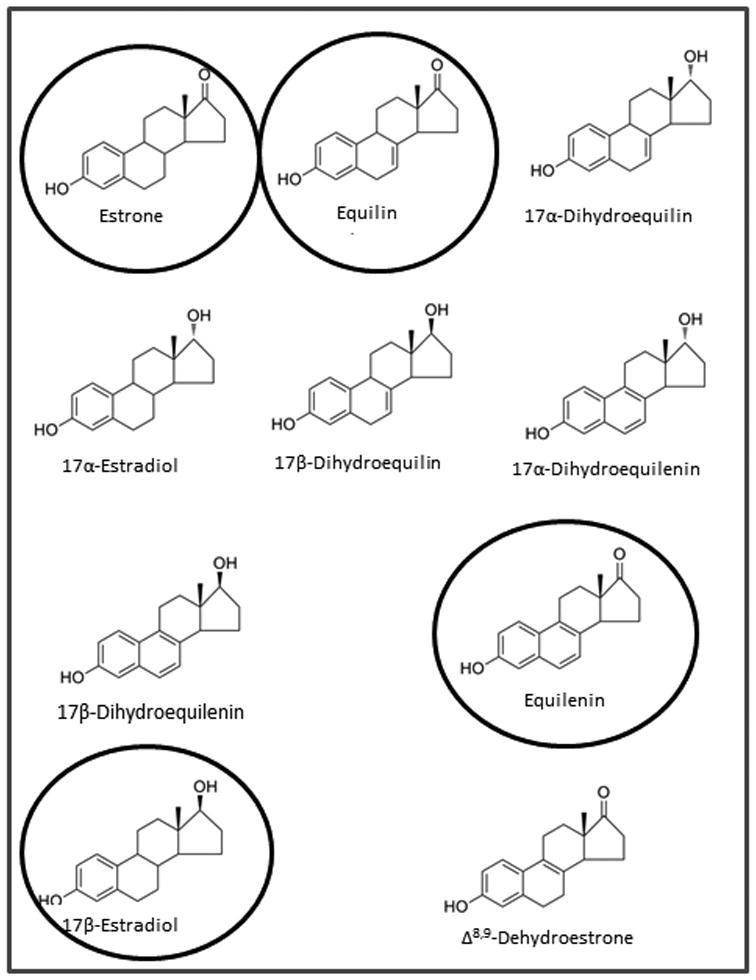
Extensive progress in the production of estrogen preparations for commercial use was made by scientists at Wyeth pharmaceuticals (then Ayerst) Canada who extracted conjugated estrogens from pregnant horse's urine69. In 1942, Food and Drug Administration (FDA) approval70 was obtained in the United States for the clinical use of conjugated equine estrogens (premarin)for the treatment of menopausal symptoms and related conditions. There was an initial worldwide acceptance of CEE in the 1960s, however increased risks of developing endometrial cancer led to a decline in prescriptions for postmenopausal women71, 72. A new generation of interest in the use estrogen therapy in the treatment of osteoporosis in the 1980s led to clinical studies of women receiving either estrogen alone or estrogen plus progestin therapies. Women on estrogen and progestin treatment had lower incidence of endometrial cancer73, 74 indicating that progestin blocked the proliferative effect of estrogens on the endometrial lining. As a result, CEE was approved for the treatment and prevention of osteoporosis; women with an intact uterus were given progestin in addition to the estrogens. CEE is made up of conjugated estrogens and the tablet consists of at least 10 estrogens (fig 1) which include estrone (59.2%), equilin (26.9%),17α-dihydroequilin (16.3%) , 17α-estradiol (4.32%) , 17β-dihydroequilin (1.76%), 17α-dihydroequilenin (1.76%), 17β-dihydroequilenin (3.36%), equilenin (2.4%), 17β-estradiol (0.8%), and Δ8,9-dehydroestrone (4.16%). Generic synthetic versions of CEE are not currently approved by the FDA based on inadequacies noted on their active ingredients, bioequivalence, safety and effectiveness75.
Figure 1. Structures of the estrogenic constituents of premarin.
Estradiol, equilin, estrone and equilenin were used in our experimental studies.
Effect of Conjugated Equine Estrogens on breast cancer cells
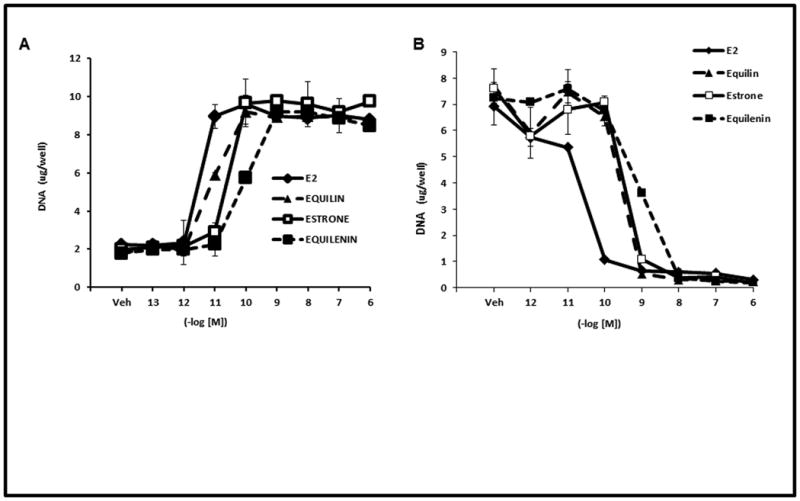
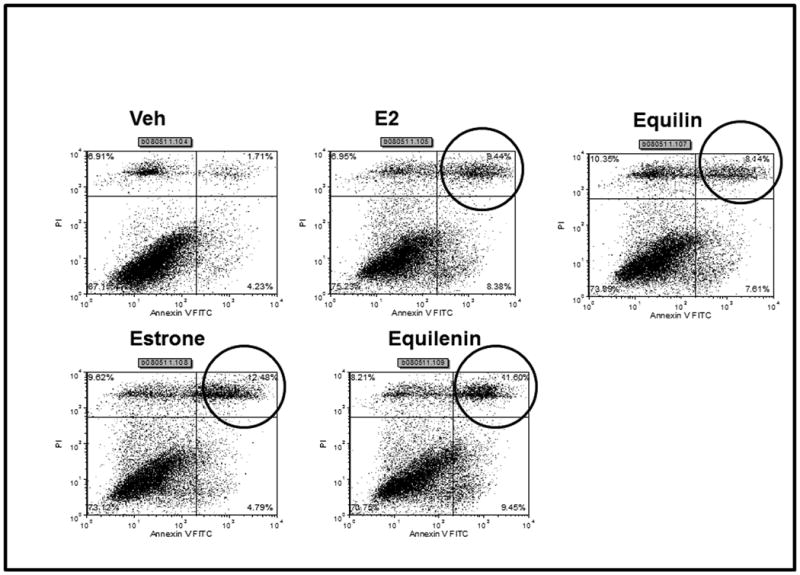
Long term concentrations of estrogen deprived MCF-7 breast cancer cells undergo apoptosis upon treatment with physiologic estradiol61. Based on the preliminary results of the WHI CEE study, we decided to elucidate the biological properties of the main estrogens in CEE in two different models of breast cancer cells. Estrogens have been shown to regulate the growth of ER positive MCF-7 breast cancer cells. To study the biological activity of the actual estrogens namely equilin, estrone and equilenin, we tested their ability to induce proliferation in MCF7:WS8 cells which contain ER and have retained estrogen responsiveness for a sustained period of continuous cell culture76. MCF-7 cells were grown in estrogen free media for 3 days and treated with various concentrations of the equilin, estrone and equilenin and their effects were compared to E2 (fig 2A). All the three estrogens were able to induce cell growth of MCF-7 cells in a dose dependent manner to the maximum level as E2. Equilin and estrone induced cell proliferation with maximum stimulation occurring at 0.1nM, whereas equilenin reached maximal stimulation at 1nM as compared to 0.01nM for E2. Next we investigated the growth properties of the equilin, estrone and equilenin in long term estrogen deprived MCF7:5C cells in comparison to E2. Fig 2B shows that equilin, estrone and equilenin drastically inhibited the growth of the MCF7:5C cells at comparable concentrations to E2. Maximum growth inhibition was achieved with E2 at 0.1M, while equilin and estrone reach maximum growth inhibition at 1nM and equilenin at 10nM after 7 days of treatment. To determine if the observed estrogen induced growth inhibition of the MCF7:5C cells was due to apoptosis, MCF7:5C cell were either the control, E2, equilin, estrone or equilenin for 72 hours and the level of apoptosis was measured using annexin V staining. E2, equilin, estrone and equilenin all show increased apoptotic staining compared to the control treated cells (Fig 3). The ability of the conjugated estrogens to inhibit the growth and induce apoptosis in the MCF7:5C cells and not the parental MCF7 cells suggest that these biological properties are dependent on the duration of deprivation of estrogen in the breast cancer cells.
Figure 2. Cell proliferation assay analysis of the biological properties of active steroids in CEE in breast cancer cells.
(A) MCF7 cells were grown in E2 stripped media for 3 days and treated for 7 days with various concentrations of E2, equilin, estrone and equilenin and compared to the Veh (control). (B) Equilin, estrone and equilenin drastically inhibited the growth of MCF7:5C cells in a similar manner as E2. The experiments were completed in triplicates and performed as previously described 61
Figure 3. Effects of estradiol and active estrogens in CEE on apoptosis in MCF7:5C cells.
MCF7:5C cells were seeded in 100mm plates and treated with Veh (control), 1nM E2, 1nM Equilin, 1nM estrone and 1uM equilenin for 72 hours and cells were stained with FITC-annexin V and propidium iodide and analyzed by flow cytometry and performed as previously described61. The upper right box of vehicle treated (Veh) cells have low apoptotic cells (1.71%), whereas all for estrogens, this fraction is increased (circled upper right hand box).
Molecular mechanisms of estrogen induced apoptosis
To decipher the precise series of events that precede estrogen induced apoptosis, differential gene expression in response to E2 was interrogated using affymetrix based microarray analysis63. Specific genes were identified for MCF7:5C which indicate that E2 induced endoplasmic reticulum stress (ERS) and inflammatory stress responses that lead to apoptosis. Identified ERS genes indicated that E2 inhibited protein folding leading to accumulation of unfolded proteins and widespread inhibition of protein translation with subsequent induction of cell death. In response to severe ERS, Bcl-2 interacting mediator of cell death (Bim; BCL211) was induced. Further evidence of the involvement of the mitochondrial pathway in E2 induced apoptosis was reported by Lewis and colleagues61 who showed increased expression of several proapoptotic proteins including, Bax, Bak, Bim, Noxa, Puma and p53 in E2 treated MCF7:5C cells. Reversal of the apoptotic effect of E2 in these cells was observed with blockade of Bax and Bim expression using short interfering RNAs (siRNAs). Involvement of the Fas/Fasl death signaling (extrinsic) pathway in the apoptotic effect of E2 has been investigated. Osipo et al77 demonstrated that E2 induced regression of tamoxifen stimulated breast cancer tumors, by activating the death receptor Fas and suppressing the antiapoptotic/prosurvival factors NF-kB and HER2/neu. Similarly, the growth of raloxifene resistant MCF7 cells in vitro and in vivo was attenuated by E2 by increasing Fas expression and reduced NF-kB activity78. Studies are currently ongoing to determine the sequence of events that occur before E2 induces apoptosis in the MCF7:5C cells. The resolution of the crystal structure provided insight into the activation of the ER by E2 and silencing by antiestrogens79, 80 and is providing the insight into the “trigger” mechanism for the ER complex. The shape the ligands make with the ER is imperative to their ability to induce apoptosis in the MCF7:5C cells. E2 is sealed within the hydrophobic pocket of the ligand binding domain of the ER by helix 12 and coactivators bind leading to activation of apoptotic genes. On the other hand, 4-hydroxytamoxifen (4OHT) pushes back helix 12, prevents coactivator binding and this may responsible for its ability to block estrogen induced apoptosis in the MCF7:5C cells. Knockdown of coactivator AIB1/SRC3 in MCF7:5C cells led to the loss of apoptosis inducing effect of E2 suggesting that AIB1is a significant control hub E2 in induced apoptosis in these breast cancer cells81. Structure function studies show that the shape of the estrogen82 can modulate the shape of the estrogen-ER complex to induce apoptosis83. Hydroxylated triphenylethylenes (TPE) which are structurally similar to 4OHT and have estrogenic properties in MCF7 cells, have been shown to block E2 induced apoptosis84. The antiestrogenic shape they make with the ER may be responsible for the delayed apoptotic effect of the TPEs in the MCF7:5C cells. These pharmacologic studies are currently under investigation and will be the focus of further reports.
Discussion
Before the clinical use of antiestrogen therapy, high dose estrogens were effective in the induction of tumor regression in metastatic breast cancer30, 32. In more recent times, estrogen therapy show significant clinical benefit in postmenopausal women who have undergone extensive anti-hormone treatment85. Development of tamoxifen stimulated tumors in athymic mice following a five year treatment with tamoxifen suggest that the development of anti-hormone resistance over years of treatment reconfigures the survival mechanism of breast cancer so that estrogen is no longer a potent mitogen that stimulates cell proliferation but rather becomes a death signal. Preclinical data clearly show that long term estrogen deprivation of ER positive MCF-7 breast cancers and subsequent treatment of the cells with E2 causes apoptosis of these cells. Creation of an estrogen deprived environment either by withdrawal of estrogen treatment 32 or by exhaustive anti-hormone therapy increases sensitivity of breast tumors to estrogen therapy which subsequently induces tumor regression. Similarly CEE alone reduces the incidence of breast cancer in hysterectomised postmenopausal women. This protective effect is not observed in women who received addition progesterone therapy, suggesting that the progestin may play a potential role in the increase in breast cancer seen in postmenopausal women who received combined hormonal therapy
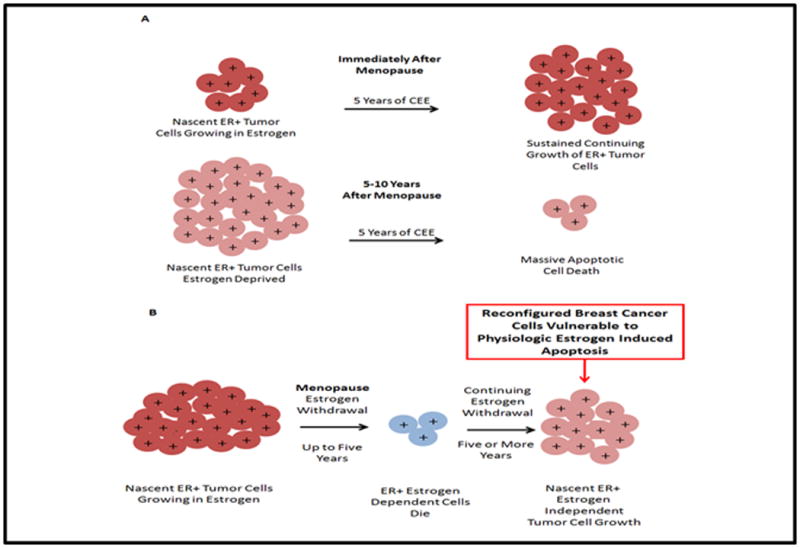
To explain the aforementioned clinical data, laboratory studies show that estrogens in the CEE were able to cause proliferation of MCF7 cells after growing these cells in an estrogen free medium for 3 days. This cell population is adapted to an environment rich in estrogen, so naturally all the cells grow with a “resupply” of natural steroidal estrogens. However, these same estrogens induce apoptosis to a similar extent as E2 in MCF-7 cells that have been deprived of estrogen treatment for many years. The ability of estrogen therapy to treat or prevent tumors is related to the menopausal status of a woman and how long they have been physiologically deprived of estrogen. In the Stoll data32(Table 1), the rate of remission of advanced breast cancer was significantly less in women who were less than 5 years postmenopausal(9%), and there was a 35% remission rate in women who were more than 5 years postmenopausal. It is important to stress that majority of the women in the WHI CEE trial were above 60 years and the mean age at screening was 63.6 years. Here, the overall result was a reduction in breast cancer and mortality. There is a need for an “estrogen holiday” before starting estrogen therapy. Induction of menopause in a woman gradually deprives the cells of estrogen. However immediate treatment with estrogens may cause growth of nascent ER positive breast tumors which may increase breast cancer risk (fig 4A). The cells vulnerable to death with estrogens in CEE, have been selected because estrogen deprivation at menopause causes estrogen dependent nascent breast cancers to die, but all do not die. Remaining cells that survive learn to grow without estrogen (fig 4B). These cells will continue to grow to produce breast cancer unless exogenous estogens induces apoptotic death. Therefore 5 years of CEE treatment immediately after menopause will cause sustained continuing growth of ER positive tumor cells. Because nascent ER+ tumor cells have been estrogen deprived in women who are 5 to 10 years postmenopausal, 5 years of CEE therapy induces massive apoptotic cell death and subsequent tumor cell death and an enhanced patient survival.
Figure 4. The success of estrogen therapy is dependent on menopausal status of a woman.
A. Treatment of women immediately after menopause with CEE results in sustained growth of nascent ER positive tumors, whereas treatment 5. years after menopause causes apoptotic cell death. B. Estrogen withdrawal in postmenopausal women causes ER positive dependent cells to die but some cells continue to grow independent of estrogen
Conclusion
High dose estrogen treatment is effective in causing tumor regression in metastatic breast cancer. The mechanism for this treatment was a paradox and unknown for 60 years but is now being deciphered63. Objective tumor remission was seen in women over 5 years postmenopausal30, 32. Estrogen therapy administered to women in their late 60s causes a sustained decrease in breast cancer incidence and a decrease in mortality26. The question was, why? Long term estrogen deprivation (LTED) for ER positive breast cancer cells is the key. We have created LTED breast cancer cell lines and for the first time, described the mechanism of estrogen-induced apoptosis. This new biology of estrogen induced apoptosis and can be now used to explain the effects of ET in reducing breast cancer incidence and mortality for women in the 60s.
Acknowledgments
The content of this article was presented by Dr. Jordan at The North American Menopause Society (NAMS)/Pfizer - Wulf H. Utian Endowed Lecture on October 6, 2012, in Orlando, Florida. An endowment to NAMS from Pfizer established this annual lectureship, with faculty selected by the NAMS Scientific Program Committee.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest and source of funding: We have no conflicts of interest. This work (VCJ) was supported by the Department of 1090 Defense Breast Program under Award number W81XWH-06-1-0590 Center of Excellence;the Susan G Komen for the Cure Foundation under Award number SAC100009 and the Lombardi Comprehensive Cancer 1095 Center Support Grant (CCSG) Core Grant NIH P30 CA051008. The views and opinions of the author(s) do not reflect those of the US Army or the Department of Defense.
References
- 1.American Cancer Society. Breast Cancer Facts & Figures 2011-2012. Atlanta: American Cancer Society Inc.; 2011. [Google Scholar]
- 2.Beatson G. On the treatment of inoperable cases of carcinoma of the mamma:suggestions for a new method of treatment, with illustrative cases. Lancet. 1896;2:162–165. [PMC free article] [PubMed] [Google Scholar]
- 3.Boyd S. On oophorectomy in cancer of the breast. BMJ. 1900;2:1161–1167. [Google Scholar]
- 4.Lathrop AE, Loeb L. Further Investigations on the Origin of Tumors in Mice: III. On the Part Played by Internal Secretion in the Spontaneous Development of Tumors. J Cancer Res. 1916;1:1–19. [PubMed] [Google Scholar]
- 5.Allen E, Doisy EA. An ovarian hormone: Preliminary report on its localization, extraction and partial purification, and action in test animals. JAMA. 1923;81:819–821. doi: 10.1001/jama.250.19.2681. [DOI] [PubMed] [Google Scholar]
- 6.Jordan VC, Brodie AMH. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72:7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obiorah I, Jordan VC. Progress in endocrine approaches to the treatment and prevention of breast cancer. Maturitas. 2011;70:315–321. doi: 10.1016/j.maturitas.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.WHI: Design of the Women's Health Initiative Clinical Trial and Observational Study. Controlled Clin Trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 9.Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev. 2012;11 doi: 10.1002/14651858.CD004143.pub4. [DOI] [PubMed] [Google Scholar]
- 10.Karas RH. Animal models of the cadiovasular effects of exogenous hormones. Am J Cardiol. 2002;90:22F–25F. doi: 10.1016/s0002-9149(02)02421-9. [DOI] [PubMed] [Google Scholar]
- 11.Manson JE, Hsia J, Johnson KC, et al. Estrogen plus Progestin and the Risk of Coronary Heart Disease. New Engl J Med. 2003;349:523–534. doi: 10.1056/NEJMoa030808. [DOI] [PubMed] [Google Scholar]
- 12.Cushman M, Kuller LH, Prentice R, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292:1573–1580. doi: 10.1001/jama.292.13.1573. [DOI] [PubMed] [Google Scholar]
- 13.Wassertheil-Smoller S, Hendrix S, Limacher M, et al. Effect of estrogen plus progestin on stroke in postmenopausal women: The women health initiative: a randomized trial. JAMA. 2003;289:2673–2684. doi: 10.1001/jama.289.20.2673. [DOI] [PubMed] [Google Scholar]
- 14.Hsia J, Criqui MH, Rodabough RJ, et al. for the Women's Health Initiative Investigators. Estrogen Plus Progestin and the Risk of Peripheral Arterial Disease. Circulation. 2004;109:620–626. doi: 10.1161/01.CIR.0000115309.63979.92. [DOI] [PubMed] [Google Scholar]
- 15.Shumaker S, Legault C, Rapp SR, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: The women health initiative memory study: a randomized controlled trial. JAMA. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- 16.Chlebowski R, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: The women health initiative randomized trial. JAMA. 2003;289:3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- 17.Chlebowski RT, Kuller LH, Prentice RL, et al. Breast Cancer after Use of Estrogen plus Progestin in Postmenopausal Women. New Engl J Med. 2009;360:573–587. doi: 10.1056/NEJMoa0807684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, et al. Estrogen plus Progestin and Colorectal Cancer in Postmenopausal Women. New Engl J Med. 2004;350:991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- 19.Chlebowski RT, Schwartz AG, Wakelee H, et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women's Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet. 2009;374:1243–1251. doi: 10.1016/S0140-6736(09)61526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson G, Judd HL, Kaunitz AM, et al. Effects of estrogen plus progestin on gynecologic cancers and associated diagnostic procedures: The women health initiative randomized trial. JAMA. 2003;290:1739–1748. doi: 10.1001/jama.290.13.1739. [DOI] [PubMed] [Google Scholar]
- 21.Cauley J, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: The women health initiative randomized trial. JAMA. 2003;290:1729–1738. doi: 10.1001/jama.290.13.1729. [DOI] [PubMed] [Google Scholar]
- 22.The North American Menopause Society. The 2012 Hormone Therapy Position Statement of The North American Menopause Society. Menopause. 2012;19:257. doi: 10.1097/gme.0b013e31824b970a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- Hsia J, Langer RD, Manson JE, et al. Conjugated equine estrogens and coronary heart disease: The women health initiative. Arch Int Med. 2006;166:357–365. doi: 10.1001/archinte.166.3.357. [DOI] [PubMed] [Google Scholar]
- 25.Curb J, Prentice RL, Bray PF, et al. Venous thrombosis and conjugated equine estrogen in women without a uterus. Arch Int Med. 2006;166:772–780. doi: 10.1001/archinte.166.7.772. [DOI] [PubMed] [Google Scholar]
- 26.Anderson GL, Chlebowski RT, Aragaki AK, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women's Health Initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13:476–486. doi: 10.1016/S1470-2045(12)70075-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haddow A. An Autobiographical Essay. Cancer Res. 1974;34:3159–3164. [PubMed] [Google Scholar]
- 28.Haddow A. The influence of carcinogenic substances on sarcomata induced by the same and other compounds. J Pathol Bacteriol. 1938;47:581–591. [Google Scholar]
- 29.Haddow A. Influence of certain Polycyclic Hydrocarbons on the Growth of the Jensen Rat Sarcoma. Nature. 1935;136:868. [Google Scholar]
- 30.Haddow A, Watkinson JM, Patterson E. Influence of synthetic oestrogens upon advanced malignant disease. BMJ. 1944;2:393–398. doi: 10.1136/bmj.2.4368.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haddow A, David A. Karnosky Memorial lecture: Thoughts on chemical therapy. Cancer Res. 1970;26:737–754. doi: 10.1002/1097-0142(197010)26:4<737::aid-cncr2820260402>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 32.Stoll B. Palliation by castration or by hormone administration. In: Stoll B, editor. Breast Cancer Management Early and Late. London, UK: William Herman Medical Books Ltd; 1977. pp. 133–146. Edited by London, UK, William Herman Medical Books Ltd, 1977, p.pp. 133-146. [Google Scholar]
- 33.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 34.Jordan VC. Antitumor activity of the antioestrogen ICI 46, 474 (tamoxifen) in the dimethylbenzanthracene (DMBA)-induced rat mammary carcinoma model. J Steroid Biochem. 1974;5:354. [Google Scholar]
- 35.Jordan VC. Effect of tamoxifen (ICI 46,474) on initiation and growth of DMBA-induced rat mammary carcinoma. Eur J Cancer. 1976;12:419. doi: 10.1016/0014-2964(76)90030-x. [DOI] [PubMed] [Google Scholar]
- 36.Jordan VC, Robinson SP. Species-specific pharmacology of anti-estrogens: role of metabolism. Fed Proc. 1987;46:1870–1874. [PubMed] [Google Scholar]
- 37.Cole M, Jones C, Todd I. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25:270–275. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingle J, Ahmann D, Green S, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 39.EBCTCG. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 40.ECBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 41.Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2012 doi: 10.1016/S0140-6736(12)61963-1. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf D, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy, Recent Results. Cancer Res. 1993;127:23–33. doi: 10.1007/978-3-642-84745-5_4. [DOI] [PubMed] [Google Scholar]
- 43.Burstein HJ, Prestrud AA, Seidenfeld J, et al. American Society of Clinical Oncology Clinical Practice Guideline: Update on Adjuvant Endocrine Therapy for Women With Hormone Receptor–Positive Breast Cancer. J Clin Oncol. 2010;28:3784–3796. doi: 10.1200/JCO.2009.26.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuzick J, Sestak I, Baum M, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11:1135–1141. doi: 10.1016/S1470-2045(10)70257-6. [DOI] [PubMed] [Google Scholar]
- 45.Mouridsen H, Giobbie-Hurder A, Goldhirsch A, et al. Letrozole Therapy Alone or in Sequence with Tamoxifen in Women with Breast Cancer. New Engl J Med. 2009;361:766–776. doi: 10.1056/NEJMoa0810818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones S, Seynaeve C, Hasenburg A, et al. Results of the first planned analysis of the TEAM (tamoxifen exemestane adjuvant multinational) prospective randomized phase III trial in hormone sensitive postmenopausal early breast cancer. Cancer Res. 2009;69(Suppl):15. [Google Scholar]
- 47.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 48.Ingle JN, Tu D, Pater JL, Muss HB, Martino S, Robert NJ, Piccart MJ, et al. Intent-to-treat analysis of the placebo-controlled trial of letrozole for extended adjuvant therapy in early breast cancer: NCIC CTG MA.17. Annals of Oncol. 2008;19:877–882. doi: 10.1093/annonc/mdm566. [DOI] [PubMed] [Google Scholar]
- 49.Osborne C, Coronado EB, Robinson JP. Human breast cancer in the athymic nude mouse: cytostatic effects of long-term antiestrogen therapy. Eur J Cancer Clin Oncol. 1987;23:1189–1196. doi: 10.1016/0277-5379(87)90154-4. [DOI] [PubMed] [Google Scholar]
- 50.Gottardis M, Jordan VC. Develoment of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term anti-estrogen administration. Cancer Res. 1988;48:5183–5187. [PubMed] [Google Scholar]
- 51.Gottardis MM, Wagner RJ, Borden EC, Jordan VC. Differential Ability of Antiestrogens to Stimulate Breast Cancer Cell (MCF-7) Growth in Vivo and in Vitro. Cancer Res. 1989;49:4765–4769. [PubMed] [Google Scholar]
- 52.Gottardis MM, Jiang SY, Jeng MH, Jordan VC. Inhibition of Tamoxifen-stimulated Growth of an MCF-7 Tumor Variant in Athymic Mice by Novel Steroidal Antiestrogens. Cancer Res. 1989;49:4090–4093. [PubMed] [Google Scholar]
- 53.Howell A, Robertson JFR, Abram P, et al. Comparison of Fulvestrant Versus Tamoxifen for the Treatment of Advanced Breast Cancer in Postmenopausal Women Previously Untreated With Endocrine Therapy: A Multinational, Double-Blind, Randomized Trial. J Clin Oncol. 2004;22:1605–1613. doi: 10.1200/JCO.2004.02.112. [DOI] [PubMed] [Google Scholar]
- 54.Yao K, Lee ES, Bentrem DJ, et al. Antitumor Action of Physiological Estradiol on Tamoxifen-stimulated Breast Tumors Grown in Athymic Mice. Clin Cancer Res. 2000;6:2028–2036. [PubMed] [Google Scholar]
- 55.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer cell. 2004;5:207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 56.Lonning P, Taylor P, Anker G, et al. High-dose estrogen treatment in post-menopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67:111–116. doi: 10.1023/a:1010619225209. [DOI] [PubMed] [Google Scholar]
- 57.Peethambaram P, Ingle J, Suman V, et al. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat. 1999;54:117–122. doi: 10.1023/a:1006185805079. [DOI] [PubMed] [Google Scholar]
- 58.Ellis M, Gao F, Dehdashti F, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor–positive, aromatase inhibitor–resistant advanced breast cancer: A phase 2 randomized study. JAMA. 2009;302(7):774–780. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang SY, Wolf DM, Yingling JM, et al. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90:77–86. doi: 10.1016/0303-7207(92)90104-e. [DOI] [PubMed] [Google Scholar]
- 60.Lewis J, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94:131–141. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 61.Lewis J, Meeke K, Osipo C, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–1759. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- 62.Pink JJ, Jiang SY, Fritsch M, Craig Jordan VC. An Estrogen-Independent MCF-7 Breast Cancer Cell Line Which Contains a Novel 80-Kilodalton Estrogen Receptor-related Protein. Cancer Res. 1995;55:2583–2590. [PubMed] [Google Scholar]
- 63.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. PNAS. 2011;108:18879–18886. doi: 10.1073/pnas.1115188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voehringer D. BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic Biol Med. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 65.Meredith MJ, Cusick CL, Soltaninassab S, et al. Expression of Bcl-2 increases intracellular glutathione by inhibiting methionine-dependent GSH efflux. Biochem Biophys Res Commun. 1998;248:458–463. doi: 10.1006/bbrc.1998.8998. [DOI] [PubMed] [Google Scholar]
- 66.Rudin CM, Yang Z, Schumaker LM, et al. Inhibition of Glutathione Synthesis Reverses Bcl-2-mediated Cisplatin Resistance. Cancer Res. 2003;63:312–318. [PubMed] [Google Scholar]
- 67.Lewis-Wambi J, Kim H, Wambi C, et al. Buthionine sulfoximine sensitizes anti-hormone-resistant human breast cancer cells to estrogen-induced apoptosis. Breast Cancer Res. 2008;10:R104. doi: 10.1186/bcr2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis-Wambi J, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11:206. doi: 10.1186/bcr2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watkins E. The Estrogen Elixir: A History of Hormone Replacement Therapy in America. Baltimore MD: The John Hopkins University Press; 2007. pp. 10–132. [Google Scholar]
- 70.FDA. FDA backgrounder on congugated estrogens, US Food and Drug Administration Center for Drug Evaluation and Research. 2005. [Google Scholar]
- 71.Smith DC, P R, Thompson DJ, Herrmann WL. Association of exogenous estrogen and endometrial carcinoma. N Engl J Med. 1975;293:1164–1167. doi: 10.1056/NEJM197512042932302. [DOI] [PubMed] [Google Scholar]
- 72.Ziel H, et al. Increased risk of endometrial cancinoma among users of conjugated estrogens. N Engl J Med. 1975;293:1167–1170. doi: 10.1056/NEJM197512042932303. [DOI] [PubMed] [Google Scholar]
- 73.Greenblatt R, Gambrell RD, Stoddard LD. The protective role of progesterone in the prevention of endometrial cancer. Pathol Res Pract. 1982;174:297–318. doi: 10.1016/s0344-0338(82)80072-1. [DOI] [PubMed] [Google Scholar]
- 74.Gambrell R. The prevention of endometrial cancer in postmenopausal women with progestogens. Maturitas. 1978;1:107–112. doi: 10.1016/0378-5122(78)90017-8. [DOI] [PubMed] [Google Scholar]
- 75.FDA. Memorandum, US Food and Drug Administration Center for Drug Evaluation and Research. FDA; 1997. Conjugated estrogens-Letter from Dr Janet Woodcock. [Google Scholar]
- 76.Soule HD, Vazquez J, Long A, et al. A Human Cell Line From a Pleural Effusion Derived From a Breast Carcinoma. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 77.Osipo C, Gajdos C, Liu H, et al. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst. 2003;95:1597–1608. doi: 10.1093/jnci/djg079. [DOI] [PubMed] [Google Scholar]
- 78.Liu H, Lee E, Gajdos C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–1597. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 79.Brzozowski AM, Pike ACW, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 80.Shiau AK, Barstad D, Loria P, et al. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 81.Hu ZZ, Kagan BL, Ariazi EA, Rosenthal DS, Zhang L, Li JV, et al. Proteomic analysis of pathways involved in estrogen-induced growth and apoptosis of breast cancer cells. PLoS One. 2011;6(6):e20410. doi: 10.1371/journal.pone.0020410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jordan VC, Schafer JM, Levenson AS, et al. Molecular Classification of Estrogens. Cancer Res. 2001;61:6619–6623. [PubMed] [Google Scholar]
- 83.Maximov PY, Myers CB, Curpan RF, et al. Structure–Function Relationships of Estrogenic Triphenylethylenes Related to Endoxifen and 4-Hydroxytamoxifen. J Med Chem. 2010;53:3273–3283. doi: 10.1021/jm901907u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maximov P, Sengupta S, Lewis-Wambi JS. The Conformation of the Estrogen Receptor Directs Estrogen-Induced Apoptosis in Breast Cancer: A Hypothesis. Horm Mol Biol Clin Investig. 2011;5(1):27–34. doi: 10.1515/HMBCI.2010.047. [DOI] [PMC free article] [PubMed] [Google Scholar]