

Abstract
Frontotemporal dementia (FTD) is a neurodegenerative disease with hallmark deficits in social and emotional function. Heterozygous loss-of-function mutations in GRN, the progranulin gene, are a common genetic cause of the disorder, but the mechanisms by which progranulin haploinsufficiency causes neuronal dysfunction in FTD are unclear. Homozygous progranulin knock-out (Grn−/−) mice have been studied as a model of this disorder and show behavioral deficits and a neuroinflammatory phenotype with robust microglial activation. However, homozygous GRN mutations causing complete progranulin deficiency were recently shown to cause a different neurological disorder, neuronal ceroid lipofuscinosis, suggesting that the total absence of progranulin may have effects distinct from those of haploinsufficiency. Here, we studied progranulin heterozygous (Grn+/−) mice, which model progranulin haploinsufficiency. We found that Grn+/− mice developed age-dependent social and emotional deficits potentially relevant to FTD. However, unlike Grn−/− mice, behavioral deficits in Grn+/− mice occurred in the absence of gliosis or increased expression of tumor necrosis factor-α. Instead, we found neuronal abnormalities in the amygdala, an area of selective vulnerability in FTD, in Grn+/− mice. Our findings indicate that FTD-related deficits resulting from progranulin haploinsufficiency can develop in the absence of detectable gliosis and neuroinflammation, thereby dissociating microglial activation from functional deficits and suggesting an important effect of progranulin deficiency on neurons.
Introduction
Frontotemporal dementia (FTD) is a devastating neurodegenerative disease and the leading cause of dementia in patients younger than 60 years (Vossel and Miller, 2008). Patients with FTD experience incapacitating behavioral changes, which commonly include social withdrawal and emotional blunting (Rascovsky et al., 2011). These symptoms result from dysfunction of a brain network called the salience network, which includes the amygdala, anterior cingulate cortex, insular cortex, limbic ventral striatum, and other regions (Seeley et al., 2009; Zhou et al., 2010). Although the mechanisms leading to this dysfunction are unclear, mutations in the GRN gene encoding progranulin are one of the most common causes of inherited FTD (Gass et al., 2006; Gijselinck et al., 2008). FTD-associated GRN mutations are loss of function, generally causing haploinsufficiency with reduced progranulin levels in brain and plasma (Finch et al., 2009).
Progranulin is a multifunctional protein expressed in neurons and microglia (Petkau et al., 2010). In neurons, progranulin has neurotrophic and neuroprotective properties and regulates synaptic function (Van Damme et al., 2008; Ryan et al., 2009; Tapia et al., 2011). However, considerable attention has focused on progranulin's role in microglia and its connection to neuroinflammation. Progranulin-deficient macrophages, when stimulated, have exaggerated inflammatory and phagocytic responses (Yin et al., 2010b; Kao et al., 2011), and progranulin deficiency causes susceptibility to inflammatory arthritis (Tang et al., 2011).
Homozygous progranulin knock-out (Grn−/−) mice have also suggested a role for gliosis and inflammation in progranulin-deficient FTD. Grn−/− mice develop an FTD-like pattern of behavioral disturbances, with social and emotional abnormalities and only mild or late hippocampal-dependent memory dysfunction (Kayasuga et al., 2007; Yin et al., 2010a; Ghoshal et al., 2012; Roberson, 2012). Grn−/− mice also have robust, age-dependent microgliosis and astrogliosis (Ahmed et al., 2010; Yin et al., 2010b; Ghoshal et al., 2012). These data suggest that glial overactivation and neuroinflammation resulting from progranulin deficiency could mediate FTD-related neuronal dysfunction in Grn−/− mice and patients with GRN mutations. However, the connection between gliosis and FTD-related neuronal dysfunction remains correlative, and no data indicate a causal role for microglial overactivation in progranulin-deficient mice or FTD patients.
To date, studies of murine progranulin deficiency have focused on homozygous knock-out mice, and relatively few data are available for the phenotype of Grn+/− mice. However, in humans, progranulin haploinsufficiency causes FTD, whereas homozygous progranulin deficiency causes another disorder, neuronal ceroid lipofuscinosis (Smith et al., 2012). Thus, haploinsufficiency and complete deficiency of progranulin have different disease outcomes, indicating a need for closer examination of possible FTD-related phenotypes in Grn+/− mice. Moreover, treatments for human progranulin-haploinsufficient FTD may center on raising expression from the intact allele, making it crucial to understand in detail the heterozygous murine model.
To determine whether progranulin haploinsufficiency in mice causes FTD-related behavioral deficits and whether those deficits involve gliosis and neuroinflammation, we studied behavior, physiology, neuropathology, and inflammatory mediators in multiple cohorts of Grn+/− mice at different ages. Our findings shed light on an unexpected dissociation between behavioral abnormalities and inflammatory changes with progranulin haploinsufficiency.
Materials and Methods
Generation of progranulin-deficient mice.
Mice with a floxed Grn allele (Grn+/F mice) were generated by homologous recombination in RF8 ES cells (129Sv/Jae) (Martens et al., 2012). Grn+/− mice were produced by breeding Grn+/F mice with mice expressing Cre recombinase in the germline (Tg(ACTB-cre)2Mrt/J, JAX #003376). Deletion of progranulin was observed at the mRNA and protein levels in multiple tissues (Martens et al., 2012); progranulin mRNA levels in brain and protein levels in brain and plasma were ∼50% of normal in Grn+/− mice (Fig. 1). Mice for this study were generated by breeding Grn+/− mice, except Figure 2E, for which the mice were a product of a cross between Grn+/+ and Grn+/− mice. Grn+/F mice that had not been crossed to Cre and, thus, had a floxed Grn allele but normal progranulin levels were used as controls in Figure 2F. An independent line of progranulin-deficient mice (Kayasuga et al., 2007) was used to confirm our hippocampal spine density results (see Figure 4F).
Figure 1.

Progranulin levels in progranulin-deficient mice. A–D, Progranulin mRNA levels in cortex (A), hippocampus (B), thalamus (C), and amygdala (D). Relative expression levels quantified by quantitative PCR were calculated using the ΔΔCt method and are displayed as a percentage of the average level in wild-type mice (N = 5–6 mice per genotype; age 19 months). E, F, Progranulin protein levels in cortex (E; N = 6 mice per genotype) and plasma (F; N = 17 mice per genotype), quantified by ELISA (age 12 months).
Figure 2.
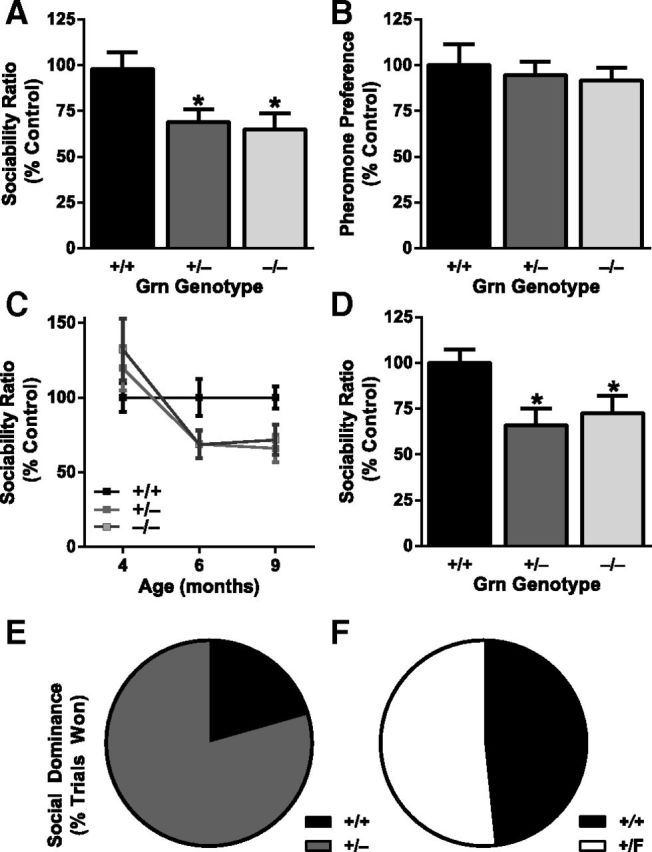
Progranulin haploinsufficiency causes social deficits. A, C, D, Three-chamber sociability test, with sociability ratio (time spent investigating another mouse divided by time spent investigating an inanimate object) expressed as percentage of control. A, On a mixed background at 6–7 months, Grn+/− mice and Grn−/− mice had lower sociability than Grn+/+ mice (ANOVA, p < 0.01). *p < 0.05 (post hoc test). N = 51–58 mice per genotype. B, No deficit in pheromone preference was observed in progranulin-deficient mice. N = 17 mice per genotype; mixed background at 7–8 months. C, A cohort of mice on a congenic C57BL/6 background was tested longitudinally at mean ages of 4, 6, and 9 months. There were no sociability deficits at 4 months, but reduced sociability was apparent at older ages (age × Grn interaction, p < 0.05). N = 17–19 mice per genotype. D, On a congenic C57BL/6 background at 9–12 months, Grn+/− mice and Grn−/− mice had lower sociability than Grn+/+ mice (ANOVA, p < 0.05). *p < 0.05 (post hoc test). N = 17–21 mice per genotype. E, F, Social interactions were further tested with the tube test of social dominance at 4–6 months of age. E, Grn+/− mice won 79% (27 of 34) of trials against Grn+/+ mice (exact p = 0.0008). F, Grn+/F controls, which have normal progranulin levels, won 52% (16 of 31) of trials against Grn+/+ mice, no different from chance. Male and female mice were used for both social tests. No sex-dependent effects were observed, so the data from males and females were combined.
Figure 4.

Progranulin-deficient mice have normal hippocampal function and spine density. A, B, Box-and-whisker plots representing the age at which early (A) and late (B) milestones were attained for each genotype. Boxes indicate the 75th and 25th percentiles, with a horizontal line at the median and a dot at the mean; whiskers represent the 10th and 90th percentiles (N = 11–21 mice per genotype). C, No differences were observed in exploratory behavior in the open field. Data are represented as total distance traveled during 15 min (congenic C57BL/6 background; N = 12–15 mice per genotype; age 9 months). D, Learning curves on the Morris water maze hidden platform task were not affected by progranulin deficiency. E, Dwell time in the target quadrant on a probe trial conducted 24 h after completion of training was not affected by progranulin deficiency. *p < 0.05 versus nontarget quadrant for each genotype. N = 14 mice per genotype; age 7 months. F, Progranulin deficiency did not affect hippocampal spine density in second-order apical dendrites of Golgi-stained CA1 pyramidal neurons (N = 9 or 10 neurons from two mice per genotype; age 12 months). G–I, Extracellular field recordings in area CA1 of acute hippocampal slices (N = 31–40 slices from 5 or 6 mice per genotype; age 12 months). G, Progranulin deficiency did not affect baseline synaptic transmission, assessed by input-output curves. H, Progranulin deficiency did not affect LTP, either early or late stages. I, Progranulin deficiency did not affect paired-pulse facilitation.
Males and females were used for all experiments, except electrophysiology and Golgi staining, which used only males. Males and females were analyzed separately and then presented together when there were no significant main effects of sex. Two cohorts of mice were tested on a mixed background that included 129Sv (on which the floxed line was originally created), FVB/N (the background strain of the Cre transgenic mice), and C57BL/6 (to which the original Grn+/− mice were crossed to expand the line for experiments). Subsequently, after 10 generations of backcrosses, a third cohort was tested on a congenic C57BL/6 background. All mice used were Cre-negative and verified Disc-1 wild-type (Disc-1 deletion is endogenous to 129 substrains) (Clapcote and Roder, 2006). Animals were housed in a pathogen-free barrier facility with a 12 h light/12 h dark cycle and ad libitum access to food (NIH-31 Open Formula Diet, #7917, Harlan) and water. All behavior experiments were conducted during daylight hours under normal room lighting conditions unless otherwise specified. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Progranulin ELISA.
Progranulin protein levels from mouse cortex and plasma were quantified using an ELISA kit (Adipogen) according to the manufacturer's protocol. To isolate protein, mouse cortex was dissected and homogenized in lysis buffer (10 mm Tris, pH 7.5, 10 mm NaCl, 3 mm MgCl2, 1 mm EGTA, and 0.05% v/v NP-40) supplemented with protease inhibitors (HALT; ThermoFisher). Samples were then spun at 16,000 × g for 10 min at 4°C. The supernatant was collected, and the concentration of soluble protein in each sample was determined using Coomassie Plus Protein Assay reagent (ThermoFisher). A total of 100 μg of protein was added to 1× diluents, and samples were run in duplicate. To isolate plasma, blood was collected from the right ventricle of the heart using a 1 ml syringe and a 23 G, ¾” needle. EDTA (250 mm) was included in the collection syringe to avoid clotting. To separate plasma from the cellular phase, samples were spun at 1000 × g for 10 min at 4°C in a swinging bucket centrifuge. Mouse plasma was then diluted 1:200 in 1× diluent and run in duplicate. Recombinant mouse progranulin supplied with the kit was used for generating a standard curve.
Three-chamber sociability.
Mice were tested for social interaction time in a three-chamber sociability test adapted from Moy et al. (2004) with minor modifications. Testing was conducted under red light. The test boxes were fabricated from white Plexiglas at the UAB machine shop and did not contain bedding. After 30–90 min of habituation to the testing room, a mouse was placed in the center chamber with the two outside chambers containing empty wire cages (Spectrum Diversified Designs). Mice were allowed to explore all three chambers for 10 min. After 10 min, a novel mouse (8-month-old, male, C57BL/6 mouse that had been habituated to being in the wire cage) and an inanimate object (matchbox car or block) were placed under the wire cages. The test mouse was allowed to explore for 10 min. The time spent interacting with the novel mouse and the inanimate object was scored either by a blinded observer with a stopwatch or a video tracking system (CleverSys). A sociability ratio was calculated for each mouse by dividing the time spent interacting with the other mouse by the time spent interacting with the inanimate object. To show data from multiple cohorts, the sociability ratio was normalized with 100% as the average sociability ratio of wild-type mice in each cohort.
Pheromone preference.
To test for pheromone preference, time spent investigating stimuli containing urine vs water was measured. Urine was collected and combined from 10 male C57BL/6 mice and subsequently frozen until testing. Urine or water (50 μl) was spotted onto small pieces of filter paper, which were then dried. The filter papers were inserted into histology cassettes to prevent direct contact, and cassettes containing urine and water were placed into opposite corners of a standard mouse cage with bedding. The test mouse was placed into the cage for 5 min and time spent within 5 cm of each stimulus was recorded with video tracking software (CleverSys).
Tube test of social dominance.
The tube test was adapted from Lindzey et al. (1961). Mice of the same sex, from different home cages, and lacking any prior experience with the test, were tested without habituation to the tube. Mice were placed, head first, into opposing sides of a clear plastic tube (3.81 cm ID × 30.5 cm in length) and released simultaneously. The trial ended when two paws of one mouse left the tube. The mouse remaining in the tube at the end of the trial was deemed the winner. A trial was aborted if the mice crossed each other or no mouse left the tube in 2 min. The tube was cleaned with 70% ethanol and dried between trials.
Fear conditioning.
Experiments were conducted in Quick Change Test chambers (Med Associates). For the acquisition of fear conditioning, mice were placed in the chambers and allowed to acclimate for 180 s. After acclimation, a conditioned stimulus (white noise at 75 Db) was given for 20 s coterminating with a 0.5 mA foot shock (2 s). This stimulus was given a total of three times with a 40 s interstimulus interval. Videos were recorded, and the time the mice spent immobile was measured by a blinded observer. The mice were then returned to their home cage. To test for cued-based conditioning, mice were tested 24 h after training. For this test, the chambers' context was made novel with a triangular, opaque white Plexiglas insert and a spearmint scent. Mice were placed in the chamber for 6 min with the white noise auditory cue given for the last 3 min. Freezing was measured before and during the cue. The chamber was cleaned with 70% ethanol between mice during the training and with isopropanol during the cued testing.
Developmental milestones.
The assessment of developmental milestones began on postnatal d 2. Pups were monitored daily for weight and attainment of physical milestones, such as incisor eruption, pinnae detachment, and opening of eyes. Further, pups were evaluated for reflexes and coordinated movements. For grasp reflex, forepaws were gently stroked with the wooden end of a swab; achievement of the milestone was registered if a grasping motion was observed. For surface righting, pups were placed in bedding with their ventral side up and timed until they successfully righted themselves. To assess cliff avoidance, pups were placed at the edge of a 6-cm-high Styrofoam platform, with head and paws hanging over the edge. The time it took to back away from the edge was measured, and achievement of the milestone was recorded if the time was <10 s. Negative geotaxis was measured by placing a pup facing downward on a coarse surface at an angle of 30°. The time it took for a pup to right itself was scored, and achievement of the milestone was recorded when the time was <10 s. To examine visual placing, pups were lowered downward by their tails toward a surface. When the pup raised their head and extended their forelimbs toward the surface, achievement of the milestone was recorded. To evaluate air righting, pups were held 30 cm above a well-bedded surface and dropped ventral side up. Achievement of the milestone was recorded on the day the pups were able to land on their feet. The bar hanging milestone was tested by allowing the mice to grasp a small bar and measuring the amount of time they could remain suspended without falling. Achievement of the milestone was scored if they could remain suspended.
Open field.
Total ambulatory distance and anxiety were measured on the open field (Med Associates). Mice were placed in the open field for 15 min trials. Activity was measured via infrared beam breaks by automated tracking software. The apparatus was cleaned with 70% ethanol between trials.
Water maze.
Spatial learning and memory were assessed with the Morris water maze as described previously (Scearce-Levie, 2011). All mice were single-housed for at least 5 d before testing. Hidden platform training consisted of 4 d of training with six trials each day (three in the morning and three in the afternoon). The platform sat 1.5 cm below the surface of the opaque water (20°C) in a fixed location. Each mouse was placed in the maze, initially facing the outer wall, at a drop-off point that changed semirandomly for each trial. A trial ended when the mouse located the platform and remained motionless on the platform for 2 s. If the mouse did not find the platform in 60 s, they were led to the platform and then removed from the pool. Performance was measured with an EthoVision video-tracking system (Noldus Information Technology). A probe trial was conducted 1 d after conclusion of training. The platform was removed and each mouse was placed into the maze opposite of where the platform resided. Each trial lasted 60 s. The latency to find the platform was analyzed using a two-way ANOVA with repeated measures.
Electrophysiology.
Electrophysiology was performed on acute hippocampal slices from 12-month-old mice. Mice were killed by decapitation and brains immediately placed in ice-cold cutting solution (110 μm sucrose, 60 μm NaCl, 3 μm KCl, 1.25 μm NaH2PO4, 28 μm NaHCO3, 2 μm CaCl2, 7 μm MgCl2, 5 μm glucose, and 0.6 μm ascorbate). Transverse slices (400 μm thick) were prepared on a Vibratome (Vibratome Company), and the hippocampus was dissected from the slice in a room temperature solution of 50% cutting solution and 50% ACSF (125 μm NaCl, 2.5 μm KCl, 1.25 μm NaH2PO4, 25 μm NaHCO3, 2 μm CaCl2, 1 μm MgCl2, 25 μm glucose). Slices were then equilibrated for 45 min in room temperature ACSF, followed by 1 h in 32°C ACSF. All solutions were saturated with 95% O2 and 5% CO2. Electrophysiological recordings were made in an interface chamber (Fine Science Tools) as described previously (Levenson et al., 2004). Hippocampal slices were constantly perfused at a rate of 1 ml/min with 30°C ACSF. Extracellular stimuli were applied to the Schaffer-collaterals along the border of areas CA1 and CA3 (Model 2200 stimulus isolator, A-M Systems) with an enameled, bipolar platinum–tungsten (92%:8%) electrode. fEPSPs were recorded in stratum radiatum with a glass recording electrode (1–3 MΩ) filled with ACSF. Clampex was used for data acquisition and Clampfit used for data analysis (Molecular Devices). Input/output curves were generating by applying a range of stimuli from 0.5 to 30 V in 1 V increments. Intensities were averaged over three sweeps. The stimulus intensity that evoked 50% of the maximal fEPSP slope was used for subsequent stimulations. Paired-pulse facilitation was measured by applying consecutive stimuli at a range of interstimulus intervals (10, 20, 50, 100, 150, 200, 250, and 300 ms). After 20 min of baseline recording, LTP was induced by applying two 100 Hz, 1 s stimulus trains, 20 s apart. Recordings were taken every 20 s, and data were averaged into 2 min intervals. Data from paired pulse facilitation and LTP were analyzed using a two-way ANOVA with repeated measures.
Spine density.
Mouse brains were processed for morphological assessment with the FD Rapid GolgiStain Kit (FD NeuroTechnologies) according to the manufacturer's protocol. Mice were killed with 100 mg/kg Fatal-Plus (Vortech Pharmaceuticals) and decapitated, and brains were removed. Hemibrains were submerged in impregnation solution for 14 d and then transferred to a cryoprotectant for an additional 7 d. Next, brains were cut into 200 μm sections with a cryostat (Leica) and mounted on gelatin-coated slides. After mounting, the slides were rinsed in ddH2O and incubated in developing solution. After development, the sections were rehydrated with increasing concentrations of ethanol and cleared with xylene. Last, slides were coverslipped using EUKITT mounting medium (Electron Microscopy Sciences).
Microscopy was performed on a MicroBrightField system (MBF Bioscience). Image stacks were taken of 10–40 μm segments of second-order apical dendrites on CA1 pyramidal neurons at 100× (oil-immersion). All image stacks were manually traced using Neurolucida and analyzed using Neurolucida Explorer.
Immunohistochemistry.
Immunohistochemistry was performed on free-floating 30 μm thick coronal sections as described previously (Palop et al., 2011). To obtain sections, mice were killed as described above and transcardially perfused with normal saline. Brains were removed, drop-fixed in 4% paraformaldehyde in phosphate buffer (80 mm Na2HPO4, 200 mm NaH2PO4, pH 7.4), and placed at 4°C for 48 h. After two PBS washes, the brains were transferred to 30% sucrose in PBS for 48 h or until they sank. The 30 μm sections were then cut on a sliding microtome with freezing stage (Leica). The sections were placed in cryoprotectant (30% ethylene glycol, 30% glycerol, 40% PBS, v/v/v) and held at −20°C until use.
Sections were washed free of cryoprotectant with PBS. Endogenous peroxidase activity was quenched with 3% H2O2 and 10% methanol in phosphate buffer, followed by blocking with 10% normal goat serum (Vector Laboratories), 1% dry milk, and 0.2% gelatin to prevent nonspecific binding. Primary antibodies were used at the following concentrations diluted in PBS: 3% normal goat serum and 0.2% gelatin: Iba1 (1:5000; Wako), GFAP (1:5000; Dako), and c-fos (1:10,000; EMD Millipore). Species-specific biotinylated secondary IgG (1:5000; Vector Laboratories) was applied, and the Vectastain Elite avidin-biotin horseradish peroxidase complex kit (Vector Laboratories) was used for amplification. Diaminobenzidine:tetrahydrochloride was added for detection and sections were mounted with cover glass using Cytoseal 60 (Electron Microscopy Sciences). For analysis of Iba1 and GFAP, photomicrographs of three sections per mouse were collected on an upright microscope (Carl Zeiss) with a 10× objective. Thresholds were set to include only GFAP or Iba1-positive pixels. The total number of GFAP or Iba1-positive pixels per image were quantified then averaged for the three images. Autofluorescent images were taken on an upright microscope (Nikon) equipped with an FITC (excitation 480/30; emission 535/40) filter cube. Pixel densities were calculated using ImageJ software.
qRT-PCR.
mRNA levels were quantified using quantitative PCR. RNA was first prepared using Trizol reagent (Invitrogen), according to the manufacturer's protocol. From the prepared RNA, cDNAs were generated with SuperScript III and random primers (Invitrogen). cDNAs were quantified using LightCycler 480 Probes master mix (Roche) and TaqMan Gene Expression Assays for TNF-α (Mm00443258_m1), progranulin (Mm00433848_m1), and β-actin (Mm00607939_s1). Amplification was performed on a Roche LightCycler 480, and data were quantified using the ΔΔCt method (User Bulletin 2, Applied Biosystems).
Novel/social environment.
Mice were exposed to a novel and social environment as previously described (Scearce-Levie et al., 2008). Briefly, two mice of the same genotype and opposite sex were placed in a cage with a new type of bedding and novel olfactory, tactile, and visual stimuli for 2 h before being killed. Control mice were left in their home cage, undisturbed for 3 d before death.
Sections were immunostained for c-fos or NeuN as described above, then scanned with a slide scanner (Electron Microscopy Sciences). Three serial sections, 300 μm apart, in which the caudate/putamen and amygdala were clearly defined (between 1.0 and 1.8 mm posterior to bregma) were chosen. For the caudate/putamen and the central amygdala (including amygdalostriatral transition area), the lateral boundary was the external capsule and the medial boundary was the globus pallidus and internal capsule (see Fig. 7A). A horizontal line extending medially from the bifurcation of the external capsule separated caudate/putamen from amygdala. The ventral border of the amygdala was defined by a line drawn between the tips of the external capsule and extending medially to either the internal capsule or optic tract, depending on the anterior/posterior location. Cells were manually counted by an observer blind to genotype. Any cell touching a line was excluded from the count. Data are represented as a sum of all cells counted through the 3 sections.
Figure 7.

Decreased neuronal activation in the amygdala of Grn+/− mice. A, Regions counted for c-Fos- and NeuN-positive neurons, including central amygdala (CeA), caudate/putamen (CPu), and dentate gyrus (DG). B–D, Active c-Fos-positive neurons were counted in various brain regions. Under resting, home-cage conditions, few neurons were c-Fos-positive. B, Fewer c-Fos-positive neurons were present in the amygdala of Grn+/− and Grn−/− mice after 2 h in a novel environment (ANOVA, p < 0.05). **p < 0.01 versus Grn+/+ mice (post hoc test). *p < 0.05 versus Grn+/+ mice (post hoc test). There was no difference in the number of c-Fos-positive neurons in the caudate/putamen (C) or dentate gyrus (D) of the hippocampus. n.s., Not significant. E, There were no differences between groups in the number of social interactions of various types during the 2 h in the novel environment. F, There were no differences in total NeuN-positive amygdala neuron counts of progranulin-deficit mice, so the decrease in c-Fos-positive neurons cannot be attributed to neuron loss, but rather to impaired activation.
Data analysis.
All experiments were conducted by observers blinded to genotype. All data represent mean ± SEM. Data were analyzed using GraphPad Prism or SPSS, and statistical comparisons were made with ANOVA with Dunnet's post hoc test for multiple comparisons unless otherwise stated. p < 0.05 was considered significant. For the tube test of social dominance, statistical comparisons were made with a two-tailed binomial test against an expected outcome (50%). Outliers more than two SDs from the mean were excluded for all behavioral assays.
Results
Progranulin haploinsufficiency causes social dysfunction in Grn+/− mice
Social withdrawal is a common symptom of FTD. To test social withdrawal in progranulin-deficient mice, we used the three-chamber sociability test (Moy et al., 2004). This test measures the sociability of a mouse when given the choice between interacting with another mouse versus an inanimate object. We tested two separate cohorts of 6- to 7-month-old wild-type (Grn+/+), Grn+/−, and Grn−/− mice on a mixed genetic background. As expected, Grn+/+ mice spent more time interacting with the other mouse than with the object (average conspecific/inanimate ratio 2.75 ± 0.23), and Grn−/− mice showed reduced sociability (Fig. 2A). Interestingly, Grn+/− mice also showed reduced sociability to a similar extent as Grn−/− mice (Fig. 2A).
This abnormality in progranulin-deficient mice could be the result of either a true social deficit or an inability to detect pheromones, an olfactory cue critical for social behavior. To distinguish between these possibilities, we determined the preference to explore a pheromone-scented stimulus over a control without pheromones. Grn+/+ mice spent more time investigating the pheromone-positive stimulus, and similar results were observed in Grn+/− and Grn−/− mice (Fig. 2B). Thus, progranulin deficiency does not impair olfactory detection of, or preference for, pheromones. We conclude that the deficits in the three-chamber test reflect social dysfunction.
We next confirmed the finding of reduced sociability in Grn+/− mice in a third cohort of mice, this time on a congenic C57BL/6 background, and found the deficits were age-dependent (Fig. 2C). When tested at 4 months, this cohort showed no abnormalities, indicating that the sociability deficits in progranulin-deficient mice are not developmental. When the same cohort was tested at 6 and at 9 months, Grn+/− and Grn−/− mice showed reduced sociability, just as on the mixed background (Fig. 2C). Group data for the sociability deficit at 9–12 months on C57BL/6 background is shown in Figure 2D. Similar deficits were observed in males and females.
To extend the evidence for social abnormalities in Grn+/− mice, we used the tube test for social dominance (Lindzey et al., 1961). In this test, a wild-type and a test mouse of the same sex enter opposite ends of a plastic tube. The mice cannot pass each other inside the tube, so one mouse must turn around to exit. In the absence of social abnormalities, each genotype should “win” 50% of trials. However, when Grn+/− mice were paired against Grn+/+ mice, Grn+/− mice won 79% of the time (27 of 34 trials; Fig. 2E). As a control, we tested mice in which one Grn allele was floxed, but not deleted by Cre (Grn+/F mice), so that progranulin expression was unaffected. Grn+/F controls did not show abnormalities, winning 52% of trials, which was not different from chance (16 of 31 trials; Fig. 2F). Thus, in multiple cohorts of mice on different genetic backgrounds, Grn+/− mice have altered social behavior.
Progranulin haploinsufficiency impairs fear conditioning in Grn+/− mice
Emotional impairment is also common in FTD patients (Rosen et al., 2004), including impairment of fear conditioning (Hoefer et al., 2008). To examine the effect of progranulin haploinsufficiency on fear conditioning, we performed classical cued fear conditioning. Cued fear conditioning requires an association between an aversive stimulus and an auditory cue and depends on the amygdala but is hippocampus-independent (Maren et al., 1996; Logue et al., 1997). The amount of time a mouse is immobile after being presented with the auditory cue 24 h post-training is a measure of fear memory. On the mixed background, both Grn+/− and Grn−/− mice had impaired fear conditioning, with a decrease in the percentage time spent immobile during the auditory cue (Fig. 3A). Similar results were obtained on a congenic C57BL/6 background (Fig. 3B). For each cohort, deficits in Grn+/− mice were equivalent to those in Grn−/− mice. As with the sociability assay, no abnormalities in fear conditioning were observed at 4 months of age (data not shown), and no significant sex effects were observed. Therefore, in multiple cohorts of mice on different genetic backgrounds, Grn+/− mice have emotional dysfunction.
Figure 3.

Progranulin haploinsufficiency impairs fear conditioning. Twelve-month-old mice from a mixed (A; N = 54–59 mice per genotype) or congenic C57BL/6 (B; N = 17–19 mice per genotype) background were tested for cued fear conditioning one day after training. A significant effect of progranulin deficiency was observed in mice on both backgrounds (ANOVA, p < 0.01). Both Grn+/− and Grn−/− mice spent significantly less time immobile than Grn+/+ mice: **p < 0.001 (post hoc test). ***p < 0.0001 (post hoc test).
Sparing of other domains in progranulin-deficient mice
We also examined a variety of other behavioral measures to determine whether the social and emotional abnormalities in Grn+/− mice reflected more global neuronal dysfunction. Progranulin-deficient mice had no detectable abnormalities in postnatal development (Fig. 4A,B), consistent with the findings that social and emotional deficits did not develop until after 4 months. We also observed no deficits in adult mice in locomotor activity (Fig. 4C).
FTD causes significant social and emotional dysfunction, but hippocampal function is relatively preserved in the early stages of disease (Kril and Halliday, 2011). To investigate hippocampal function in progranulin-deficient mice, we performed a rigorous battery of behavioral, electrophysiological, and structural analyses. First, we used the Morris water maze to test spatial memory in 7-month-old mice. Grn+/− and Grn−/− mice learned to find the hidden platform just as well as Grn+/+ mice (Fig. 4D). During the probe trial, all three groups spent more time in the target quadrant than the other quadrants (Fig. 4E). Next, we evaluated the morphology of hippocampal CA1 pyramidal neurons and found that progranulin deficiency did not affect spine density in this region (Fig. 4F). We confirmed that progranulin deficiency does not alter hippocampal spine density in an independent line of progranulin knock-out mice (Kayasuga et al., 2007) produced by another group (data not shown). Finally, we recorded electrophysiological responses at the Schaffer collateral-CA1 synapse in acute hippocampal slices. Progranulin deficiency caused no changes in basal synaptic transmission, determined by input/output curves (Fig. 4G), long-term potentiation (Fig. 4H), or paired-pulse facilitation (Fig. 4I). We conclude that the hippocampus is relatively spared by progranulin deficiency at ages when deficits are seen in more FTD-related regions.
Progranulin haploinsufficiency does not cause gliosis or lipofuscinosis in Grn+/− mice
Our analysis of Grn+/− mice indicates that they display an FTD-like phenotype with social and emotional dysfunction and relative preservation of other domains. To begin determining how progranulin haploinsufficiency causes deficits in these mice, we looked for gliosis and neuroinflammation, which in other lines was present in Grn−/− mice but absent in Grn+/− mice (Ahmed et al., 2010). As expected, Grn−/− mice had significant microgliosis by Iba1 staining in multiple brain regions, including thalamus (Fig. 5A,B), hippocampus (Fig. 5C), and cortex (Fig. 5D), but not in the amygdala (Fig. 5E). And consistent with prior studies, Grn+/− mice did not have microgliosis (Fig. 5A–E). Other measures of neuroinflammation had the same pattern of abnormalities in Grn−/− but not Grn+/− mice, including TNF-α mRNA levels (Fig. 5F–I) and astrocytosis (Fig. 5J–M). Although not surprising given prior studies, the differences in neuroinflammation between Grn−/− and Grn+/− mice become quite interesting in light of the presence of behavioral deficits in Grn+/− mice. The dissociation between functional impairment and microgliosis in Grn+/− mice suggests that neuroinflammation is likely not driving the functional deficits resulting from progranulin haploinsufficiency in these mice.
Figure 5.
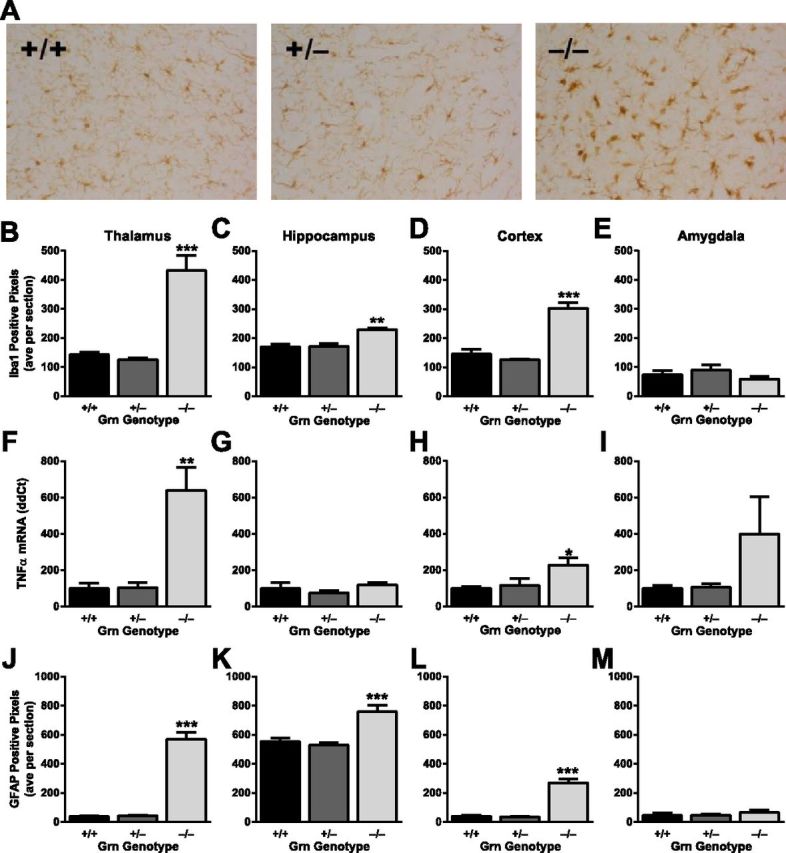
Absence of neuroinflammation in Grn+/− mice with FTD-related behavioral abnormalities. A, Representative images of Iba1 immunohistochemistry in the thalamus of control and progranulin-deficient mice. B–E, Quantification of Iba1 immunoreactivity in various brain regions (N = 6 mice per genotype; age 12 months). B, Grn−/−, but not Grn+/−, mice had microgliosis in thalamus (ANOVA, p < 0.0001). On post hoc tests, only Grn−/− mice differ from other groups: ***p < 0.0001. C, Grn−/−, but not Grn+/−, mice had microgliosis in hippocampus (ANOVA, p < 0.005). On post hoc tests, only Grn−/− mice differ from other groups: **p < 0.001. D, Grn−/−, but not Grn+/−, mice had microgliosis in cortex (ANOVA, p < 0.0001). On post hoc tests, only Grn−/− mice differ from other groups: ***p < 0.0001. E, There were no significant differences in Iba1 immunoreactivity in amygdala. F–I, Quantitative PCR analysis of TNF-α mRNA levels in various brain regions (N = 5 or 6 mice per genotype; age 19 months). F, Grn−/−, but not Grn+/−, mice had increased TNF-α in thalamus (ANOVA, p < 0.005). On post hoc tests, only Grn−/− mice differ from other groups: **p < 0.001. G, There were no significant differences in TNF-α in hippocampus. H, Grn−/−, but not Grn+/−, mice had increased TNF-α in cortex (ANOVA, p < 0.05). On post hoc tests, only Grn−/− mice differ from other groups: *p < 0.05. I, Although trending, no significant differences in TNF-α in amygdala were detected (ANOVA, p = 0.12). J–M, Quantification of GFAP immunoreactivity in various brain regions (N = 6 mice per genotype; age 12 months). J, Grn−/−, but not Grn+/−, mice had astrocytosis in thalamus (ANOVA, p < 0.0001). On post hoc tests, only Grn−/− mice differ from other groups: ***p < 0.0001. K, Grn−/−, but not Grn+/−, mice had astrocytosis in hippocampus (ANOVA, p = 0.0001). On post hoc tests, only Grn−/− mice differ from other groups: ***p < 0.001. L, Grn−/−, but not Grn+/−, mice had astrocytosis in cortex (ANOVA, p < 0.0001). On post hoc tests, only Grn−/− mice differ from other groups: ***p < 0.0001. M, There were no significant differences in GFAP immunoreactivity in amygdala.
Because homozygous GRN mutations in humans cause neuronal ceroid lipofuscinosis (Smith et al., 2012) and Grn−/− mice display autofluorescent lipofuscin granules in the hippocampus (Ahmed et al., 2010; Petkau et al., 2012), we quantified these deposits in our mice. The density of autofluorescent granules was increased not only in the hippocampus of Grn−/− mice, but also in the frontal cortex, thalamus, and amygdala (Fig. 6). However, these deposits were not increased above normal levels in Grn+/− mice (Fig. 6). These data also dissociate lipofuscin pathology from the functional deficits in Grn+/− mice and indicate that lipofuscinosis is not required for the impairments caused by progranulin haploinsufficiency.
Figure 6.

Absence of lipofuscinosis in Grn+/− mice. A, Representative images of autofluorescent lipofuscin granules in the CA3 region of the hippocampus. B–E, Quantification of autofluorescence in various brain regions (N = 6–8 mice per genotype; age 12 months). Increased autofluorescence was observed in Grn−/−, but not Grn+/− mice (ANOVA, p < 0.001; post hoc tests). Only Grn−/− mice differ from other groups: *p < 0.05; ****p < 0.0001.
Progranulin haploinsufficiency impairs amygdala function in Grn+/− mice
Given the dissociation between neuroinflammation and functional deficits, we turned our attention from microglia to neurons, which are the other major cell type expressing progranulin in the brain (Petkau et al., 2010). We exposed mice to a novel environment with various stimuli, including a conspecific of the same genotype and opposite sex for 2 h before death, then stained for the immediate early gene, c-Fos, to investigate patterns of neuronal activation (Scearce-Levie et al., 2008). We focused on the amygdala for several reasons. First, amygdala dysfunction could account for the social and emotional abnormalities we observed in progranulin-deficient mice, as regulation of social interactions and emotion are core functions of the amygdala (LeDoux, 2003; Adolphs, 2010). Specifically, amygdala dysfunction has been shown to decrease sociability in the three-chamber test (Moya et al., 2011) and impair fear conditioning (Maren et al., 1996). Second, the amygdala plays a major role in social and emotional symptoms of FTD (Liu et al., 2004; Hoefer et al., 2008; Kipps et al., 2009). Third, the amygdala is a component of the salience network, which is selectively vulnerable in FTD (Seeley et al., 2009; Zhou et al., 2010).
We counted c-Fos-positive neurons in the central amygdala and two control regions in the same sections (Fig. 7A). Under resting, home-cage conditions, there were few c-Fos-positive neurons in any of these areas (Fig. 7B–D). In Grn+/+ mice, exposure to a novel environment caused neuronal activation in the central amygdala (Fig. 7B). In contrast, both Grn+/− and Grn−/− mice had reduced activation, to a similar extent, in this area (Fig. 7B). This region of the amygdala is a primary output of other amygdala nuclei (LeDoux, 2007) and is critically involved in fear and social behavior (LeDoux, 2007; Bosch and Neumann, 2010). The impairment of neuronal activation was selective to the amygdala as no differences were observed in the adjacent caudate/putamen (Fig. 7C) or in the dentate gyrus (Fig. 7D). The reduced neuronal activation was not due to decreased interactions during exploration of the novel environment (Fig. 7E). Furthermore, the total number of amygdala neurons, assessed by NeuN immunohistochemistry, was unchanged (Fig. 7F), indicating that the observed abnormality reflects a functional impairment of neuronal activation, not neuronal loss. Thus, while Grn+/− mice do not have gliosis or lipofuscinosis, they do have neuronal dysfunction in the amygdala, a region critical for normal social and emotional function (Adolphs, 2010; Bickart et al., 2011) that is strongly affected in FTD (Rabinovici et al., 2007).
Discussion
In this study, we demonstrate that progranulin haploinsufficiency in Grn+/− mice causes neuronal dysfunction potentially related to FTD, but with mechanistically informative differences from existing Grn−/− models with complete progranulin deficiency. We found that Grn+/− mice have social and emotional dysfunction without detectable differences in hippocampal function or other behavioral abnormalities. However, neuronal dysfunction in Grn+/− mice occurs without evidence of neuroinflammation (microgliosis, increased TNF-α, and astrogliosis) or lipofuscin deposits, indicating that these abnormalities found in Grn−/− mice do not mediate the FTD-like dysfunction resulting from progranulin haploinsufficiency in Grn+/− mice. Instead, we found decreased neuronal activation in the amygdala of Grn+/− (and Grn−/−) mice, suggesting important neuronal effects from progranulin haploinsufficiency.
Behavioral abnormalities in progranulin mouse models
Most reports on the three previously published progranulin knock-out mouse lines have focused on homozygous Grn−/− mice. To our knowledge, there is just one study reporting behavioral data from Grn+/− mice, which examined only sexual behavior, aggression, and anxiety (Kayasuga et al., 2007). There were no significant abnormalities in Grn+/− mice, although they trended toward abnormal sexual behavior that was intermediate between Grn+/+ and Grn−/− mice (Kayasuga et al., 2007). Other behavioral studies included only Grn−/− mice. Of note, in one line using a gene trap construct, progranulin levels were not reduced in heterozygous mice (Petkau et al., 2012), so their phenotype does not reflect the effects of progranulin haploinsufficiency.
We found that progranulin haploinsufficiency in mice produces key features of FTD. Impaired social function is a cardinal symptom of FTD (Rascovsky et al., 2011). We observed social deficits in Grn+/− mice in multiple cohorts of mice on two different genetic backgrounds (Fig. 2). These deficits were apparent with both the three-chamber sociability test and the tube test. In previous studies of three different lines of Grn−/− mice, social dysfunction is the most consistently observed abnormality (Roberson, 2012). Although different groups used different assays, each of the four Grn−/− lines now published (including the data herein) has social deficits. One prior study showed deficits on the three-chamber sociability test that we also used (Yin et al., 2010a). Deficits in that study appeared at 1 month of age, but we found the sociability deficit to be age-dependent, with deficits emerging after 4 months (Fig. 2C). Two other Grn−/− lines had social deficits on the resident-intruder test (Kayasuga et al., 2007; Ghoshal et al., 2012; Petkau et al., 2012).
Impairment of emotion, particularly “negative” emotions such as fear, is another common feature of FTD (Werner et al., 2007; Kipps et al., 2009). We tested classical fear conditioning, which is impaired in FTD patients (Hoefer et al., 2008). Grn+/− and Grn−/− mice had impaired fear conditioning, again on both genetic backgrounds (Fig. 3).
In addition to social and emotional deficits, FTD is characterized by relative preservation, in early stages, of other cognitive domains, including learning and memory. We observed no deficits in learning and memory, hippocampal spine density, or hippocampal physiology in Grn+/− or Grn−/− mice (Fig. 4). These results corroborate previous work indicating that deficits in hippocampal-dependent learning and memory, if present, do not occur until much later than social abnormalities in Grn−/− mice (Yin et al., 2010a; Ghoshal et al., 2012). One study using a gene trap that expresses LacZ in place of progranulin found hippocampal synaptic deficits and spine loss in Grn−/− mice (Petkau et al., 2012); we did not observe these changes in two different lines of Grn−/− mice (Fig. 4F; and data not shown). It is possible that the abnormalities observed in the gene trap line result from overexpression of LacZ rather than from progranulin deficiency.
Progranulin deficiency, inflammation, and FTD
Evidence from Grn−/− mice suggested that progranulin deficiency fosters inflammatory changes that could contribute to FTD. Stimulating Grn−/− macrophages leads to exaggerated release of neurotoxic cytokines (Yin et al., 2010b) and accelerated phagocytosis (Kao et al., 2011), and cultured Grn−/− microglia also have an amplified inflammatory response (Martens et al., 2012). Grn−/− mice also display striking, age-dependent microgliosis and astrogliosis (Ahmed et al., 2010; Yin et al., 2010b; Ghoshal et al., 2012) (Fig. 5). Recent studies showed that progranulin attenuates the activation of TNF-α receptors and that loss of this anti-inflammatory effect of progranulin increases susceptibility to inflammatory arthritis in Grn−/− mice (Tang et al., 2011). Consistent with a role of progranulin in attenuating inflammation, we found increased TNF-α mRNA in the brains of Grn−/− mice (Fig. 5F–I).
However, our findings indicate that these inflammatory changes are not necessary for the FTD-like dysfunction induced by progranulin haploinsufficiency. Middle-aged Grn+/− mice had similar social and emotional dysfunction as Grn−/− mice (Figs. 2 and 3) but no apparent microgliosis, increased TNF-α, or astrogliosis (Fig. 5). Others have also found that Grn+/− mice do not develop gliosis (Ahmed et al., 2010). Because the FTD-like behavioral abnormalities did not differ between Grn+/− and Grn−/− mice, our data suggest that gliosis and increased TNF-α, although present in Grn−/− mice, are not required for associated behavioral deficits. Of course, these findings do not rule out a role for TNF signaling in the deficits in Grn+/− mice; it is possible that reduced progranulin levels in Grn+/− mice increase TNF receptor signaling without elevation of TNF-α levels.
Although our data indicate that gliosis is not necessary for deficits in mice with progranulin haploinsufficiency, microgliosis and astrogliosis are present at autopsy in FTD patients with progranulin haploinsufficiency (Mackenzie, 2007). This may reflect the fact that, although we are studying the initial sequelae of progranulin haploinsufficiency in mice, the human tissue is from end-stage disease and the gliosis may represent a reactive change to neuronal injury that developed over many years. Thus, although our data suggest that neuronal dysfunction as a direct consequence of progranulin haploinsufficiency is an initial event in the disease process and gliosis is not necessary for this early dysfunction, it is possible that, in FTD patients, gliosis exacerbates neuronal injury, perhaps creating a vicious circle leading to further reactive gliosis and additional injury. Indeed, murine progranulin deficiency predisposes to increased neuroinflammation in response to CNS injury (Martens et al., 2012). Causality is difficult to dissect in human subjects, and cell type–specific knock-out mice in which progranulin is selectively reduced in neurons or microglia will further illuminate the contribution of each cell type to the progression of FTD.
Haploinsufficiency versus complete progranulin deficiency
The early finding that Grn+/− mice lacked many neuropathological abnormalities seen in Grn−/− mice (Ahmed et al., 2010) suggested that Grn−/− mice may be superior to Grn+/− mice for studying FTD. The recent discovery that patients completely lacking progranulin due to homozygous GRN mutations develop a different disease, neuronal ceroid lipofuscinosis (Smith et al., 2012), calls for reevaluating this idea. Complete progranulin deficiency produces a more aggressive disorder affecting completely different brain regions than progranulin haploinsufficiency (the siblings with homozygous GRN mutations developed a syndrome of vision loss, seizures, and cerebellar ataxia in their 20s). Thus, we should expect that some phenotypes of Grn−/− mice will not be applicable to FTD. The lipofuscinosis in Grn−/− mice (Ahmed et al., 2010; Petkau et al., 2012) (Fig. 6) is probably one example. Further work is needed to determine whether the neuroinflammatory changes in Grn−/− mice are mechanistically related to progranulin haploinsufficient FTD, or only to neuronal ceroid lipofuscinosis.
Grn+/− mice have advantages and disadvantages for studying FTD. The lack of TDP-43 pathology in Grn+/− mice makes them an incomplete model of FTD resulting from progranulin deficiency and limits their use for addressing how progranulin impacts TDP-43. However, this is not an irredeemable weakness. There are now several examples of dissociation between the functional effects of proteins involved in neurodegenerative disease and their neuropathological aggregates, including β-amyloid plaques (Mucke et al., 2000), tau tangles (Santacruz et al., 2005), and huntingtin inclusion bodies (Arrasate et al., 2004). Thus, the lack of TDP-43 aggregates does not necessarily preclude using Grn+/− mice to study how progranulin haploinsufficiency produces social and emotional dysfunction, which should be an important goal for future research.
The chief advantage of Grn+/− mice over Grn−/− mice is that one progranulin allele remains intact, which may enable their use for evaluating potential therapeutics. Increasing progranulin levels from the normal allele is perhaps the most straightforward strategy for treating FTD resulting from GRN mutations. Several progranulin expression-raising compounds have been identified, including amiodarone and the histone deacetylase inhibitor, SAHA (Capell et al., 2011; Cenik et al., 2011), which are both already FDA-approved for other indications. It may also be possible to target specific miRNAs to increase progranulin levels (Jiao et al., 2010; Wang et al., 2010). The presence of multiple functional outcome measures relevant to FTD in Grn+/− mice may make them a useful tool for preclinical studies of potential therapies for treating patients with progranulin mutations.
Footnotes
The work was supported by the Consortium for Frontotemporal Dementia Research (E.D.R., E.J.H., F.-B.G., and R.V.F.), the National Institutes of Health (Grants NS054811 and NS075487 to E.D.R., Grant AG023501 to R.V.F., Grant AG034793 to L.H.M., and Grant NS057098 to the Alabama Neuroscience Blueprint Core), and the Cellular & Synaptic Physiology and Cellular & Molecular Neuropathology Cores of the Alabama Neuroscience Blueprint Core at University of Alabama Birmingham (with particular thanks to Jing Wang, Felecia Hester, and David Sweatt). We thank James Black, Dheepa Sekar, and Miriam Roberson for colony maintenance and tissue processing; Zhiyong Li, Alicia Hall, and Andrew Arrant for help with behavior; and Laura Mitic for comments on an earlier draft of the manuscript.
The authors declare no competing financial interests.
References
- Adolphs R. What does the amygdala contribute to social cognition? Ann N Y Acad Sci. 2010;1191:42–61. doi: 10.1111/j.1749-6632.2010.05445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bickart KC, Wright CI, Dautoff RJ, Dickerson BC, Barrett LF. Amygdala volume and social network size in humans. Nat Neurosci. 2011;14:163–164. doi: 10.1038/nn.2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch OJ, Neumann ID. Vasopressin released within the central amygdala promotes maternal aggression. Eur J Neurosci. 2010;31:883–891. doi: 10.1111/j.1460-9568.2010.07115.x. [DOI] [PubMed] [Google Scholar]
- Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, Gijselinck I, Bittner T, Carlson AM, Sasse F, Kunze B, Steinmetz H, Jansen R, Dormann D, Sleegers K, Cruts M, Herms J, Van Broeckhoven C, Haass C. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Dewey CM, Xian X, Wei S, Yu K, Niu W, Coppola G, Coughlin SE, Lee SE, Dries DR, Almeida S, Geschwind DH, Gao FB, Miller BL, Farese RV, Jr, Posner BA, Yu G, Herz J. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. 2011;286:16101–16108. doi: 10.1074/jbc.M110.193433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapcote SJ, Roder JC. Deletion polymorphism of Disc1 is common to all 129 mouse substrains: implications for gene-targeting studies of brain function. Genetics. 2006;173:2407–2410. doi: 10.1534/genetics.106.060749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45:395–408. doi: 10.1016/j.nbd.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat. 2008;29:1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- Hoefer M, Allison SC, Schauer GF, Neuhaus JM, Hall J, Dang JN, Weiner MW, Miller BL, Rosen HJ. Fear conditioning in frontotemporal lobar degeneration and Alzheimer's disease. Brain. 2008;131:1646–1657. doi: 10.1093/brain/awn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Herl LD, Farese RV, Gao FB. MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS One. 2010;5:e10551. doi: 10.1371/journal.pone.0010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, Eisenhut RJ, Martens LH, Nakamura A, Huang A, Bagley JA, Zhou P, de Luis A, Neukomm LJ, Cabello J, Farese RV, Jr, Kenyon C. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci U S A. 2011;108:4441–4446. doi: 10.1073/pnas.1100650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai R, Iwakura Y, Nishihara M. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain Res. 2007;185:110–118. doi: 10.1016/j.bbr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Kipps CM, Nestor PJ, Acosta-Cabronero J, Arnold R, Hodges JR. Understanding social dysfunction in the behavioural variant of frontotemporal dementia: the role of emotion and sarcasm processing. Brain. 2009;132:592–603. doi: 10.1093/brain/awn314. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM. Pathological staging of frontotemporal lobar degeneration. J Mol Neurosci. 2011;45:379–383. doi: 10.1007/s12031-011-9528-0. [DOI] [PubMed] [Google Scholar]
- LeDoux J. The emotional brain, fear, and the amygdala. Cell Mol Neurobiol. 2003;23:727–738. doi: 10.1023/A:1025048802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J. The amygdala. Curr Biol. 2007;17:R868–R874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lindzey G, Winston H, Manosevitz M. Social dominance in inbred mouse strains. Nature. 1961;191:474–476. doi: 10.1038/191474a0. [DOI] [PubMed] [Google Scholar]
- Liu W, Miller BL, Kramer JH, Rankin K, Wyss-Coray C, Gearhart R, Phengrasamy L, Weiner M, Rosen HJ. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology. 2004;62:742–748. doi: 10.1212/01.wnl.0000113729.77161.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114:49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Retrograde abolition of conditional fear after excitotoxic lesions in the basolateral amygdala of rats: absence of a temporal gradient. Behav Neurosci. 1996;110:718–726. doi: 10.1037//0735-7044.110.4.718. [DOI] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, Farese RV., Jr Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–3959. doi: 10.1172/JCI63113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya PR, Fox MA, Jensen CL, Laporte JL, French HT, Wendland JR, Murphy DL. Altered 5-HT2C receptor agonist-induced responses and 5-HT2C receptor RNA editing in the amygdala of serotonin transporter knockout mice. BMC Pharmacol. 2011;11:3. doi: 10.1186/1471-2210-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. In: Roberson ED, editor. Alzheimer's disease and frontotemporal dementia: methods and protocols. Totowa, NJ: Humana; 2011. pp. 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, Neal SJ, Orban PC, MacDonald JL, Hill AM, Lu G, Feldman HH, Mackenzie IR, Leavitt BR. Progranulin expression in the developing and adult murine brain. J Comp Neurol. 2010;518:3931–3947. doi: 10.1002/cne.22430. [DOI] [PubMed] [Google Scholar]
- Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, Gregg J, Lu G, Feldman HH, Mackenzie IR, Raymond LA, Leavitt BR. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45:711–722. doi: 10.1016/j.nbd.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Seeley WW, Kim EJ, Gorno-Tempini ML, Rascovsky K, Pagliaro TA, Allison SC, Halabi C, Kramer JH, Johnson JK, Weiner MW, Forman MS, Trojanowski JQ, Dearmond SJ, Miller BL, Rosen HJ. Distinct MRI atrophy patterns in autopsy-proven Alzheimer's disease and frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen. 2007;22:474–488. doi: 10.1177/1533317507308779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED. Mouse models of frontotemporal dementia. Ann Neurol. 2012;72:837–849. doi: 10.1002/ana.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen HJ, Pace-Savitsky K, Perry RJ, Kramer JH, Miller BL, Levenson RW. Recognition of emotion in the frontal and temporal variants of frontotemporal dementia. Dement Geriatr Cogn Disord. 2004;17:277–281. doi: 10.1159/000077154. [DOI] [PubMed] [Google Scholar]
- Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scearce-Levie K. Monitoring spatial learning and memory in Alzheimer's disease mouse models using the Morris water maze. In: Roberson ED, editor. Alzheimer's disease and frontotemporal dementia: methods and protocols. Totowa, NJ: Humana; 2011. pp. 191–205. [DOI] [PubMed] [Google Scholar]
- Scearce-Levie K, Roberson ED, Gerstein H, Cholfin JA, Mandiyan VS, Shah NM, Rubenstein JL, Mucke L. Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 2008;7:344–354. doi: 10.1111/j.1601-183X.2007.00357.x. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, Syed NM, Lai Y, Lin EA, Kong L, Su J, Yin F, Ding AH, Zanin-Zhorov A, Dustin ML, Tao J, Craft J, Yin Z, Feng JQ, Abramson SB, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332:478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia L, Milnerwood A, Guo A, Mills F, Yoshida E, Vasuta C, Mackenzie IR, Raymond L, Cynader M, Jia W, Bamji SX. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci. 2011;31:11126–11132. doi: 10.1523/JNEUROSCI.6244-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Miller BL. New approaches to the treatment of frontotemporal lobar degeneration. Curr Opin Neurol. 2008;21:708–716. doi: 10.1097/WCO.0b013e328318444d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Wilfred BR, Madathil SK, Tang G, Hu Y, Dimayuga J, Stromberg AJ, Huang Q, Saatman KE, Nelson PT. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am J Pathol. 2010;177:334–345. doi: 10.2353/ajpath.2010.091202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner KH, Roberts NA, Rosen HJ, Dean DL, Kramer JH, Weiner MW, Miller BL, Levenson RW. Emotional reactivity and emotion recognition in frontotemporal lobar degeneration. Neurology. 2007;69:148–155. doi: 10.1212/01.wnl.0000265589.32060.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, Ding A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010a;24:4639–4647. doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010b;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, Kramer JH, Weiner M, Miller BL, Seeley WW. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain. 2010;133:1352–1367. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]