

Abstract
The development of insulin resistance is tightly linked to fatty liver disease and is considered a major health concern worldwide, although their mechanistic relationship remains controversial. Activin has emerging roles in nutrient homeostasis, but its metabolic effects on hepatocytes remain unknown. In this study, we investigated the effects of increased endogenous activin bioactivity on hepatic nutrient homeostasis by creating mice with inactivating mutations that deplete the circulating activin antagonists follistatin-like-3 (FSTL3) or the follistatin 315 isoform (FST315; FST288-only mice). We investigated liver histology and lipid content, hepatic insulin sensitivity, and metabolic gene expression including the HepG2 cell and primary hepatocyte response to activin treatment. Both FSTL3-knockout and FST288-only mice had extensive hepatic steatosis and elevated hepatic triglyceride content. Unexpectedly, insulin signaling, as assessed by phospho-Akt (a.k.a. protein kinase B), was enhanced in both mouse models. Pretreatment of HepG2 cells with activin A increased their response to subsequent insulin challenge. Gene expression analysis suggests that increased lipid uptake, enhanced de novo lipid synthesis, decreased lipolysis, and/or enhanced glucose uptake contribute to increased hepatic triglyceride content in these models. However, activin treatment recapitulated only some of these gene changes, suggesting that increased activin bioactivity may be only partially responsible for this phenotype. Nevertheless, our results indicate that activin enhances hepatocyte insulin response, which ultimately leads to hepatic steatosis despite the increased insulin sensitivity. Thus, regulation of activin bioactivity is critical for maintaining normal liver lipid homeostasis and response to insulin, whereas activin agonists may be useful for increasing liver insulin sensitivity.
Hepatic steatosis results from ectopic accumulation of triglycerides (TGs) and other lipids in hepatocytes and is tightly associated with insulin resistance and diabetes. Hepatic steatosis is a component of a group of disorders known as nonalcoholic fatty liver disease (NAFLD) and is considered the earliest manifestation of this spectrum that in some subjects can lead to more severe forms of liver injury such as nonalcoholic steatohepatitis and cirrhosis (1). The incidence of fatty liver is increasing in association with the well-documented acceleration of obesity, diabetes, and metabolic syndrome, suggesting a mechanistic connection between insulin resistance, hyperglycemia, increased fatty acid intake, and hepatic steatosis (1).
It remains unclear, however, whether the association between hepatic steatosis and insulin resistance is causal, that is, whether accumulation of TG and other lipids in hepatocytes causes insulin resistance or whether whole-body insulin resistance and hyperglycemia of diabetes leads to accumulation of TGs in liver (1). Normally, insulin inhibits glucose production and promotes fatty acid synthesis, whereas liver insulin resistance leads to hepatic steatosis through the recently described bifurcation in the hepatic insulin signaling pathway in which the cascade regulating lipid synthesis remains insulin sensitive while gluconeogenic regulation becomes insulin-resistant (2–4). This would suggest that whole-body and/or liver insulin resistance leads to hepatic steatosis. On the other hand, accumulation of hepatic triacylglycerol through increased de novo lipogenesis leads to insulin resistance, at least in part due to serine phosphorylation of insulin receptor substrate proteins (5, 6). Furthermore, evidence dissociating hepatic TG accumulation from insulin resistance is derived from mouse models that develop hepatic steatosis in the absence of insulin resistance (7, 8) as well as from human diseases such as glycogen storage disease or citrin deficiency that cause steatosis without insulin resistance (9, 10). Therefore, the relationship between insulin resistance, hepatic steatosis, and fatty acid metabolism remains to be more clearly defined.
Activin is a member of the TGFß superfamily that was originally discovered based on its role in reproductive biology but is now recognized for its actions in embryonic tissue differentiation as well as in regulating homeostasis of various adult organs and systems as well as emerging roles in obesity, diabetes, NAFLD, and the modulation of glucose homeostasis (11). For example, activin was shown to be associated with obesity (12) and nonalcoholic steatohepatitis (13) in human subjects. Activin A concentration was significantly elevated in the serum of NAFLD patients in comparison with healthy controls (13) and stimulated proliferation in adipocyte (14) and β-cell lines (15). Activin treatment decreased lipolysis in adipocytes and increased intracellular TG storage by decreasing expression of key lipolytic genes, such as adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) (14). Taken together, these observations suggest that activin may have direct actions in liver that regulate lipid homeostasis.
The bioavailability of activin, and the related TGFβ family members myostatin and growth and differentiation factor 11 are regulated by the secreted antagonists follistatin (FST) and FST-like-3 (FSTL3) (16, 17). Three protein isoforms of FST have been identified with distinct biochemical properties and distributions (16). Although global deletion of Fst was neonatally lethal (18), we recently demonstrated that expression of only the FST288 isoform was sufficient to rescue this lethality (FST288-only mouse) (19). We also demonstrated that global deletion of Fstl3 resulted in a phenotype that included enlarged pancreatic islets, β-cell hyperplasia, and decreased visceral fat mass (20). Interestingly, glucose tolerance and insulin sensitivity were enhanced in the FSTL3-knockout (KO) or were unchanged in the FST288-only mouse (20, 21) whereas severe hepatic steatosis developed in both mouse models.
The presence of hepatic steatosis along with enhanced or unaltered insulin sensitivity in mice with reduced activin antagonism suggests the hypothesis that enhanced hepatic activin bioactivity accelerates accumulation of liver TG by enhancing hepatocyte response to insulin, particularly in the de novo lipogenesis pathway. This would result in increased hepatic TG content in the absence of systemic or hepatic liver insulin resistance, hyperglycemia, or elevated circulating lipids. We tested this hypothesis by examining hepatic response to acute insulin treatment in vivo as well as the response of primary mouse hepatocytes and the HepG2 hepatoma cell line to acute insulin treatment after activin pretreatment. Our results indicate that activin directly enhances hepatocyte response to insulin and activates de novo lipogenesis. Moreover, this occurs in the absence of systemic insulin resistance or hyperglycemia, suggesting that alteration of hepatic lipid biosynthesis, utilization, or degradation is primarily responsible for hepatic steatosis.
Materials and Methods
Mice
Global FSTL3-KO mice were produced as previously described (20) and maintained in the original 129S4/SvJae × C57BL/6J × FVB mixed background. FST288-only mice were created as previously described (19) and were maintained in the 129S4/SvJae × C57BL/6J mixed background. To control for the mixed backgrounds of these lines, studies used wild-type (WT) and homozygote male littermates selected from heterozygous breeding pairs. Male mice were studied between 7 and 11 months of age, when their phenotype became more distinct. All animal studies were carried out according to procedures approved by the Baystate Medical Center Institutional Animal Car and Use Committee.
RT-PCR procedure
Total RNA was extracted from tissues or cells using columns from Macherey Nagel (Düren, Germany) and reverse transcribed as preciously detailed (20), and expression of specific genes were quantitated by SYBR PCR (Stratagene, La Jolla, California). A cDNA standard pooled from liver cDNA was run in each PCR for each target, and each target was interpolated from the standard and then normalized to mouse Rpl19 expression (or human RPL19 for HepG2) to control for RNA quality. Primers used for mRNA expression studies are listed in Supplemental Table 1 (published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org).
Cell culture and transfection
Mouse hepatocytes were isolated as previously described (22) from 12-week-old C57BL6 male mice. HepG2 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 100 U/ml of penicillin and streptomycin. HepG2 cells in a 24-well plate were transfected with reporter plasmids for SMA and mothers against decapentaplegic 2/3 (SMAD2/3) (0.2 μg) (CAGA-LUC) using Lipofectamine reagent (Invitrogen, Grand Island, New York). Twelve hours after transfection, culture medium was replaced by DMEM without serum, supplemented with activin or FST288 at concentrations indicated, and incubated for 48 hours. Primary hepatocytes were cultured overnight in serum-free medium before the initiation of treatment. For gene expression experiments, cells were cultured with the indicated treatments for 48 hours. For determination of insulin sensitivity, cells were pretreated with activin A for 4 hours and subsequently treated with 5nM insulin for 10 minutes.
Insulin injection response study
Insulin (0.9 U) was diluted to a volume of 10 μL in PBS. Mice were injected ip with 0.9 U/kg body weight of insulin. Mice were killed, and livers were dissected and snap-frozen 10 minutes after injection.
Tissue/cell lysis and immunoblotting
Liver tissue and cells were lysed in cell lysis buffer (Sigma, St. Louis, Missouri). Protein concentration was determined using the bicinchoninic acid assay (Pierce Scientific, Rockford, Illinois). Equal amounts of protein (15–25 μg) were loaded into each lane of 10% gel (Invitrogen), electrophoresed, and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, Massachusetts). Membranes were blocked in 5% milk for 1 hour and then incubated with a primary antibody at 4°C overnight. Antibodies were used at the following concentrations: Akt (1:1000), phospho-Akt (1:1000), β-actin (1:1000), and sterol regulatory element binding protein 1c (SREBP1c) (1:150). Anti-Akt, anti–phospho-Akt (Ser473), and anti–β-actin were purchased from Cell Signaling (Danvers, Massachusetts). Anti-SREBP1c was purchased from AbCam (Cambridge, Massachusetts). Membranes were then incubated with horseradish peroxidase-linked goat antimouse or goat antirabbit secondary antibodies from Jackson ImmunoResearch Laboratories (West Grove, Pennsylvania), and signal was measured using ECL (Bio-Rad, Hercules, California).
Histology
Liver tissue from 7- to 11-month-old old mice was collected and fixed in 10% formaldehyde and embedded in paraffin. Four-micron sections were stained with hematoxylin and eosin or periodic acid Schiff's reagent (Sigma) and visualized using ×10 magnification on a light microscope.
Glycogen and TG extraction and quantification
Liver TGs were extracted using Folch's method (23). In brief, liver tissue was collected and immediately homogenized in 8:4:3 chloroform/methanol/0.9% NaCl. The organic layer was evaporated and reconstituted in chloroform. TG concentrations were determined using a kit (TR0100; Sigma), and values were normalized to the weight of the extracted liver. Liver glycogen was extracted by harvesting and immediately homogenizing liver tissue in 10% perchloric acid. Samples were centrifuged at 1000g, and supernatants transferred to a new tube containing an equal volume of absolute ethanol. Tubes were left overnight in −20°C, centrifuged at 3000g, and pellet was reconstituted in H2O. Glycogen concentration was measured by adding 1 mL of sample to 1 mL of 5% phenol, with subsequent addition of 100% H2SO4 followed by analysis at 490 nm. Sample values were then interpolated against a pure glycogen standard (Sigma).
Statistics
Group means were compared using Student's t test, and results with P < .05 were considered significant.
Results
Deletion of FSTL3 or FST315 results in hepatic steatosis
By 7 months of age, both FSTL3-KO and FST288-only mice developed widespread hepatic steatosis compared with WT littermates (Figure 1, A–D). However, the lipid droplets tended to be larger in FST288-only mice so that intracellular organelles were displaced, whereas the droplets in FSTL3-KO mice were mostly smaller and appeared less disruptive to hepatocyte structure. TG levels in liver extracts were significantly elevated in FSTL3-KO mice (66.6 ± 4.94 vs 43.21 ± 3.61 mg TG/g liver for FSTL3-KO vs WT, respectively, P < .01) and even more so in FST288-only mice (85.97 ± 18.63 and 31.97 ± 3.74 mg TG/g liver for FST288-only vs WT, respectively, P < .05) (Figure 1E), consistent with the histological appearance. Liver glycogen was significantly elevated by 2-fold in FSTL3-KO mice relative to WT littermates, whereas there was no difference in FST288-only mice compared with their WT littermates (Figure 1F). These observations indicate that the loss of either FSTL3 or FST315 results in ectopic hepatic TG accumulation in the absence of whole-body insulin resistance (20, 21) or elevated circulating TGs or free fatty acids (20). Moreover, they suggest that the mechanism and/or degree of hepatic steatosis might be different in these two models of enhanced activin bioactivity.
Figure 1.

Hepatic steatosis and TG accumulation in 7- to 11-month-old FSTL3-KO and FST288-only mice. A–D, Hematoxylin and eosin staining of liver sections reveals relatively homogenous TG distribution in FSTL3-KO and larger lipid droplets that disturb cytoplasmic organelle arrangement in FST288-only mice, both compared with WT littermates. E, Hepatic TG content is increased in both FSTL3- and FST-deficient mice. F, Hepatic glycogen is significantly increased in FSTL3-KO mice relative to WT littermates. *, P < .05; **, P < .01 (n = 3–7).
Loss of FSTL3 or FST315 antagonists improves acute hepatic insulin sensitivity
Although whole-body insulin sensitivity was not adversely affected by hepatic steatosis, accumulation of TG or its metabolites in hepatocytes has been reported to inhibit insulin signaling (5). We therefore determined the acute hepatic response to insulin by comparing phosphorylation of Akt 10 minutes after insulin injection because Akt is a critical node in the insulin cascade that regulates lipogenesis (24). Akt phosphorylation at Ser473 was significantly elevated 3-fold in livers of FSTL3-KO and FST288-only mice compared with WT littermates (Figure 2A). We also found that protein levels of full-length SREBP1c, a major lipogenic transcription factor regulated by Akt, was also significantly elevated in both FSTL3-KO and FST288-only mice compared with WT littermates (Figure 2B), with no detectable alteration in postprandial circulating insulin levels (21). These results suggest that hepatic TG accumulation may be induced by amplification of the lipogenic actions of insulin resulting from increased activin bioactivity after antagonist loss.
Figure 2.

Enhanced hepatic insulin response in FSTL3-KO and FST288-only mice. A, Livers were dissected 10 minutes after ip insulin injection. Insulin signaling in hepatic tissue, as assessed by immunoblotting for phospho-Akt (pAkt) normalized to total Akt, was enhanced in both FSTL3-KO and FST288-only livers. B, SREBP1c protein, as assessed by immunoblotting and normalization to β-actin, was significantly increased in both FSTL3-KO and FST288-only livers. *, P < .05; **, P < .01 (n = 4–8).
Activin enhances acute insulin response in cultured HepG2 hepatoma cells
Increased phospho-Akt in both FSTL3-KO and FST288-only mice suggested that increased activin bioactivity could directly sensitize hepatocytes to insulin. To test this hypothesis, we first determined that HepG2 hepatoma cells respond to activin using a luciferase reporter. We found that activin treatment dose-dependently increased luciferase response, whereas an excess of FST had no effect (Figure 3A), indicating that these cells respond to activin but don't release endogenous activin basally. We also verified that both SMAD2 and -3 second messengers were phosphorylated within 30 minutes of activin treatment (Figure 3B). Therefore, HepG2 cells make a good model for assessing activin's effects on insulin signaling.
Figure 3.
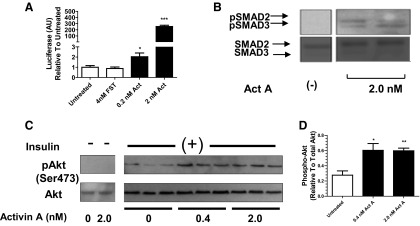
HepG2 cells respond to activin A with enhanced insulin signaling. A, HepG2 cells were transfected with a SMAD3 CAGA-Luc reporter and treated with activin A. Luciferase activity was dose-dependently increased. B, Both SMADs 2 and 3 were phosphorylated in response to activin. C, HepG2 cells were pretreated with activin A for 4 hours and subsequently treated with insulin or untreated for 10 minutes. Insulin-stimulated phospho-Akt was enhanced at both doses of activin A, whereas no phospho-Akt was detected in the absence of insulin. D, Densitometric analysis of immunoblot in C showing significant enhancement of phospho-Akt after activin A pretreatment. *, P < .05; **, P < .01; ***, P < .001. Abbreviations: Act, activin A; AU, arbitrary units; pAkt, phospho-Akt.
Pretreatment of HepG2 cells with 0.4nM activin A for 4 hours significantly increased Akt phosphorylation 2-fold in response to 10 minutes exposure to insulin compared with non-pretreated cells (Figure 3, C and D), and this level of phosphorylation was not enhanced at the 2.0nM activin A dose. These results indicate that exposure to activin A sensitizes HepG2 cells to a subsequent insulin challenge, supporting a role for activin in enhancing insulin signaling in FSTL3-KO and FST288-only livers.
Altered gene expression associated with fatty liver in FSTL3-KO and FST288-only mice
To investigate potential mechanisms for TG accumulation in livers of FSTL3-KO and FST288-only mice, we compared expression of genes important for uptake, breakdown, or de novo synthesis of lipids as well as other metabolic genes. FST288-only but not FSTL3-KO mice had significantly increased expression of low-density lipoprotein (LDL) receptor (LDLR) and lipoprotein lipase (LPL) expression (Figure 4A and Table 1), which together mediate the uptake and catabolism of circulating LDL (25), suggesting that the different histological appearance of hepatic TG in these two models may derive from at least partially distinct mechanisms of TG accumulation with uptake from the circulation being a component of TG accumulation in FST288-only mice but not necessarily FSTL3-KO mice.
Figure 4.

Gene expression in FSTL3-KO or FST288-only mouse liver or activin-treated HepG2 cells. A, Analysis of hepatic expression of genes involved with lipid uptake, de novo synthesis, lipolysis, insulin response, glucose uptake, or metabolism. Results are expressed relative to WT littermate gene expression. B, Gene expression in HepG2 cells after exposure to activin A (0.2nM or 2.0nM) for 48 hours to mimic chronic exposure in mouse liver. C, Gene expression in hepatocytes isolated from 12-week-old male C57BL6 mice treated with 2nM activin A for 48 hours. *, P < .05 (n = 4–5 for each gene).
Table 1.
Summary of Gene Expression Changes Shown in Figure 4, A–C, for FSTL3-KO and FST288-Only Liver, HepG2 Cells, or Primary Mouse Hepatocytes
| Gene | FSTL3-KO | FST288-Only | HepG2 | Primary Hepatocyte |
|---|---|---|---|---|
| LDLR | NC | + | − | − |
| SREBP1C | NC | NC | − | NC |
| ACC1 | + | + | NC | NC |
| FAS | NC | + | − | − |
| PNPLA3 | − | − | − | − |
| ATGL | NC | NC | + | − |
| GLUT2 | + | + | NC | NC |
| PEPCK | NC | + | + | + |
| G6PASE | NC | NC | − | NC |
| GCK | NC | NC | − | − |
| PGC1α | − | − | + | NC |
| PPARγ | NC | NC | + | NC |
Abbreviations: NC, no change; +, increased, −, decreased.
SREBP1c enhances transcription of a group of genes involved in lipid synthesis including acetyl coenzyme A carboxylesterase (ACC1) (26) and fatty acid synthase (FAS) (27). Although we found elevated SREBP1c protein levels in both FSTL3-KO and FST288-only mice, SREBP1c mRNA expression was not altered in either mouse line, consistent with regulation of SREBP1c at the protein level (28). However, ACC1 expression was significantly elevated in both lines, whereas FAS expression was elevated in FST288-only mice, consistent with increased de novo synthesis of lipids in both mouse lines contributing to hepatic steatosis. Expression of palatin-like phospholipase domain-containing protein 3 (Pnpla3), a gene in which a polymorphism has been closely linked to human NAFLD (29, 30), was significantly reduced in both mouse lines. Adipose triglyceride lipase (ATGL) expression was not significantly altered in either line. These results suggest that lipid synthesis may be enhanced, whereas lipolysis may be suppressed in these mouse lines, contributing to their overall lipid accumulation.
Expression of the predominant glucose transporter found in liver tissue, GLUT2, was modestly but significantly elevated in both mutant mouse lines (Figure 4A), suggesting enhanced glucose uptake with subsequent conversion into lipid might also contribute to TG accumulation. Expression of gluconeogenic genes were differentially affected by antagonist loss with phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) being increased in FST288-only mice, although only PEPCK was significant, whereas these genes were unaffected in FSTL3-KO mice. Glucokinase (GCK) expression was slightly but not significantly reduced in both mouse models. PGC1α expression was significantly suppressed in both mouse lines but PPARγ expression was not significantly altered. Taken together, these gene expression results indicate that portions of fatty acid uptake, synthesis, and metabolism pathways are altered in both FSTL3-KO and FST288-only mice but that the expression patterns are somewhat distinct, suggesting that the underlying mechanisms that result in fatty liver may not entirely overlap in the 2 mouse models.
Deletion of the activin antagonists FSTL3 or FST315 suggests that increased activin bioactivity might be responsible for the gene expression changes observed in livers of these mice. We therefore examined the effects on gene expression of 48 hours activin (0.2nM and/or 2.0nM) treatment of both HepG2 hepatoma cells and primary mouse hepatocytes to mimic the chronic activin exposure in the mouse models (Figure 4, B and C). Pnpla3 expression was decreased in both cell types by activin exposure, consistent with changes observed in mutant mouse liver. Likewise, PEPCK expression was increased by activin treatment in HepG2 cells and primary hepatocytes as well as in mouse liver. In contrast, G6P was decreased in HepG2 cells and unaltered in primary hepatocytes and mutant livers perhaps because HepG2 cells were derived from a human liver tumor where G6P is regulated differently. Unlike liver mRNA expression, LDLR was suppressed and unaltered by activin treatment in primary hepatocytes and HepG2 cells, respectively. Expression of ACC was unaltered by direct activin treatment, suggesting the elevation in ACC expression in mutant mice was an indirect effect of the mutation. Interestingly, FAS expression was significantly reduced by activin in both cell types, a change that is opposite that found in liver. ATGL expression was significantly enhanced in HepG2 cells but decreased in mouse hepatocytes, whereas the glucose transporter Glut2 was unaltered by activin treatment. Gck expression was significantly reduced in both cell types by activin treatment, and both PGC1α and PPARγ expression were significantly enhanced by activin treatment only in HepG2 cells These results suggest that direct activin action on hepatocytes may be involved with changes in expression of some genes involved with glucose and lipid metabolism but cannot explain all of the expression changes observed in the FSTL3-KO and FST288-only mouse livers. However, the consistent alterations in expression of both PNPLA3 and PEPCK in response to activin treatment in vitro or elevated activin bioavailability in vivo support a role for activin in regulating expression of these genes as well as their connection to fatty liver.
Discussion
To examine the physiological roles of the activin antagonists FSTL3 and FST315, we created 2 mouse models, one in which Fstl3 was globally deleted and the other containing a knock-in mutation that deleted an alternatively spliced mRNA preventing production of the full-length circulating form of FST (FST315) while preserving expression of FST288 (19, 20). We previously demonstrated that whole-body glucose tolerance and insulin sensitivity were enhanced in FSTL3-KO mice (20, 21), whereas these parameters were unaltered in FST288-only mice (21). Paradoxically, hepatic steatosis developed in both mouse models despite the absence of whole-body insulin resistance. However, it remained possible that TG accumulation resulted from insulin resistance of the liver itself leading to dysregulation of lipid metabolism and accumulation of TGs.
In this study, we demonstrated that in both mouse models, TG accumulation is pervasive and significantly increased compared with WT mice. However, insulin signaling via Akt was actually enhanced in both mouse models, as was SREBP1c protein concentration, suggesting that increased lipogenesis was at least partly responsible for ectopic liver TG accumulation. We then demonstrated that activin pretreatment increased Akt activation in HepG2 cells, indicating that increased activin bioactivity resulting from antagonist loss likely contributes to the fatty liver phenotype of these mice. Examination of altered gene expression in liver and activin-treated hepatocytes for the most part supports this concept. Nevertheless, our results demonstrate that hepatic steatosis can develop in settings of enhanced or unaltered whole-body insulin sensitivity along with enhanced hepatic insulin sensitivity and that hepatic steatosis is not sufficient for the development of whole-body insulin resistance, at least in these genetic models. Furthermore, these studies identify potential new roles for activin in enhancing insulin sensitivity and in regulating hepatic lipid metabolism, the alteration of which can induce hepatic steatosis.
Hepatic steatosis develops due to an imbalance favoring TG synthesis and uptake over TG utilization in hepatocytes, often in the context of systemic insulin resistance (1). Although many mouse models of NAFLD were also insulin-resistant (31), others maintained their insulin sensitivity (7, 32). Both of our mutant lines demonstrated increased insulin sensitivity and hepatic expression of SREBP1c, FAS, and ACC, genes involved in lipogenesis (33). Insulin promotes lipid storage, and therefore it is likely that the hepatic steatosis in these mice resulted from the activin-induced enhancement of insulin signaling rather than alteration of insulin levels, which were not different in FSTL3-KO (20) or FST288-only (21) mice relative to littermate controls.
FST288-only livers had elevated expression levels of genes involved in LDL uptake, but FSTL3 deletion had no effect on these genes. Furthermore, this elevation was not recapitulated in activin-treated cells, suggesting that the regulation of cholesterol uptake is not a direct effect of activin bioavailability. Moreover, the appearance of hepatic steatosis in these 2 models was distinct, and development of this pathology occurred earlier in FST288-only mice (data not shown), collectively suggesting that the mechanisms leading to fatty liver do not entirely overlap in these two models. Although both FST and FSTL3 are both extracellular activin antagonists with similar binding and neutralization profiles in vitro (16), differences in expression and biochemical properties (34, 35) may contribute to partially distinct activity profiles in vivo.
A mutation in PNPLA3 (I148M) is strongly linked to NAFLD through genetic association studies (29). PNPLA3 is highly expressed in adipose and liver, is transcriptionally regulated by insulin (36), and is localized to lipid droplets (37). Although the identified I148M substitution reduces hydrolase activity (38), genetic inactivation of Pnpla3 in mice does not alter TG content in liver (39, 40), whereas overexpression in mouse liver leads to enhanced TG concentrations (38). Therefore, at present, it is not clear whether increased or decreased expression/activity of PNPLA3 is responsible for inducing hepatic steatosis in mice or humans. Interestingly, in both FSTL3-KO and FST288-only mice, both of which develop pervasive hepatic steatosis, Pnpla3 expression is significantly reduced in liver, whereas activin treatment directly inhibits PNPLA3 expression in HepG2 cells and primary hepatocytes. These results are consistent with an activin-mediated reduction in expression and/or activity of PNPLA3 as having a direct role in mouse and human hepatic steatosis and, thus, NAFLD.
It is possible that the absence of whole-body insulin resistance in our mutant mouse lines reflects additional effects of deleting circulating FST315 or FSTL3 that compensate for insulin resistance caused by hepatic steatosis. It was recently demonstrated that inhibition of TGFβ family growth factors that use activin receptor IIB (ActRIIB) for signaling, including activins myostatin (MSTN) and growth differentiation factor 11, through administration of a soluble ActRIIB-Fc fusion protein increases muscle mass and reduces adiposity through enhancing a brown fat-like phenotype in white fat that included enhanced UCP1 and PGC1α gene expression (41, 42). Blockade of ActIIRB signaling by treatment with a monoclonal antibody also activated brown fat through enhancement of PGC1α expression leading to increased mitochondrial respiration as well as uncoupled respiration, suggesting increased energy flux and decreased lipid storage (43). Moreover, genetic deletion of MSTN expression inhibits diabetes through enhancing muscle glucose metabolism resulting in reduced blood glucose, insulin, and TG concentrations as well as improved insulin sensitivity of the muscle itself (44). Deletion of circulating activin/MSTN antagonists like FST315 and FSTL3, as in this study, would therefore be expected to increase bioactivity of these ligands and, thus, induce phenotypes opposite of those described above for ligand inhibition such as increased glucose and insulin concentrations as well as increased whole-body insulin resistance. We previously reported that FSTL3-KO mice have free fatty acid, TG, aspartate aminotransferase, alanine aminotransferase, leptin, and adiponectin levels that are not different from WT littermates along with enhanced whole-body glucose tolerance and insulin sensitivity (20). Glucose tolerance and insulin sensitivity were unchanged in FST288-only mice compared with WT controls (21). Given that increased bioactivity of activin or MSTN would be expected to have deleterious effects on glucose and insulin homeostasis and that no such effects were observed in our mice, it would seem unlikely that secondary effects of loss of FST315 or FSTL3 compensates for insulin resistance caused by hepatic steatosis.
The response to insulin was enhanced in both mouse livers and HepG2 cells after activin exposure, yet PEPCK expression was not suppressed. In fact, PEPCK expression was actually increased in FST288-only liver as well as in HepG2 cells and primary hepatocytes. The in vitro response suggests that activin might have dual actions in hepatocytes, serving to enhance the response to insulin on the one hand while stimulating PEPCK mRNA expression on the other, with the outcome in vivo dependent on which of these 2 effects is dominant. Additional studies will be required to identify the precise downstream mechanisms for activin's role in regulating both gene expression and insulin action in liver.
Nutrient deprivation in humans has been shown to increase FST and decrease activin A in the circulation (45). Furthermore, administration of glucose and insulin each resulted in an elevation of circulating activin A levels in human patients (46). In this study, we demonstrate that activin A acts as an insulin sensitizer in hepatocytes and stimulates the expression of genes controlling lipid uptake and de novo lipid synthesis. Taken together, these data suggest that activin A acts on hepatocytes in synergy with insulin to promote TG accumulation, which, when unregulated, leads to hepatic steatosis in the absence of insulin resistance.
Supplementary Material
Acknowledgments
We appreciate the technical assistance with histological experiments from Brooke Bentley (Pioneer Valley Life Sciences Institute, Springfield, Massachusetts).
This work was supported in part by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grant 5 R01 DK075058 (to A.L.S.).
Disclosure Summary: N.A.U., L.M.B., M.L.B., and A.L.S. have nothing to declare.
Footnotes
- ActRIIB
- activin receptor IIB
- Akt
- protein kinase B
- FST
- follistatin
- FSTL3
- FST-like-3
- LDL
- low-density lipoprotein
- KO
- knockout
- MSTN
- myostatin
- NAFLD
- nonalcoholic fatty liver disease
- SMAD
- SMA and mothers against decapentaplegic
- SREBP1c
- sterol regulatory element binding protein 1c
- TG
- triglyceride
- WT
- wild-type.
References
- 1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107(8):3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6(1):77–86 [PubMed] [Google Scholar]
- 4. Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353 [DOI] [PubMed] [Google Scholar]
- 6. Perseghin G, Scifo P, De Cobelli F, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48(8):1600–1606 [DOI] [PubMed] [Google Scholar]
- 7. Chakravarthy MV, Pan Z, Zhu Y, et al. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005;1(5):309–322 [DOI] [PubMed] [Google Scholar]
- 8. Stefan N, Häring HU. The metabolically benign and malignant fatty liver. Diabetes. 2011;60(8):2011–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bandsma RH, Prinsen BH, van Der Velden Mde S, et al. Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res. 2008;63(6):702–707 [DOI] [PubMed] [Google Scholar]
- 10. Komatsu M, Yazaki M, Tanaka N, et al. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol. 2008;49(5):810–820 [DOI] [PubMed] [Google Scholar]
- 11. Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocr. 2009;202:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zaragosi LE, Wdziekonski B, Villageois P, et al. Activin A plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes. 2010;59:2513–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yndestad A, Haukeland JW, Dahl TB, et al. A complex role of activin A in non-alcoholic fatty liver disease. Am J Gastroenterol. 2009;104(9):2196–2205 [DOI] [PubMed] [Google Scholar]
- 14. Magnusson B, Svensson PA, Carlsson LM, Sjöholm K. Activin B inhibits lipolysis in 3T3–L1 adipocytes. Biochem Biophys Res Commun. 2010;395(3):373–376 [DOI] [PubMed] [Google Scholar]
- 15. Demeterco C, Beattie GM, Dib SA, Lopez AD, Hayek A. A role for activin A and betacellulin in human fetal pancreatic cell differentiation and growth. J Clin Endocrinol Metab. 2000;85:3892–3897 [DOI] [PubMed] [Google Scholar]
- 16. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology. 2006;147(7):3586–3597 [DOI] [PubMed] [Google Scholar]
- 17. Schneyer AL, Sidis Y, Gulati A, Sun JL, Keutmann H, Krasney PA. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology. 2008;149:4589–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374:360–363 [DOI] [PubMed] [Google Scholar]
- 19. Kimura F, Sidis Y, Bonomi L, Xia Y, Schneyer A. The follistatin-288 isoform alone is sufficient for survival but not for normal fertility in mice. Endocrinology. 2010;151:1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukherjee A, Sidis Y, Mahan A, et al. FSTL3 deletion reveals roles for TGF-β family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci U S A. 2007;104:1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown ML, Bonomi L, Ungerleider N, et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity (Silver Spring). 2011;19(10):1940–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li WC, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–196 [DOI] [PubMed] [Google Scholar]
- 23. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509 [PubMed] [Google Scholar]
- 24. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96 [DOI] [PubMed] [Google Scholar]
- 25. Loeffler B, Heeren J, Blaeser M, et al. Lipoprotein lipase-facilitated uptake of LDL is mediated by the LDL receptor. J Lipid Res. 2007;48(2):288–298 [DOI] [PubMed] [Google Scholar]
- 26. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277(11):9520–9528 [DOI] [PubMed] [Google Scholar]
- 27. Bennett MK, Lopez JM, Sanchez HB, Osborne TF. Sterol regulation of fatty acid synthase promoter. Coordinate feedback regulation of two major lipid pathways. J Biol Chem. 1995;270(43):25578–25583 [DOI] [PubMed] [Google Scholar]
- 28. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86(11):839–848 [DOI] [PubMed] [Google Scholar]
- 29. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Browning JD, Cohen JC, Hobbs HH. Patatin-like phospholipase domain-containing 3 and the pathogenesis and progression of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52(4):1189–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8(1):35–44 [DOI] [PubMed] [Google Scholar]
- 32. Brown JM, Betters JL, Lord C, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51(11):3306–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koo SH, Dutcher AK, Towle HC. Glucose and insulin function through two distinct transcription factors to stimulate expression of lipogenic enzyme genes in liver. J Biol Chem. 2001;276(12):9437–9445 [DOI] [PubMed] [Google Scholar]
- 34. Tortoriello DV, Sidis Y, Holtzman DA, Holmes WE, Schneyer AL. Human follistatin-related protein: a structural homologue of follistatin with nuclear localization. Endocrinology. 2001;142:3426–3434 [DOI] [PubMed] [Google Scholar]
- 35. Sidis Y, Tortoriello DV, Holmes WE, Pan Y, Keutmann HT, Schneyer AL. Follistatin-related protein and follistatin differentially neutralize endogenous vs. exogenous activin. Endocrinology. 2002;143:1613–1624 [DOI] [PubMed] [Google Scholar]
- 36. Huang Y, He S, Li JZ, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A. 2010;107(17):7892–7897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Welte MA. Proteins under new management: lipid droplets deliver. Trends Cell Biol. 2007;17(8):363–369 [DOI] [PubMed] [Google Scholar]
- 38. He S, McPhaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52(3):1134–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Basantani MK, Sitnick MT, Cai L, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52(2):318–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koncarevic A, Cornwall-Brady M, Pullen A, et al. A soluble activin receptor type IIb prevents the effects of androgen deprivation on body composition and bone health. Endocrinology. 2010;151:4289–4300 [DOI] [PubMed] [Google Scholar]
- 42. Cadena SM, Tomkinson KN, Monnell TE, et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010;109:635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fournier B, Murray B, Gutzwiller S, et al. Blockade of the activin receptor IIb activates functional brown adipogenesis and thermogenesis by inducing mitochondrial oxidative metabolism. Mol Cell Biol. 2012;32(14):2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McPherron AC, Guo T, Wang Q, Portas J. Soluble activin receptor type IIB treatment does not cause fat loss in mice with diet-induced obesity. Diabetes Obes Metab. 2012;14(3):279–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vamvini MT, Aronis KN, Chamberland JP, Mantzoros CS. Energy deprivation alters in a leptin- and cortisol-independent manner circulating levels of activin A and follistatin but not myostatin in healthy males. J Clin Endocrinol Metab. 2011;96(11):3416–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Andersen GO, Ueland T, Knudsen EC, et al. Activin A levels are associated with abnormal glucose regulation in patients with myocardial infarction: potential counteracting effects of activin A on inflammation. Diabetes. 2011;60(5):1544–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.